
Abstract
Injury to the brain (e.g., stroke) results in a disruption of neuronal connectivity and loss of fundamental sensori-motor functions. The subsequent recovery of certain functions involves structural rearrangements in areas adjacent to the infarct. This remodeling of the injured brain requires trafficking of macromolecular components including cholesterol and phospholipids, a transport carried out by apolipoproteins including apolipoprotein D (apoD). We investigated the changes in the levels of apoD mRNA and protein, and its cellular localization during a recovery period up to 30 days after experimental stroke in the rat brain. In the core of the brain infarct, apoD immunoreactivity but not mRNA increased in dying pyramidal neurons, indicative of cellular redistribution of lipids. During 2 to 7 days of recovery after stroke, the apoD levels increased in the peri-infarct and white matter areas in cells identified as mature oligodendrocytes. The apoD expressing cells were conspicuously located along the rim of the infarct, suggesting a role for apoD in tissue repair. Furthermore, housing animals in an enriched environment improved sensori-motor function and increased the apoD levels. Our data strongly suggest that apoD is involved in regenerative processes and scar formation in the peri-infarct area presumably by enhancing lipid trafficking.
Keywords
Introduction
Stroke leads to dysfunction and death of brain tissue, which disrupts neuronal circuitry and cell signaling in brain areas adjacent and remote to damaged tissue (Carmichael, 2006; Wieloch and Nikolich, 2006). During recovery after stroke, brain repair and plasticity partially restore neurologic functions. This post-stroke recovery has been attributed to structural and functional adaptations of the injured brain involving activation of alternative neuronal pathways and axonal sprouting (Carmichael, 2006; Dancause et al, 2005). In the experimental setting, post-stroke recovery can be enhanced by housing rats or mice in cages enriched with toys and several rodent cage mates (Johansson and Ohlsson, 1996; Nygren and Wieloch, 2005). The enhanced recovery by the enriched environment has been attributed to dendritic arborizations, increased spine density and is associated with dynamic changes of genes encoding synaptic proteins and growth factors (Johansson, 2004; Keyvani et al, 2004).
Cellular remodeling and growth require synthesis and supply of critical macromolecular components, including lipids for membrane and myelin formation such as cholesterol. In this context, lipoproteins serve as carriers for the delivery of lipids (Posse De Chaves et al, 2000). Moreover, lipid components (cholesterol) released by degenerating axons are reutilized for regeneration via a lipoprotein-mediated shuttling of lipids involving apolipoproteins (Boyles et al, 1989; Goodrum et al, 1994). Furthermore, impaired lipoprotein-derived export of cholesterol impedes remyelination after nerve crush injury (Goodrum and Pentchev, 1997). The role of apolipoproteins after central nervous system (CNS) injury has not been well characterized.
Apolipoprotein D belongs to the lipocalin family of proteins involved in the transport of hydrophobic substances including cholesterol, heme-related molecules, and arachidonic acid (Rassart et al, 2000). In the CNS, apoD expression is mainly localized to oligodendrocytes and neuronal populations (Ong et al, 1997; Rassart et al, 2000). Apolipoprotein D mRNA is elevated at sites of regenerating peripheral nerves (Boyles et al, 1990; Spreyer et al, 1990), and CNS injury increases expression of apoD mRNA and protein levels (Franz et al, 1999). Moreover, gene profiling analysis of ischemic brain tissue showed upregulation of genes related to lipid transport, including ApoD (Rickhag et al, 2006). Furthermore, recent studies have shown that overexpression of an apoD homolog confers stress resistance and enhanced lifespan while loss of function have opposite effects suggesting that apoD may play a protective role (Sanchez et al, 2006; Walker et al, 2006).
The aim of this study was to investigate the mRNA and protein levels of apoD in the late post-stroke recovery period in the rat subjected to middle cerebral artery (MCA) occlusion (MCAO). To determine whether changes in the levels of apoD correlated with functional recovery, the mRNA expression and protein levels of apoD were assessed in the brain of rats housed in normal cages during the post-stroke period with those housed in an enriched environment.
Materials and methods
Transient Middle Cerebral Artery Occlusion
Adult male Wistar rats (Harlan, Netherlands) were housed under diurnal light conditions (12 h light/12 h dark cycle) with free access to food and water. All experiments were approved by the ethical committee at Lund University. The rats were subjected to middle cerebral artery occlusion (MCAO) for 2 h using the intraluminal filament model as described previously (Rickhag et al, 2006). Briefly, rats were anaesthetized by inhalation of 3.5% halothane in O2/N2O (30:70). During surgery, anesthesia was maintained by insufflation of 1.5% halothane through a nose mask. A catheter was placed in the tail artery to monitor blood gas parameters and a rectal probe was inserted to control the body temperature. The common carotid artery was exposed by an incision in the middle of the neck and the internal and external artery was made accessible. Ligation of the external carotid artery was followed by a small incision in the common carotid close to the bifurcation. A filament was introduced into the internal carotid and advanced until it blocked the origin of the MCA. This was followed by recovery of the animals from anesthesia during the occlusion period. Reperfusion was initiated 2 h later by retraction of the filament. All physiologic parameters of animals included in the study were within normal ranges and included animals showed typical ischemia-induced behavioral deficits, rotational asymmetry, and dysfunctional limb placement. After various time points of recovery (1, 2, 4, 7, 14, and 30 days, n = 3 for all time points), the animals were perfusion-fixed and processed for immunohistochemistry. Sham-operated animals served as controls.
Permanent Middle Cerebral Artery Occlusion
Spontaneous hypertensive rats (Møllegaard Breeding Center, Denmark) were subjected to permanent distal MCAO and at 48 h of recovery placed in either standard cages or in an enriched environment for 12 days (Ohlsson and Johansson, 1995). Fourteen days after ischemia, sensori-motor functions were tested using the rotating pole test followed by decapitation. Brains were then processed for quantitative real-time polymerase chain reaction (PCR) analyses and Western blotting.
In Situ Hybridization Histochemistry
Spontaneous hypertensive rats were subjected to permanent MCAO (pMCAO) and killed at various time points of recovery (2, 3, 7, 12, and 30 days, n = 5 for each time point). Isolated brains were cut on a cryostat in coronal sections and mounted on Superslide glasses (Super-Frost®Plus, Menzel-Gläser, Germany). After fixation for 15 mins with 4% paraformaldehyde, sections were rinsed in phosphate-buffered saline and dehydrated in ethanol. An oligonucleotide specific to apoD mRNA (Rickhag et al, 2006) was 3′-labeled with α-[35S]dATP using terminal deoxynucleotidyl transferase (Amersham Biosciences, Uppsala, Sweden). Hybridization was performed in a humidified chamber for 18 h at 42°C using a hybridization cocktail containing 50% formamide and 107 c.p.m./mL of labeled oligonucleotide. After hybridization, the sections were washed at 55°C in 1 × SSC for 3 × 15 mins followed by 1 × SSC at room temperature for 15 mins. Furthermore, the sections were rinsed in water and dehydrated in ethanol. The radioactive slides were then exposed to BioMax MR films (Kodak) and the intensity from the autoradiograms was photographed.
