
Abstract
We examined the ability of tempol, a catalytic scavenger of peroxynitrite (PN)-derived free radicals, to reduce cortical oxidative damage, mitochondrial dysfunction, calpain-mediated cytoskeletal (α-spectrin) degradation, and neurodegeneration, and to improve behavioral recovery after a severe (depth 1.0 mm), unilateral controlled cortical impact traumatic brain injury (CCI-TBI) in male CF-1 mice. Administration of a single 300 mg/kg intraperitoneal dose of tempol 15 mins after TBI produced a complete suppression of PN-mediated oxidative damage (3-nitrotyrosine, 3NT) in injured cortical tissue at 1 h after injury. Identical tempol dosing maintained respiratory function and attenuated 3NT in isolated cortical mitochondria at 12 h after injury, the peak of mitochondrial dysfunction. Multiple dosing with tempol (300 mg/kg intraperitoneally at 15 mins, 3, 6, 9, and 12 h) also suppressed α-spectrin degradation by 45% at its 24 h post-injury peak. The same dosing regimen improved 48 h motor function and produced a significant, but limited (17.4%, P<0.05), decrease in hemispheric neurodegeneration at 7 days. These results are consistent with a mechanistic link between PN-mediated oxidative damage to brain mitochondria, calpain-mediated proteolytic damage, and neurodegeneration. However, the modest neuroprotective effect of tempol suggests that multitarget combination strategies may be needed to interfere with posttraumatic secondary injury to a degree worthy of clinical translation.
Introduction
In a recent work, we have shown that production of the reactive oxygen species peroxynitrite (PN) is an early upstream posttraumatic event that appears to lead to mitochondrial damage and functional failure, calcium-mediated calpain overactivation, and neurodegeneration following traumatic brain injury (TBI) (Deng et al, 2007). Therefore, antioxidants that can scavenge PN or its derivative free radicals may exert a neuroprotective effect.
Nitroxide antioxidants, such as tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl), are generally described in the literature as potent metal-independent superoxide dismutase mimics with better membrane permeability and potency (Krishna et al, 1996). Nitroxides have been shown to catalytically convert superoxide radical (O2−) to hydrogen peroxide and oxygen just like superoxide dismutase. However, it has been shown more recently that tempol can also catalytically decompose the PN-derived free radicals nitrogen dioxide (•NO2) and carbonate (•CO3) with a very rapid rate constant (Carroll et al, 2000), as illustrated in Figure 1. Thus, because of its multiple antioxidant properties, and its membrane permeability, tempol or similar nitroxide compounds may have considerable therapeutic value in protection against PN-mediated oxidative damage.
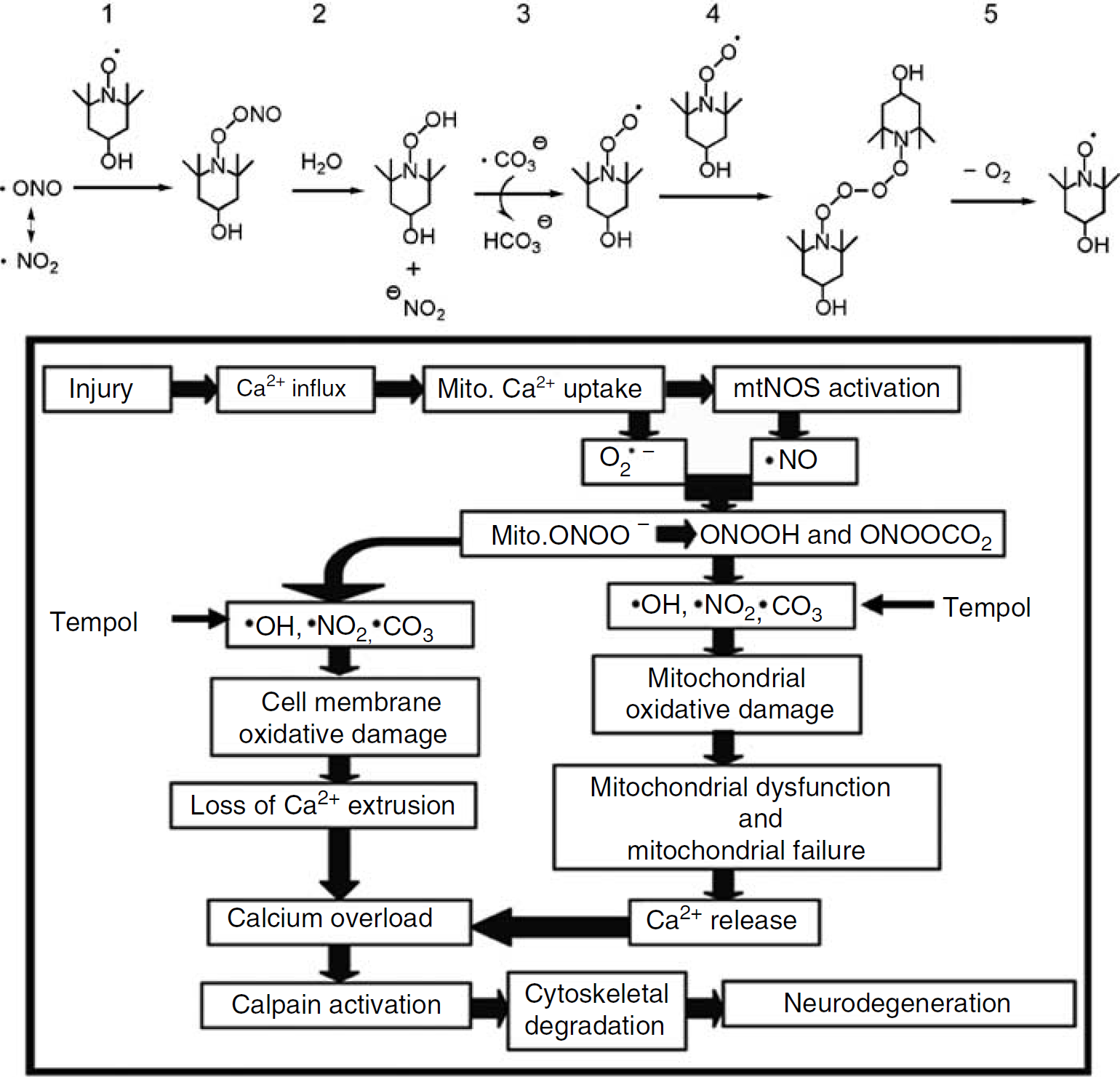
Top: chemical structure of tempol and the chemistry of its catalytic scavenging of PN-derived nitrogen dioxide (•NO2) and carbonate radicals (•CO3) (Carroll et al, 2000). Step 1, tempol reacts with the nitrogen dioxide radical to form a nitrosylated tempol; Step 2, reaction with water leads to release of nitrite anion (−NO2) and formation of a tempol peroxide intermediate; Step 3, reaction of the latter tempol intermediate with carbonate radical, which gives off bicarbonate (HCO3−) and a peroxylated tempol; Step 4, two peroxylated tempols combine; Step 5, the latter species reacts and breaks apart generating 1 mol oxygen and a regenerated tempol, which can then react with another mol nitrogen dioxide and carbonate radicals. Bottom: hypothetical interrelationship between PN-induced oxidative damage in neuronal mitochondria and the compromise of Ca2+ homeostasis, calpain-mediated proteolysis, and neurodegeneration. Tempol, by targeting the upstream PN-derived free radicals, should decrease oxidative damage and respiratory dysfunction in brain mitochondria, which in turn would ameliorate Ca2+ overload, reduce calpain-mediated proteolysis, and decrease neurodegeneration.