Real-Time Quantitative Polymerase Chain Reaction Assay
Spontaneous hypertensive rats subjected to pMCAO and 12 days of housing in standard (n = 4) or enriched (n = 4) environment were killed. From each brain, infarct core including white matter areas (corpus callosum) was dissected and total RNA was extracted using RNeasy Lipid Tissue Kit (Qiagen Sciences, MD, USA) in accordance with manufacturer's instructions. Spectrophotometric analysis and agarose gel electrophoresis was used to assess integrity and quality of isolated RNA. One microgram of RNA was reverse-transcribed using a Superscript II reverse transcription kit (Invitrogen, CA, USA). The 20-μL transcription mix for each sample contained 1.0 μg total RNA, 4 μL 5 × first-strand buffer, 2 μL dithiothreitol (0.1 mol/L), 1 μL dNTP mix (10 mmol/L), 1 μL oligo-(dT)12 to 18 primer (0.5 μg/μL), and 1 μL Superscript II (Invitrogen, 200 units/μL). Total RNA plus primer and dNTP mix were heated to 65°C for 5 mins and quickly chilled on ice. First-strand buffer and dithiothreitol were added to the reaction mixture and then incubated at 42°C for 2 mins. This was followed by addition of Superscript II and incubation at 42°C for 50 mins. To inactivate reaction, the reaction mixture was then heated to 70°C for 15 mins.
TaqMan PCR assay was performed using the ABI Prism 7700 sequence detection system (Applied Biosystems Inc.). The PCR reaction for each mRNA was performed in triplicate (ApoD) and duplicate (GAPDH) on cDNA samples in a 96-well plate (MicroAmpTM, Applied Biosystems). The reaction mixture (25 μL) consisted of diluted cDNA aliquots, 900 nmol/L of forward and reverse primers (TAG Copenhagen, Denmark), 200 nmol/L of the TaqMan probe (TAG Copenhagen, Denmark) and 1 × TaqMan® Universal PCR Master Mix (Applied Biosystems). Polymerase chain reaction amplification was performed according to the following protocol: 2 mins incubation at 50°C and then heated to 95°C for 10 mins, followed by 40 cycles of 15 secs at 95°C and 1 min at 60°C. Relative changes in gene expression were determined by using the 2-ΔΔCt method (Livak and Schmittgen, 2001). All expression values were normalized to the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) because its mRNA levels do not vary after focal ischemia (Carmichael et al, 2005; Medhurst et al, 2000). The primers and TaqMan probe for apoD were designed in the software Primer Express in accordance with the accompanying guidelines. The oligonucleotide sequences were designed to span the junction of two adjoining exons to avoid contamination of genomic DNA. In addition, the primers were designed as close as possible to the 3′-coding region of the target gene. To confirm absence of genomic contamination, the PCR products were run on an agarose gel to confirm that the amplified product had the proper size (amplified product for apoD: 76 bp). The forward and reverse primers and TaqMan probe for GAPDH was from the article (Medhurst et al, 2000) and the following sequences were used for apoD, FW: 5′-CGGTGGCATCAACGAGAAG-3′, RV: 5′-TTGAGGGTGAAGCCAAACAGA-3′ and TaqMan probe: 5-'FAM-TCCAGCTTGGCTGGCTCTGACATGTT-TAMRA-3′. The TaqMan probes for apoD and GAPDH were synthesized with the fluorescent reporter dye FAM (6-carboxy-fluorescein) attached to the 5′ end and a quencher dye TAMRA (6-carboxy-tetramethyl-rhodamine) to the 3′ end.
Immunohistochemistry
At 1, 2, 4, 7, 14, and 30 days of recovery, rats subjected to tMCAO were anaesthetized and transcardially perfused with physiologic saline followed by buffered 4% paraformaldehyde. The brains were isolated, post-fixed in paraformaldehyde overnight, and then placed in 25% sucrose solution. In a vibratome, coronal sections (30 μm) were generated and these free-floating sections were stored in a cryoprotective solution kept at −20°C. For bright-field immunohistochemistry, a standard peroxidase-based method using 3,3′-diaminobenzidine (DAB) was applied. Briefly, free-floating sections were rinsed in phosphate-buffered saline followed by incubation in 3% H2O2 and 10% methanol to remove endogenous peroxidase activity. After preincubation with 5% normal swine serum using 0.25% Triton X-100 in phosphate-buffered saline for 1 h, sections were incubated with rabbit polyclonal apoD antibody (Ong et al, 2002) at a dilution of 1:5,000 overnight. The second day, sections were incubated with biotinylated swine anti-rabbit secondary antibody (1:200; Dako Cytomation, Glostrup, Denmark) for 85 mins followed by incubation with avidin—biotin complex (Vector Laboratories, Burlingame, CA, USA) for 1 h. By using 3,3′-diaminobenzidine as a chromogen, the reaction was visualized and further enhanced by NiCl2. After being rinsed, sections were mounted, air-dried overnight, dehydrated, immersed in xylene, and finally coverslipped using Pertex (Histolab, Gothenburg, Sweden).
Phenotypic characterization of cells expressing apoD was performed using dual-labeling immunofluorescence. For colocalization of apoD with neurons, sections were incubated with primary antibodies for NeuN (mouse monoclonal 1:100; Chemicon, Temecula, CA, USA) and apoD (rabbit polyclonal 1:2,000). For colocalization of apoD with mature oligodendrocytes, sections were incubated with primary antibodies for GSTπ (mouse monoclonal 1:100; BD Biosciences) and apoD (rabbit polyclonal 1:2,000). GSTπ is a marker for mature oligodendrocytes (Tansey and Cammer, 1991). The sections were incubated with primary antibodies (NeuN/apoD or GSTπ/apoD) overnight in phosphate-buffered saline containing 5% normal swine serum and 5% normal goat serum with 0.25% Triton X-100. On the second day, sections were rinsed and incubated with Cy3-conjugated goat anti-mouse (1:200; Jackson ImmunoResearch Laboratories, USA) and biotinylated swine anti-rabbit secondary antibody (1:200; Dako Cytomation). Then, sections were incubated with Alexa 488-conjugated avidin (1:200; Invitrogen, Molecular Probes, Eugene, Oregon, USA) and finally mounted and coverslipped with PVA-DABCO (Sigma-Aldrich AB, Stockholm, Sweden) to preserve the fluorophores. Analysis was performed with a fluorescence microscope (Leica BX60) with a FITC/CY3 filter at × 40 magnification. Three-dimensional reconstructions from fluorescent micrographs were obtained using a LSM 510 confocal microscope (Zeiss, Jena, Germany).