As shown in one of our previous studies, the early increase of PN-mediated oxidative damage in mitochondrial proteins parallels that in the injured cortical tissue, which also coincides with the early time course of mitochondrial dysfunction following injury (Singh et al, 2006). This strongly suggests that mitochondria are the primary source and target of PN, which is triggered on mitochondrial calcium uptake (Singh et al, 2006). Moreover, as also indicated in our time-course study on cellular changes triggered by TBI in the cortical impact (CCI) model, the peak of oxidative damage at 1 h after injury preceded both the peak of calpain activity at 24 h and neurodegeneration, measured by silver staining, at 48 h (Deng et al, 2007). Therefore, the current experiments were conducted to investigate the ability of tempol in reducing posttraumatic PN-induced oxidative damage, mitochondrial dysfunction, and calpain-mediated cytoskeletal degradation to establish a mechanistic linkage between these successive pathophysiological events. Moreover, the neuroprotective effect of tempol was accessed by the measurement of neurologic outcome and neurodegeneration at 7 days. The rationale for these studies is also shown in Figure 1.
Materials and methods
All the surgical, injury, and animal care protocols described below have been approved by the University of Kentucky Institutional Animal Care and Use Committee, and are consistent with the animal care procedures set forth in the guidelines of the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Mouse Model of Controlled Cortical Impact (Focal) Traumatic Brain Injury
Young adult male CF-1 mice (Charles River, Portage, MI, USA) weighing 29 to 31 g were used in this study. The mice were anesthetized with isoflurane (3.0%), shaved, and then placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). Throughout the surgery, the mice were provided with constant isoflurane (SurgiVet 100 Series) and oxygen (SurgiVet, O2 flowmeter, 0 to 4 L/min). The head was positioned in the horizontal plane of a stereotaxic frame with the nose bar set at zero. First, we produced a 4 mm craniotomy lateral to the sagittal suture, and centered between lambda and bregma. A cortical contusion was produced on the exposed cortex (the anterior—posterior coordinate for the epicenter of the injury was bregma −2.0 mm) using a pneumatically controlled impactor device (Precision System Instruments TBI-0200 Impactor, Lexington, KY, USA), as previously described (Deng et al, 2007; Hall et al, 2005; Singh et al, 2006). To prevent immediate posttraumatic hypothermia, the injured mice were placed in a Hova-Bator incubator (model 1583, Randall Burkey Co., Boerne, TX, USA) set at 37°C for 20 to 30 mins after the injury. Consciousness (return of righting reflex and mobility) was regained within 5 to 10 mins after the CCI and 10 to 15 mins after weight-drop injury. In a previous study (Kupina et al, 2003), rectal temperatures taken 2 h after injury indicated TBI male mice to be approximately 2°C cooler than sham-injured mice (35.4°C±0.3°C versus 37.7°C±0.1°C, mean±s.e.m.). By 24 and 48 h after injury in that study, TBI mice remained about 1°C cooler compared with sham-injured mice (36.9°C±0.2°C versus 38.0°C±0.2°C, mean±s.e.m.).
The injured mice were allowed to survive from 1, 12 or 24 h or 7 days depending on their experimental group. For immunoblotting studies (measurements of oxidative damage marker and cytoskeletal degradation, see below), the N for each treatment group was 8 animals; for mitochondrial studies, the N for each treatment group was 6 (3 mice were pooled together to create one N); for behavioral and neurodegenerative studies, the N for each treatment group was 12 based on our experience with the variability seen with these measurements in previous studies (Deng et al, 2007; Hall et al, 2005).
Tempol Preparation and Dosing
Tempol was purchased from Sigma-Aldrich (Milwaukee, WI, USA) and freshly prepared in 0.9% saline before abdominal intraperitoneal injection. In our dose—response study, we tested different dosages at 3, 10, 30, 100, and 300 mg/kg (n=8), which well covered the dosage range that has been explored in related neuropathological and pilot studies (Behringer et al, 2002; Beit-Yannai et al, 1996). Saline (0.9%) was used as control for the tempol treatment group. Another control group was composed of sham animals that were subjected to craniotomy but no contusion injury and treated with either saline or tempol.