Cell Counting Analysis
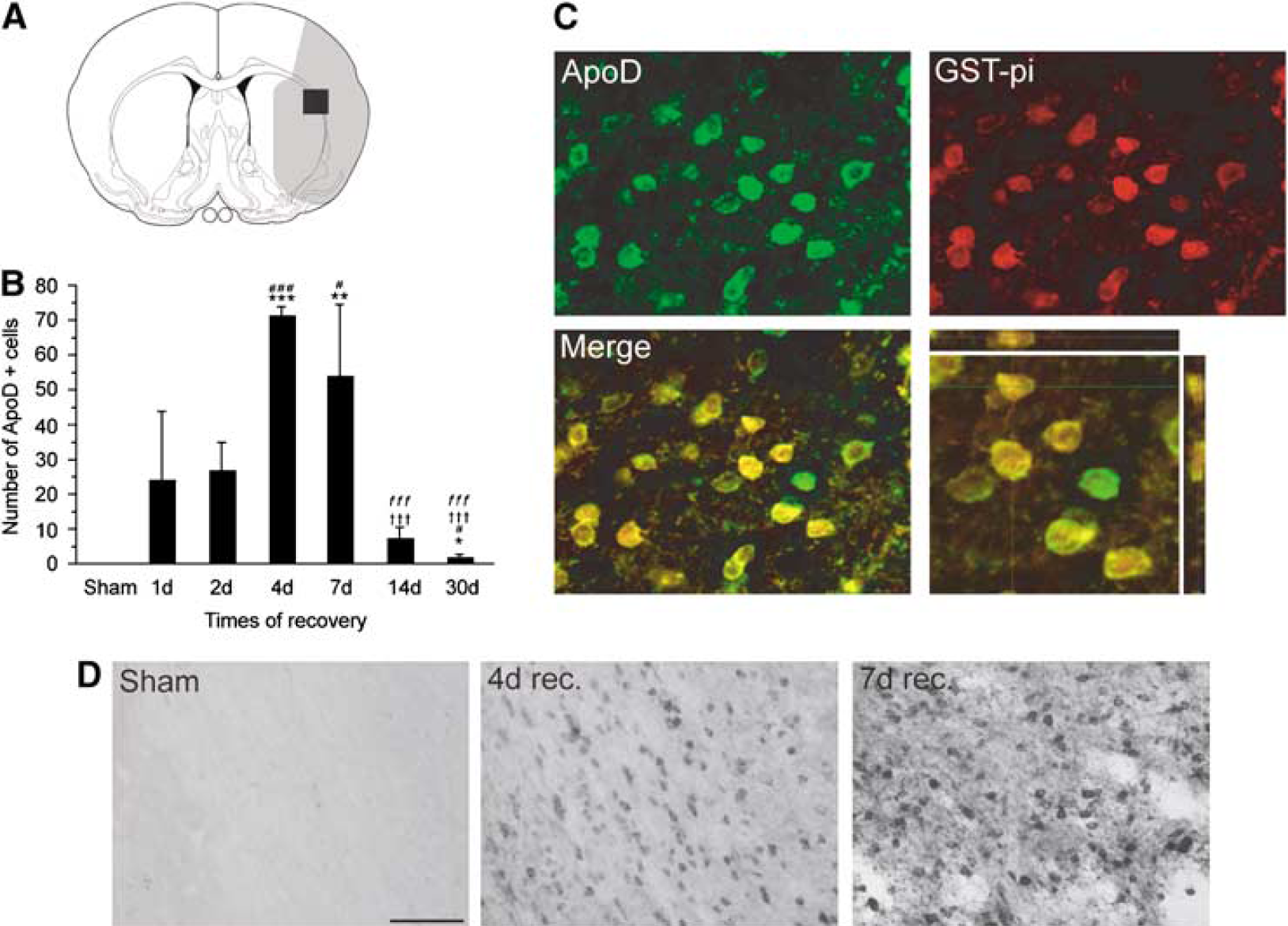
Quantification of apoD+ cells was analyzed at a coronal level corresponding to the center of the striatum (0.2 mm anterior of bregma). The number of apoD+ cells was counted in the lateral part of the corpus callosum ipsilateral to the damage (Figure 4A). Cells with morphologic features identical to cells double-positive for apoD and GSTπ (Figures 2A to 2C) were counted.
Accumulation of apoD+ cells in the lateral part of the corpus callosum at multiple time points of recovery after 2 h transient MCAO. (
Western Blotting
Spontaneous hypertensive rats were subjected to pMCAO. Two days after the ischemia, animals were placed in enriched (n = 3) or standard environment (n = 3) for 12 days, after which animals were killed. The frontal pole of each animal was dissected out, which included peri-infarct area and white matter. Unoperated animals from both standard (n = 2) and enriched (n = 2) environment served as control in this experimental setup. Tissue samples were harvested in cell lysis buffer (20 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerolphosphate, 1 mmol/L Na3VO4, 1 μg/mL leupeptin, and 1 mmol/L phenylmethylsulphonyl fluoride). After incubation on ice for 15 mins and centrifugation at 18,000 g at 4°C for 10 mins, whole-protein concentrations were determined by the Bradford assay (Pierce, IL, USA) using bovine serum albumin as a standard. Lysates were diluted in sodium dodecyl sulfate sample buffer (final concentrations: 62.5 mmol/L Tris—HCl (pH 6.8), 2% sodium dodecyl sulfate, 10% glycerol, 50 mmol/L dithiothreitol, 0.1% bromphenol blue) and the mixture was boiled for 5 mins. Ten micrograms of protein were separated on a 10% sodium dodecyl sulfate polyacrylamide gel. Blocking was performed onto polyvinyldifluoride membranes using blocking buffer (20 mmol/L Tris, 136 mmol/L NaCl, pH 7.6, 0.1% Tween 20, and 5% nonfat dry milk), and detected using primary monoclonal antibody against the apoD (Ong et al, 2002) (diluted 1:2,000). After incubation overnight at 4°C, signals were obtained by binding of a secondary anti-rabbit horseradish peroxidase-linked antibody (New England Biolabs, Sweden; diluted 1:3,000) and visualized by exposing the membrane to a CCD camera (Raytest, Gmbh, Germany) using a chemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK).
Membranes were stripped in respective buffer (62.5 mmol/L Tris (pH 6.7), 100 mmol/L β-mercaptoethanol, 2% sodium dodecyl sulfate) at 70°C for 30 mins and reprobed for α-tubulin (clone B-5-1-2; Sigma, Steinheim, Germany; diluted 1:10,000). Immunoblotting was performed as described above using an anti-mouse horseradish peroxidase-linked secondary antibody (Cell Signaling, MA, USA; diluted 1:2,000). After densitometric analysis, apoD expression was calculated as percentage of α-tubulin expression assumed to be stable in all treatment groups.
Rotating Pole Test
Sensori-motor function of ischemia-lesioned animals was evaluated by the ability to traverse a rotating pole. The rotating pole test assesses coordination and integration of movements as described previously (Ohlsson and Johansson, 1995). The pole (length 1,500 mm, diameter 40 mm, and elevation 700 mm) rotates either to the left or to the right at various speeds (3 or 10 rotations per minute). The ability of injured animals to cross this pole was graded according to a scoring system from 6 to 0 assessing behavioral dysfunction: 6—the animal traverses pole without any foot slips; 5—the animal traverses pole with few foot slips; 4—the animal crosses pole with 50% slipping of the foot steps; 3—the animal crosses the pole while jumping with both hindlimbs; 2—the animal falls off during crossing; 1—the animal remains embraced to the pole unable to cross; 0—the animal falls off immediately. According to this scoring system, a normal rat should perform with a scoring of 5 and 6.
Statistics
Expression changes between control and ischemic animals in the real-time quantitative PCR study were statistically analyzed by one-way analysis of variance (ANOVA) with post hoc Bonferroni—Dunn test. The ΔCt for enriched environment animals (n = 4) was compared with the ΔCt for standard housed animals (n = 4) and control animals (n =3) using the 2-ΔΔCt method (Livak and Schmittgen, 2001). A P-value of less than 0.05 was set for statistical significance level. Behavioral performances were analyzed using the non-parametric Mann—Whitney U-test and P < 0.05 was considered significant. For the cell counting analysis, one-way ANOVA with post hoc Fisher's test was used and again the level of significance was set to P < 0.05.
Results
Apolipoprotein D mRNA Levels Increase in the Rat Brain After Permanent Middle Cerebral Artery Occlusion
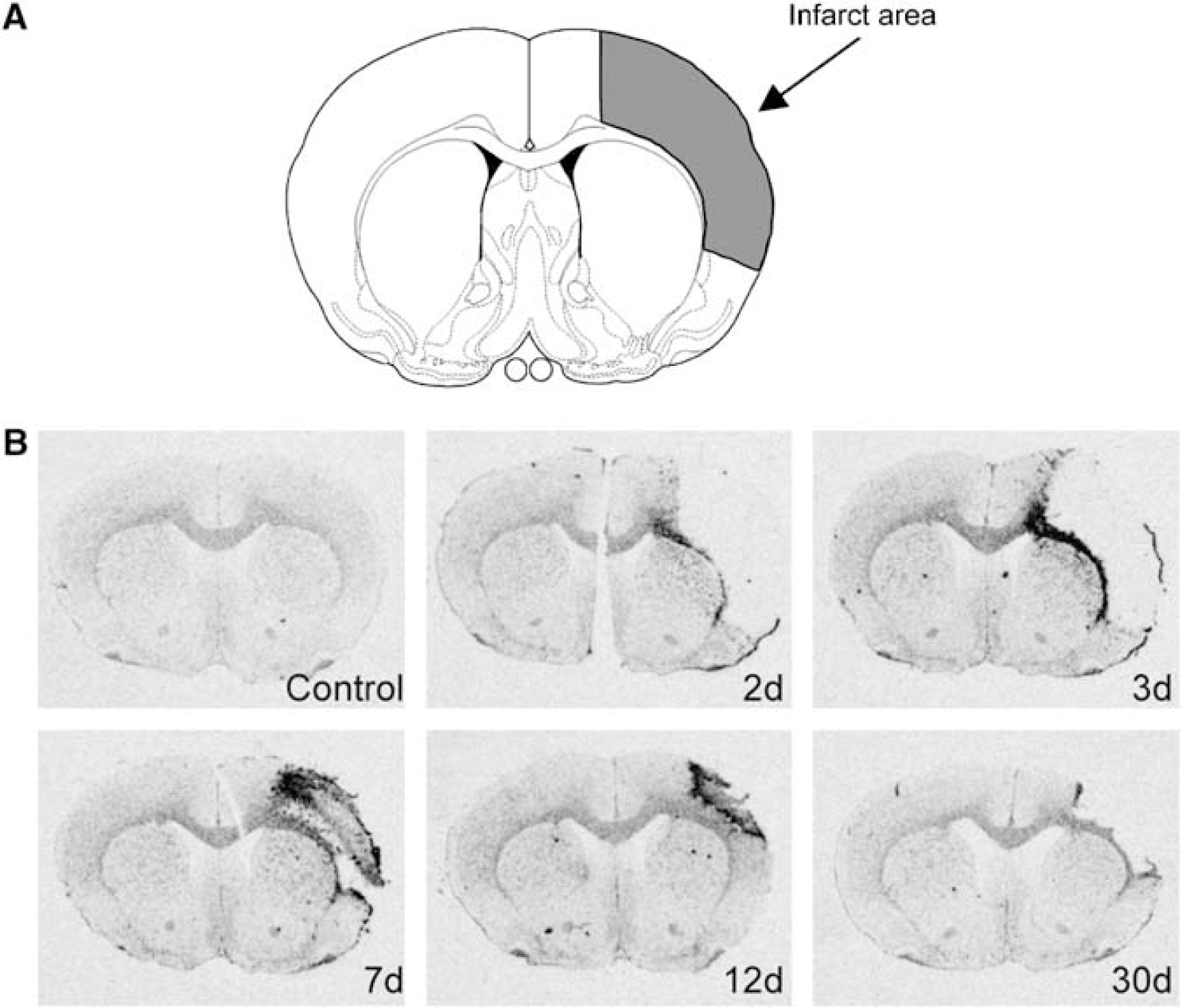
Permanent MCAO resulted in a robust cortical infarction (Figure 1A). In unoperated control animals, apoD mRNA expression was observed in the white matter areas, especially the corpus callosum, whereas low expression was found in the cortico-striatal areas. After MCAO, increased mRNA levels for apoD were observed in the ipsilateral corpus callosum and peri-infarct area at 2 and 3 days after the insult (Figure 1B). Later, after 1 week of recovery, the upregulation of apoD mRNA was now evident in an area around the infarct, whereas expression decreased in the subjacent white matter (corpus callosum). The mRNA signal in the infarct zone was attenuated by 12 days of recovery, and now demarcated the border between brain tissue and the infarct cavity. After 30 days of recovery, only a minor increase of apoD mRNA was observed in the peri-infarct area compared with control animals. Three animals in each experimental group were analyzed and showed similar changes in the distribution of apoD mRNA.