Immunoslot-blotting Analysis of Oxidative Damage (3-nitrotyrosine)
At selected time points (1, 6, 12, or 24 h), the sham, or injured vehicle-treated, or injured tempol-treated mice were deeply overdosed with sodium pentobarbital (200 mg/kg intraperitoneally). After decapitation, the ipsilateral cortical area of interest was rapidly dissected on an ice-chilled stage, as previously described (Deng et al, 2007). Immediately after dissection, samples were transferred into precooled Triton lysis buffer (1% Triton, 20 mmol/L Tris-HCL, 150 mmol/L NaCl, 5 mmol/L EGTA, 10 mmol/L EDTA, and 10% glycerol) with protease inhibitors (Complete Mini Protease Inhibitor Cocktail tablet). Samples were then briefly sonicated and vortexed at 14,000 r.p.m. for 30 mins at 4°C and the supernatants were collected for protein assay. Protein concentration was determined by Bio-Rad DC Protein Assay, with sample solutions diluted to contain 1 mg/mL of protein for immunoblotting.
To measure 3-nitrotyrosine (3NT), an aliquot of each ipsilateral cortical protein or isolated mitochondrial protein sample (2 μg) was diluted with 200 μL of tris-buffered saline (TBS) and transferred to a Protran (0.2 μm) nitrocellulose membrane by a Minifold II vacuum slot-blot apparatus (Schleicher & Schuell, Dassel, Germany) (Deng et al, 2007). After the samples were loaded into the slots, they were allowed to filter through the membrane by gravity (no vacuum). Each slot was then washed with 200 μL TBS, which was allowed to filter through the membrane again. The membranes were then disassembled from the apparatus and incubated in a TBS blocking solution with 5% milk for 1 h at room temperature. For the detection of 3NT, a rabbit polyclonal anti-nitrotyrosine antibody (Upstate Biotechnology, MA, USA) was used at a dilution of 1:2,000 in TBS-Tween-20 (TBST) blocking solution with 5% milk, overnight at 4°C. A goat anti-rabbit secondary antibody conjugated to an infrared dye (1:500, IRDye800CW, Rockland, Gilbertsville, PA, USA) was then applied for 1 h at room temperature. After drying, the membranes were imaged and densitometrically (quantified using a Li-Cor Odyssey Infrared Imaging System (Lincoln, NE, USA).
Preliminary experiments established protein concentration curves to ensure that quantified blots were in the linear range. From these results, we selected 2 μg as the amount to be used based on the observation that this amount gave blots that were well within the linear range. A standardized protein loading control was included on each blot to normalize the band densities so that comparisons could be made across multiple blots. All the samples were run in duplicate, which were then averaged. No primary antibody controls were run to verify that the oxidative damage staining was specific.
Mitochondrial Percoll Gradient Purification and Respiration Measurement
Brain cortical mitochondria were isolated by Percoll gradient purification, as previously described (Singh et al, 2006, 2007). After decapitation, the ipsilateral cortical tissue was quickly removed and pooled from three mice. Fresh mitochondria were prepared for each experiment and used within 4 h.
Mitochondrial respiratory rates were measured using a Clarke-type electrode in a continuously stirred, sealed, thermostatically controlled chamber (Oxytherm System, Hansatech Instruments Ltd, Kings Lynn, UK) maintained at 37°C, as previously described (Singh et al, 2006, 2007). Twenty-five to 40 μg of isolated mitochondrial protein was placed in the chamber containing 250 μL of KCl-based respiration buffer (125 mmol/L KCl, 2 mmol/L MgCl2, 2.5 mmol/L KH2PO4, 0.1% bovine serum albumin, and 20 mmol/L HEPES at pH 7.2) and allowed to equilibrate for 1 min. This was followed by the addition of complex-I substrates, 5 mmol/L pyruvate and 2.5 mmol/L malate, to monitor state II respiratory rate. Two boluses of 150 μmol/L ADP were added to the mitochondria to initiate state III respiratory rate for 2 mins followed by the addition of 2 μmol/L oligomycin to monitor state IV respiration rate for an additional 2 mins. For the measurement of uncoupled respiratory rate, 2 μmol/L p-trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP) was added to the mitochondria in the chamber and oxygen consumption was monitored for another 2 mins. This was followed by the addition of 10 mmol/L succinate to monitor complex-II-driven respiration. The respiratory control ratio (RCR) was calculated by dividing the state III oxygen consumption (defined as the rate of respiration in the presence of ADP, second bolus addition) by the state IV oxygen consumption (rate obtained in the presence of oligomycin).