Distribution of Apolipoprotein D Immunoreactivity in Intact Brain
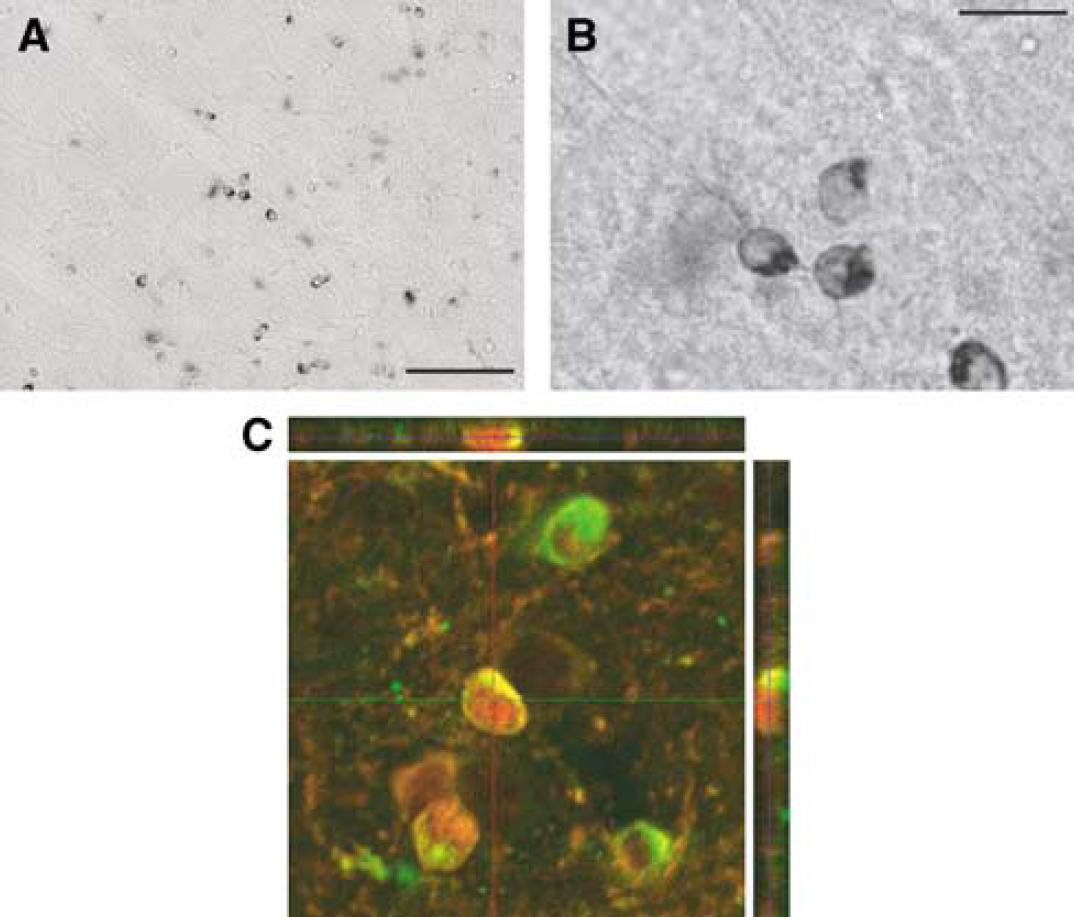
In sham-operated animals, a scattered distribution of apoD immunoreactive (apoD-ir) cells was found in all layers of the neocortex (Figure 2A). In these cells, immunoreactivity was located in the cytoplasm (Figure 2B). In addition, perivascular cells (pia mater) showed high immunoreactivity while only low levels were seen in white matter areas including the corpus callosum. In cortex, apoD-ir was typically localized to the perinuclear areas and close to the plasma membrane (Figure 2B). The cells were often arranged in beadlike rows and sometimes in pairs.
The majority of these cells co-expressed GSTπ, indicative of mature oligodendrocytes (Tansey and Cammer, 1991), (Figure 2C). Sporadic expression of apoD was found in S100β+ cells (data not shown). S100β has been used as a marker of astrocytes but is also expressed transiently in the oligodendroglial lineage (Deloulme et al, 2004). Furthermore, phenotypic characterization of apoD+ cells showed no colocalization for apoD with glial fibrillary acidic protein (marker of astrocytes) or NG2 (marker of pericytes/oligodendrocyte precursors).
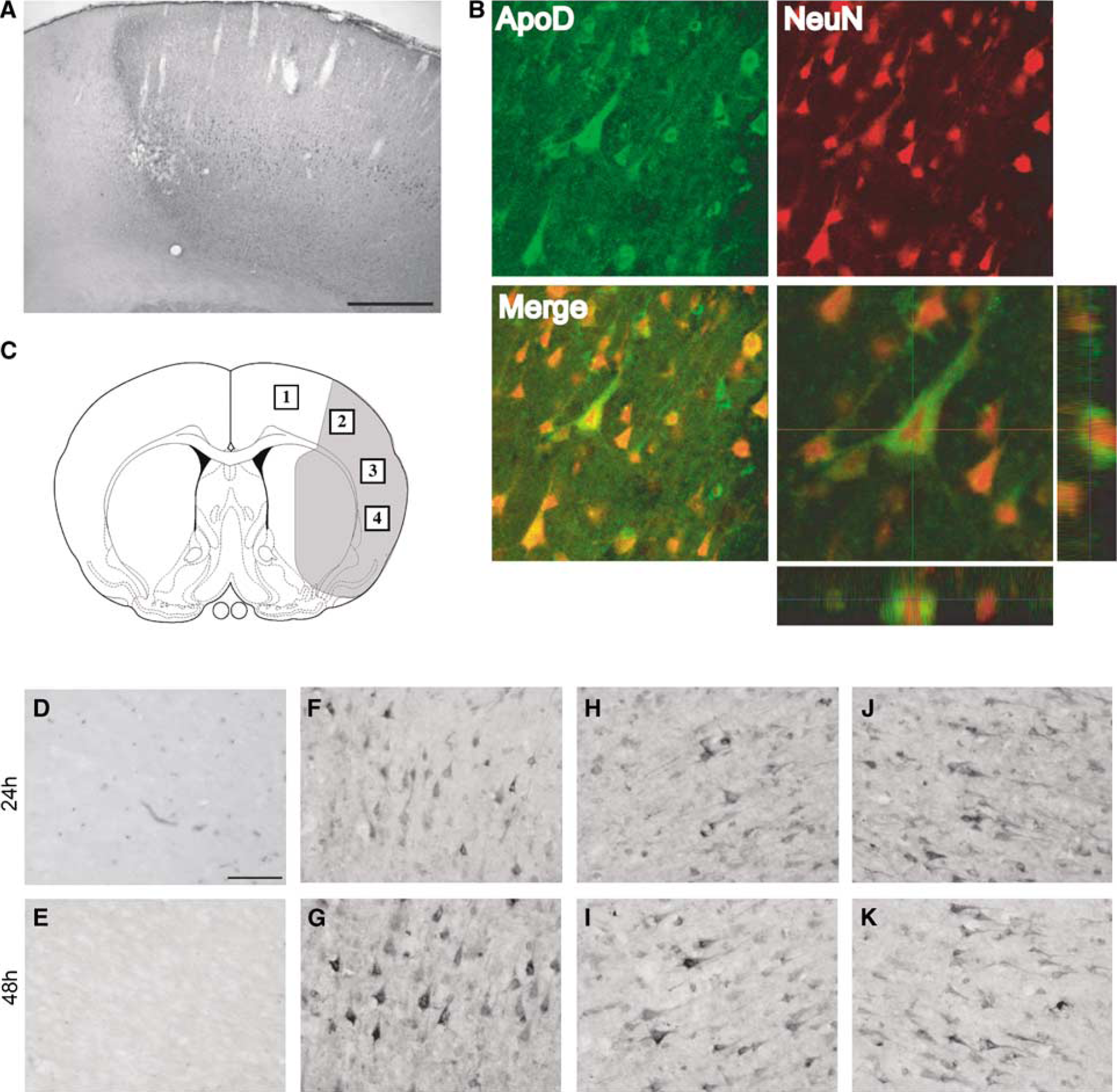
Apolipoprotein D Immunoreactivity is Increased in Neurons After Transient Middle Cerebral Artery Occlusion
At the end of 2 h tMCAO, apoD levels were elevated and found in neurons and glial cells. Twenty-four and forty-eight hours after tMCAO, a dramatic increase of apoD+ cells co-localized with NeuN was found in cortex ipsilateral to the occlusion, in areas that appears to develop into the final infarct (Figures 3A and 3B). These apoD+/NeuN+ cells were triangular shaped and located in the cortical layers III and IV. No apoD-ir was detectable in ipsilateral unaffected neurons of the frontal-cingulate cortex or in the contralateral cortex.