A parallel aliquot of the isolated mitochondria, which were not subjected to respiratory rate analyses, was used for immunoblot measurement of 3NT by immunoslot-blotting, as described above.
Western-Blotting Analysis of Calpain-Mediated Cytoskeletal Degradation
To measure calpain-mediated α-spectrin proteolysis, aliquots of each cortical sample (5 μg) were run on an SDS—polyacrylamide gel electrophoresis precast gel (3% to 8% Tris-Acetate Criterion XT Precast gel; Bio-Rad, Hercules, CA, USA) and then transferred to a nitrocellulose membrane using a semi-dry electro-transferring unit set at 15 V for 15 mins, as previously described (Deng et al, 2007). The membranes were incubated in a TBS blocking solution with 5% milk for 1 h at room temperature. For the detection of α-spectrin and its breakdown products, a mouse monoclonal anti-α-spectrin antibody (Affiniti FG6090) was used at a dilution of 1:5,000 in TBST blocking solution with 5% milk, overnight at 4°C. A goat anti-mouse secondary antibody conjugated to an infrared dye (1:5,000, IRDye800CW; Rockland, Gilbertsville, PA, USA) was then applied for 1 h at room temperature. After drying, the membranes were imaged and densitometrically quantified using the Li-Cor Odyssey Infrared Imaging System (Lincoln, NE, USA). A standardized protein loading control was included on each blot to normalize the band densities so that comparisons could be made across multiple blots. This was made up of pooled brain tissue protein collected from previously run TBI mice that gave strong bands corresponding to the 280 kDa parent α-spectrin, and the 150 and 145 kDa breakdown products. The amount of protein in the loading control had been previously determined and shown to be within the linear range as measured with the Li-Cor Odyssey Infrared Imaging System. After the transfer, the gels were stained with Coomassie blue to verify even transfer. All samples were run in duplicate and averaged.
Neuroscore Motor Function Test
The neuroscore motor function test was used to assess the neurologic status of the sham and TBI mice at 48 h and 7 days after injury. This test is modified from a previously described series of tests used for assessing the effects of TBI in rat (McIntosh et al, 1989), and adapted for mouse as previously described (Raghupathi et al, 1998). In the present study, we employed the Neuroscore with the following two components: grid walk (front limb and hind limb) and cage top (front limb and hind limb).
De Olmos Silver Staining Analysis of Neurodegeneration
After the completion of the 7 days postinjury neuroscore test, neurodegeneration was examined using the de Olmos aminocupric silver histochemical technique, as previously described (de Olmos et al, 1994; Hall et al, 2005; Switzer, 2000). At 7 days, the mice (N=12) were overdosed with sodium pentobarbital (200 mg/kg intraperitoneally) and transcardially perfused with 0.9% sodium chloride, followed by a fixative solution containing 4% paraformaldehyde. After decapitation, the heads were stored in a fixative solution containing 15% sucrose for 24 h, after which the brains were removed, placed in fresh fixative, and shipped for histologic processing to Neuroscience Associates Inc. (Knoxville, TN, USA). Brains used for this study were embedded into one gelatin block (Multiblock Technology, Neuroscience Associates Inc.). The block was then frozen and thirteen 35 μm coronal sections were taken 420 μm apart between 1.1 mm anterior and 4.4 mm posterior to bregma, stained with the de Olmos silver stain to reveal degenerating neurons and neuronal processes, and then counterstained with nuclear fast red. The brain sections were photographed on an Olympus Provis A70 microscope at × 1.25 magnification using an Olympus Magnafire digital camera and the image was analyzed by Image-Pro Plus (4.0). The percentage area of silver staining for each brain section was calculated by dividing the area of the ipsilateral hemispheric silver staining in each section by the area of the contralateral hemispheric area and multiplying by 100. The volume of silver staining in the ipsilateral hemisphere as a percentage of the contralateral hemispheric volume was estimated by the equation %V=tΣ%a(s), where %V is the percentage silver stain volume, t is the distance between the sections analyzed (420 μm), and Σ%a(s) is the sum of the percentage area of silver staining in all the sections examined (13 for each brain) (Deng et al, 2007; Hall et al, 2005).
Cortical Lesion Volume Measurement
In addition to measuring the volume of neurodegeneration with the de Olmos silver staining method, the brain sections were photographed on an Olympus Provis A70 microscope at × 1.25 magnification using an Olympus Magnafire digital camera and the cortical lesion volume was analyzed by ImageJ 1.37v (NIH), as previously described (Pandya et al, 2007). The percentage area of ipsilateral cortical tissue area for each brain section was calculated by dividing the area in each section by the area of the contralateral cortex and multiplying by 100. The volume of the ipsilateral cortical lesion volume as a percentage of the contralateral cortical volume was estimated by the sum of the percentage area of spared tissue of all the sections multiplied by the distance between the sections analyzed (420 μm).
Statistical analysis
For treatment group analyses, we used Statview 5.0 to perform a one-way analysis of variance (ANOVA), followed by Student-Newman—Keuls (SNK) post hoc analysis to determine the significance of differences between the noninjured sham group and the injured vehicle-treated versus injured tempol-treated groups. For the multiple section analyses of neurodegeneration at different brain levels, we used Statview 5.0 to perform a repeated measures ANOVA followed by SNK post hoc analysis to determine the significance of differences between individual sections in the noninjured sham group and the injured vehicle-treated versus injured tempol-treated groups. For all ANOVAs, a P<0.05 was required to establish a statistically significant difference across the groups. In addition, for the post hoc SNK analysis, the program determined significance based on a correction for multiple comparisons among the groups.
Results
Dose—Response Study of the Effect of Tempol on Protein Nitration
The level of protein nitration in cortical tissue homogenate was assessed by quantitative antibody-based immunoslot-blotting. Figure 2 shows the results of a dose—response analysis examining ipsilateral cortical 3NT levels after TBI (N=8). Five different single intraperitoneal dosages were administered to the mice 15 mins after cortical contusion. All mice were euthanized 1 h after injury (45 mins after tempol) when PN-induced oxidative damage was at its peak (Deng et al, 2007). The ANOVA showed a significant difference across the experimental groups (F(6, 49)=3.397; P<0.01). Post hoc analysis revealed that the injured mice treated with vehicle (0.9% saline) showed about a 90% increase in 3NT level (P=0.007). In comparison, the three highest dosages of tempol (30, 100, and 300 mg/kg) significantly reduced the 3NT level compared with the injured mice treated with vehicle. However, there was no significant difference among the three highest dosage groups. Nevertheless, the highest dose of tempol treatment in the current study (300 mg/kg) was able to completely prevent any posttraumatic increase in 3NT level over the sham baseline level. Therefore, this dosage was chosen for subsequent studies. Another control group, in which sham was treated with tempol (n=8) showed no difference from sham vehicle-treated group (data not shown).