The distribution of apoD+ cortical neurons from the peri-infarct area and different subregions of the MCA territory is presented in Figure 3. In the non-ischemic areas, no apoD+ neurons were observed after 24 and 48 h of recovery (Figures 3D and 3E). A robust increase in apoD-ir was observed in the peri-infarct area (penumbra) (Rickhag et al, 2006) (Figures 3F and 3G). Already 24 h after tMCAO, a clear increase was detectable and by 48 h this neuronal expression of apoD was further enhanced. The neurons had a preserved cellular integrity with a rounded nucleus that indicates a normal cellular structure. Furthermore, apical dendrites of a few pyramidal neurons were stained positive for apoD.
In contrast, the majority of apoD+ neurons from selected infarct core regions appeared collapsed with condensed pyknotic somata and condensed nuclei, still staining strongly for apoD (Figures 3H to K).
The Number of apoD+ Oligodendrocytes Increases in the Peri-Infarct Area
Compared with sham-operated animals, a significant increase of apoD+, mature oligodendrocytes were detected after tMCAO in the ipsilateral hemisphere. Representative photomicrographs of the time course of expression show the absence of apoD+ cells in the lateral part of corpus callosum in sham-operated animals (Figure 4D). After 4 days of recovery, a profound increase of apoD+ cells was found in corpus callosum that continued until day 7 after tMCAO. Figure 4 shows the number of apoD+ cells (Figure 4B) found in the lateral area of the corpus callosum (Figure 4A) at 1, 2, 4, 7, 14, and 30 days of recovery. As indicated in Figure 4B, an increase in apoD+ cells was already evident in the lateral part of the corpus callosum after 24 and 48 h. A further significant increase was observed in the same area 4 and 7 days after the insult. ApoD was expressed transiently and the number of apoD+ oligodendrocytes decreased at 14 and 30 days after the ischemia. On the basis of colocalization with GSTπ, the majority of apoD+ cells were identified as mature oligodendrocytes (Figure 4C).
Figure 5 shows the spatio-temporal representation of apoD+ GSTπ+ double-labeled cells in the white matter and peri-infarct areas after transient MCAO. The number of these cells increased in the corpus callosum within the first 2 days after the insult. The peak expression was achieved after 4 to 7 days of recovery and involved activation in the entire boundary zone between intact tissue and infarct area. ApoD expression was detectable along the rim of the infarct with a pronounced region in the corpus callosum. By 14 and 30 days after insult, only few apoD+ cells remained in the infarct zone border.
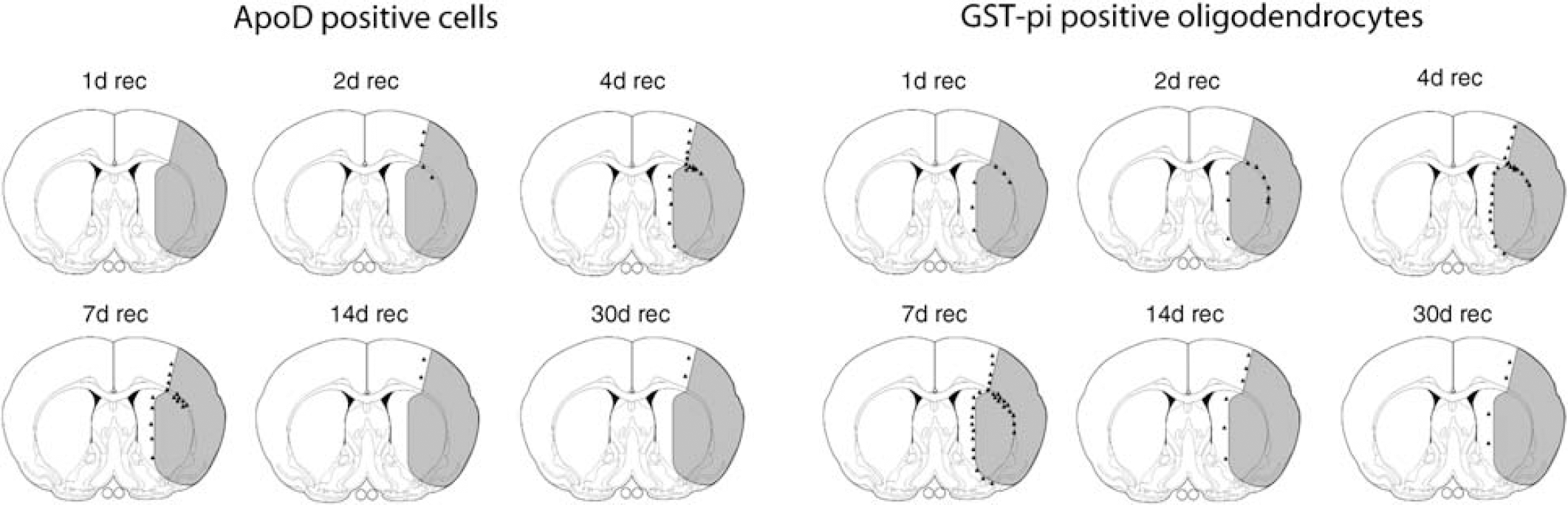
Schematic illustration showing accumulation of apoD+ cells in the white matter and peri-infarct area after transient MCAO (0 to 30 days of recovery). Apolipoprotein D accumulation originates in the lateral part of the corpus callosum and spreads along the boundary zone of the infarct. Peak accumulation occurs after 4 to 7 days of recovery and comprises the entire boundary zone of the infarct. The majority of apoD+ cells colocalized with GSTπ+ cells. The spatial and temporal distribution of GSTπ+ cells correlate with the apoD pattern. A subpopulation of oligodendrocytes expresses apoD in white matter and peri-infarct areas after ischemic brain injury.
Enriched Environment Enhances Functional Recovery and Apolipoprotein D mRNA and Protein Levels After Middle Cerebral Artery Occlusion
To study the correlation between apoD levels and functional recovery after MCAO, rats were subjected to pMCAO and after 48 h of recovery transferred for housing in standard or enriched environment for 12 days (Ohlsson and Johansson, 1995). The coordination and integration of movements were assessed using the rotating pole test at 14 days of recovery. Rats housed in an enriched environment (n = 7) showed improved sensori-motor function compared with rats housed in standard cages (n = 7). Rats housed in enriched environment had a median score of 5 at 10 turns/min of the rotating pole to the left, Figure 6A, and a score of 6 at 10 turns/min to the right, Figure 6B. Rats housed in standard cages had a median score of 3 at either test respectively.
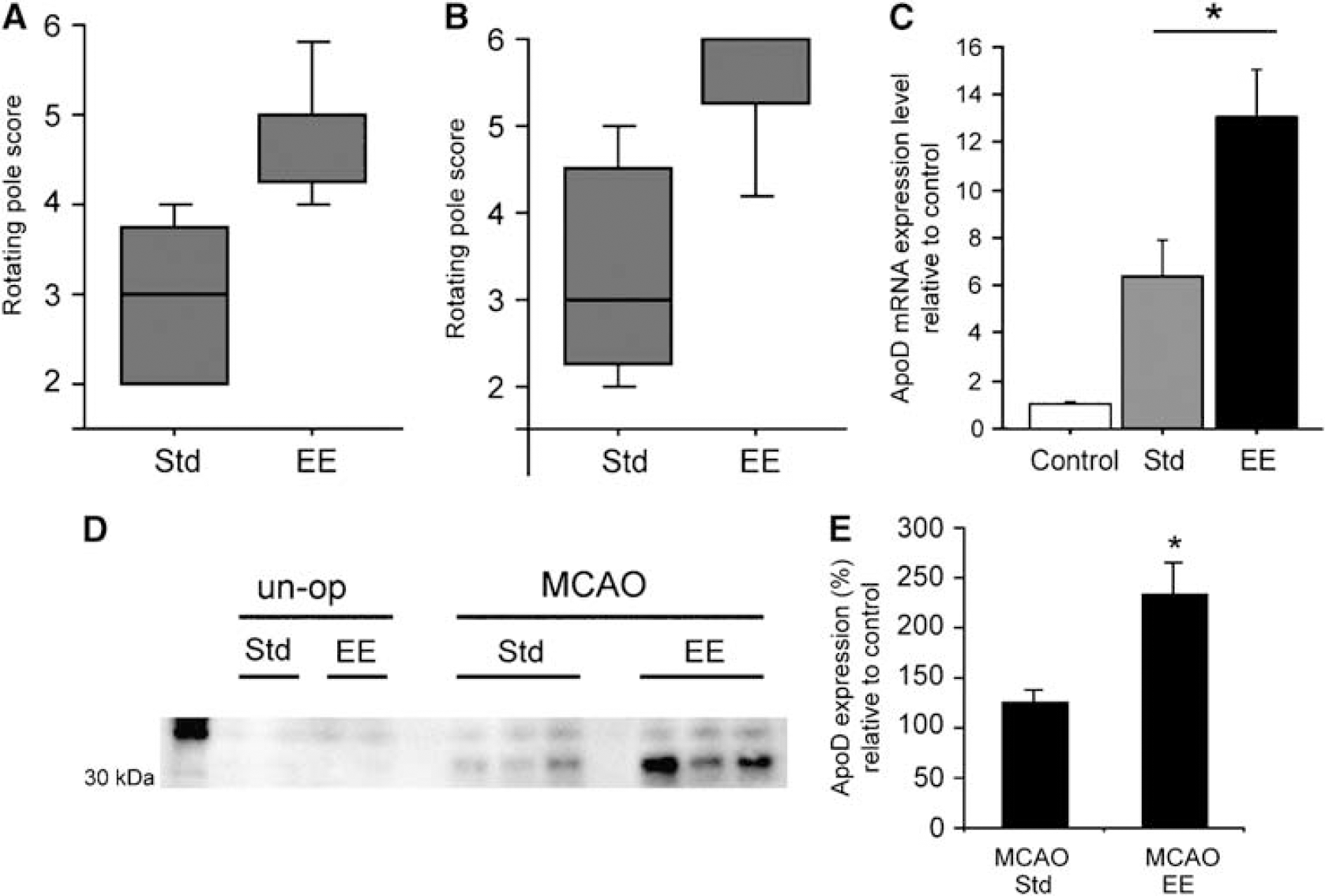
Evaluation of sensori-motor function after brain ischemia and different housing conditions using the rotating pole test. After 14 days of recovery from pMCAO, rats housed in standard (n = 7) or enriched environment (n = 7) were tested on the rotating pole at 10 turns/min to
To investigate the effect of housing conditions on the mRNA levels after pMCAO, the infarct area including the corpus callosum was dissected out from frozen brains. A prominent upregulation of apoD mRNA was detected in the infarct zone by quantitative PCR analysis, confirming the in situ hybridization data (Figure 6C). Importantly, a more than twofold increase in the levels of apoD mRNA was found in ischemic rats housed in an enriched environment (13±2) compared with standard housed rats (6±1.5) relative to control levels (Figure 6C).
Analysis of apoD protein levels with Western blotting yielded similar results. The ipsilateral frontal lobe from control and ischemic animals was dissected including the peri-infarct area and white matter. Figure 6D shows a representative Western blot of samples from unoperated and ischemic animals. Apolipoprotein D protein levels were significantly increased in animals housed in an enriched environment compared with standard housed animals after pMCAO as shown by densitometric analysis.
Discussion
During reperfusion after MCAO, cell death in ischemic tissue, develops rapidly over the first 6 h of reperfusion (Rickhag et al, 2006). Although sparse neuronal degeneration may occur at later time points, the processes in the tissue occurring at 1 day of recovery and onwards, are due to compensatory processes including neuronal plasticity and cell repair as well as inflammation. The dynamic changes seen in the levels and in the cellular distribution of apoD therefore reflects these important post-ischemic events, and we will discuss our findings from this perspective.
Apolipoprotein D After Focal Brain Ischemia
Apolipoprotein D mRNA in control brains was mainly found in white matter corpus callosum and nucleus accumbens as reported earlier (Thomas et al, 2001). We also found that after MCAO, apoD levels increase in the white matter and in the peri-infarct areas at 2 days of recovery, in line with our earlier in situ hybridization findings from experiments using a tMCAO model (Rickhag et al, 2006). After peripheral nerve injury, apoD levels increase significantly in regenerating areas and likely contribute to nerve regeneration (Spreyer et al, 1990). During rat brain development, enhanced expression of apoD coincides with the period of active myelination (Ong et al, 1999). During regeneration, the injured brain cells show features characteristic of early brain development, such as activation of gene programs that promote processes fundamental for adaptation and recovery (Cramer and Chopp, 2000). The increased expression of apoD mRNA takes place concomitant with changes in the expression of Gap-43, and c-jun, genes induced in sequential waves, which correlate with axonal outgrowth after focal ischemia (Carmichael et al, 2005). Axonal regeneration may play a pivotal role in restoration of brain circuitry (Gregersen et al, 2001). The factors activating the apoD gene after focal ischemia are not obvious, but because expression is initiated after the tissue starts degenerating, factors released from degenerating brain tissue may have a strong influence on expression. Apolipoprotein D is expressed in terminally differentiated fibroblasts (Provost et al, 1991), regulated by among other steroids and interleukin-1α. Hence, the increased expression of apoD could be due to neurosteroids and interleukins released from astrocytes/microglia cells during and after stroke (Barone and Feuerstein, 1999). At 3 days of reperfusion, strong apoD expression is induced in the ipsilateral white matter and in the peri-infarct zone. Later, at 7 to 12 days of reperfusion, the expression subsides in the white matter, whereas at the same time increasing in the border zone between the infarct cavity and parenchyma.
In control, sham-operated animals, apoD immunoreactivity is seen in small, scattered cells and is colocalized with the oligodendrocytic marker GSTπ, corroborating earlier findings using electron microscopy (Ong et al, 2002). Neuronal levels of apoD albeit weak, were seen in some neurons in layers II to IV and evident as dots on the cell soma.
Apolipoprotein D in Degenerating Neurons
Prominent apoD immunoreactivity was found in NeuN+ cells that were identified as neurons. The levels increased abruptly at 1 day of reperfusion and peaked at 2 days, but were still seen in few cells at 4 days. Apolipoprotein D was observed in pyramidal neurons restricted to degenerating tissue, but not in the non-ischemic peri-infarct area. Similar findings of increased apoD immunoreactivity have been observed in neurons after kainic acid-induced injury and traumatic brain injury (Franz et al, 1999; Ong et al, 1997). The fact that apoD mRNA is virtually absent at 1 to 2 days of recovery after MCAO (Rickhag et al, 2006) in the areas that will develop an infarct cavity, strongly suggest that the increased levels of apoD in neurons in these regions and at these time points of recovery are due to other processes than de novo synthesis of apoD. From our data, we cannot delineate the source of apoD in neurons, but apoD could have been transferred from glia cells or accumulated in neurons due to changes in intraneuronal lipid trafficking.
The accumulation of apoD in dying neurons suggests an important protective function of the protein, similar to that of apoE (Kamada et al, 2003). Also, a stress-induced expression of apoD in dying neurons supports its role as a scavenger of toxic substances (Rassart et al, 2000). Studies in Drosophila show that deletion of a homolog of apoD, GLaz reduces lifespan and stress resistance, whereas overexpression has the opposite effects (Sanchez et al, 2006; Walker et al, 2006), suggesting a protective role of apoD. Along the same line is the finding that apoD binds toxic arachidonic acid and heme-related products (Rassart et al, 2000). In addition, apoD could be an important source of cholesterol and lipid reutilization in regenerative processes by sequestering these components released by the dying neurons.
Apolipoprotein D in Oligodendrocytes Accumulating in White Matter and Peri-Infarct Areas
The loss of apoD in degenerating neurons over 4 days of recovery was accompanied by an increase in oligodendrocytes residing in white matter and peri-infarct areas. The majority of these cells were mature oligodendrocytes as judged by the colocalization with GSTπ (Tansey and Cammer, 1991). We did not see any colocalization with glial fibrillary acidic protein, but a small population of apoD+ cells was S-100β+. Also we did not see any colocalization with NG2+ cells, suggesting that apoD is mainly increased in mature oligodendrocytes in models of MCAO. A marked accumulation of apoD+ oligodendrocytes was evident by 4 days of recovery, particularly in the lateral part of the corpus callosum along the rim of the entire infarct zone. This pattern of apoD remained at 7 days, but then subsided at 14 and 30 days (Figure 4). Still, at these late time points, some expression of apoD+ cells was found in the boundary zone between intact and damaged tissue, corroborating the location of apoD mRNA, and these cells were all GSTπ+. Factors inducing apoD in mature oligodendrocytes could be inflammatory mediators such as interleukin-1 released from microglia/macrophages (Clausen et al, 2005) and astrocytes (Liberto et al, 2004) in the peri-infarct area.
Oligodendrocytes degenerate in the acute phase after MCAO as demonstrated by increased tau immunoreactivity early after MCAO (Irving et al, 1997). However, in the later recovery phase, they likely contribute to the regenerative response of surviving brain tissue. Oligodendrocytes proliferate and accumulate in the peri-infarct area during the first week after focal ischemia (Gregersen et al, 2001; Mandai et al, 1997). Also, oligodendrocytes localized to the boundary zone at 2 days after MCAO, express cell-cycle proteins. The peri-infarct recruitment of oligodendrocytes is similar to that seen in models of chemically induced de-myelination (Gensert and Goldman, 1997), suggesting that mature oligodendrocytes may directly participate in remyelination or re-engage a developmental program promoting myelination after CNS injury (Cramer and Chopp, 2000).
After peripheral nerve injuries, degenerating axons release their contents of myelin cholesterol and free fatty acids which are sequestered by macrophages and reutilized by Schwann cells during regeneration (Goodrum et al, 1994, 1995). This conservation and recycling of cholesterol and fatty acids are accomplished via a lipoprotein-mediated process involving apoD. We propose that the apoD+ oligodendrocytes in the peri-infarct area may promote repair after ischemic injury by enhancing axonal regeneration and/or scar formation in the border between intact and damaged tissue, by promoting trafficking of cholesterol and lipids necessary for membrane biogenesis including myelin sheaths.
Improved Functional Recovery is Correlated with Increased Levels of Apolipoprotein D
The role of apoD in the recovery process after experimental stroke is highlighted by the findings that both the mRNA and protein levels are robustly increased in white matter and peri-infarct areas of rats housed in an enriched environment. Animals with increased levels of apoD showed improved motor function assessed by the rotating pole test, a test for coordinated sensori-motor function (Nygren and Wieloch, 2005; Ohlsson and Johansson, 1995). Enriched environment stimulates biologic processes associated with remodeling of neuronal circuits in the injured brain, such as changes in spine density, arborizations of dendrites, expression patterns of plasticity genes, and cell genesis (Johansson, 2004). Because lipids are also crucial for synaptogenesis, increased levels of apoD by housing in enriched environment could enhance these remodeling processes. For example, increased trafficking of cholesterol by apoD in the post-ischemic phase may promote a faster tissue recovery by enhancing synaptogenesis or normalizing neuronal transmission, in addition to remyelination (Goritz et al, 2005). Because the apoD+ cells accumulate along the border between the infarct cavity and the intact tissue, apoD may also contribute to scar formation.
Conclusions
The present investigation shows an involvement of apoD in stress and recovery processes in the post-ischemic brain. We propose that apoD in degenerating neurons protects tissue from injury, whereas apoD+ oligodendrocytes in the peri-infarct area support axonal regeneration. Here, we envisage that apoD provides cholesterol and lipids for membrane biogenesis promoting neuritogenesis, synaptogenesis as well as astrocytic scar formation at the infarct border. A detailed understanding of the role of apoD in recovery processes may provide novel approaches to treat CNS injuries including stroke.
Footnotes
Acknowledgements
We thank Carin Sjölund and Kerstin Beirup for excellent technical assistance.