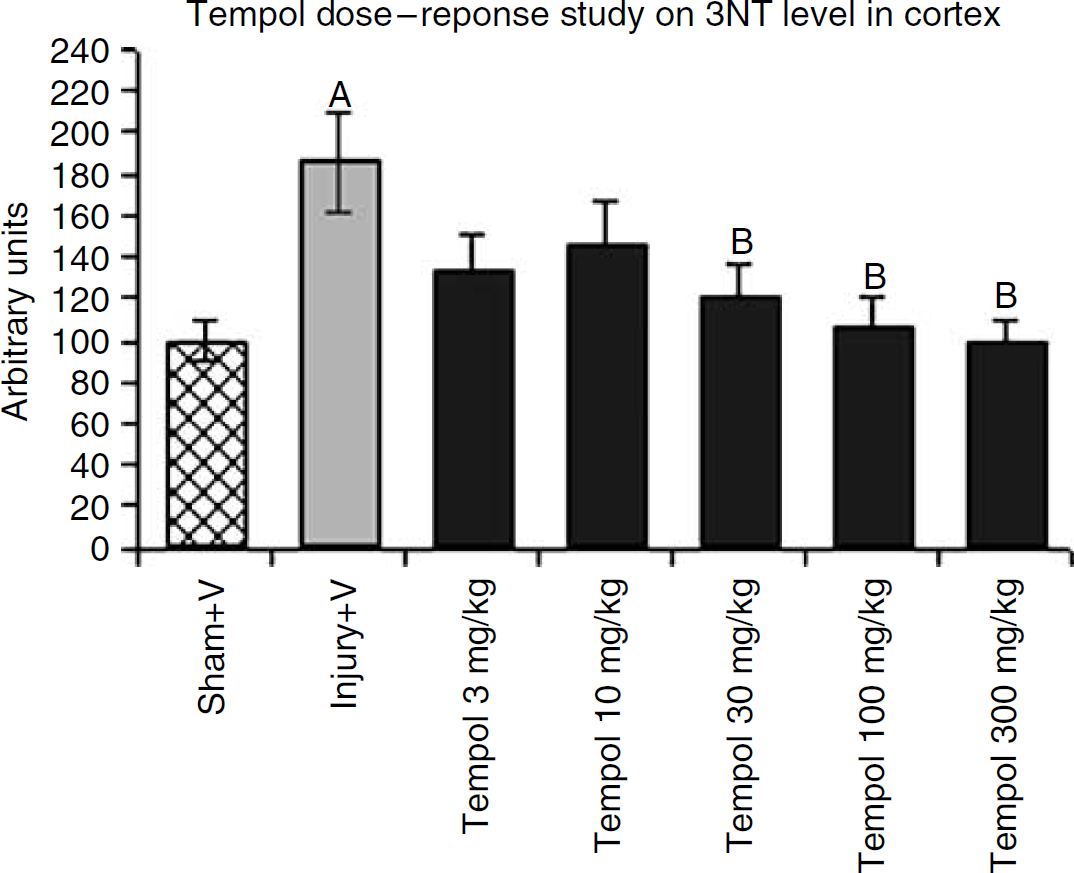
Tempol treatment studies using quantitative slot-blotting in ipsilateral cortical traumatic brain injury tissues showing dose-related attenuation of protein nitration (3-nitrotyrosine, 3NT). A single dose of tempol was administered intraperitoneally to animals for 15 mins followed by killing at 1 h after TBI. The three highest doses at 30, 100, and 300 mg/kg significantly reduced 3NT level, a specific marker for PN-induced oxidative damage, in ipsilateral cortical tissues. N=8 animals per treatment group; values=mean+s.e.; one-way ANOVA and Student-Newman—Keuls post hoc test: A=P<0.01 versus sham, B=P<0.01 versus vehicle.
Effect of Tempol on Mitochondrial Respiration and Oxidative Damage
Parallel aliquots of isolated cortical mitochondria were examined in the mitochondrial respiratory assay or for 3NT levels by quantitative immunoslot-blotting. Mitochondrial oxygen consumption was measured by a Clarke-type electrode. In this study, mice (n=6) were treated with a single intraperitoneal dose of either vehicle or tempol (300 mg/kg) 15 mins after injury, followed by euthanasia 12 h after TBI. This time point was selected as we had previously shown that it represents the peak of mitochondrial dysfunction in the CCI model (Singh et al, 2006). Cortical mitochondria were isolated and purified followed by incubation in the constantly stirred sealed chamber. The oxygen consumption rate at each step was recorded by the slope of the oxygen respiration trace on substrate addition. Figure 3A indicates the respiratory rates compared between the sham vehicle-treated, injured vehicle-treated, and injured tempol-treated groups. Two doses of ADP after pyruvate and malate initiated mitochondrial respiration. There was a significant difference across the sham, injured vehicle-treated, and injured tempol-treated groups (F(2, 15)=4.339; P<0.05). Post hoc analysis showed that injured vehicle-treated mitochondria displayed significantly decreased complex-I-driven state III respiration compared with sham, indicating impaired mitochondrial respiratory function. In contrast, mitochondria isolated from the tempol-treated mice showed a complete maintenance of normal state III respiration, which was significantly higher than the vehicle-treated group. There was no significant difference detected in state IV respiration among groups on inhibition of ATP synthase by oligomycin addition.
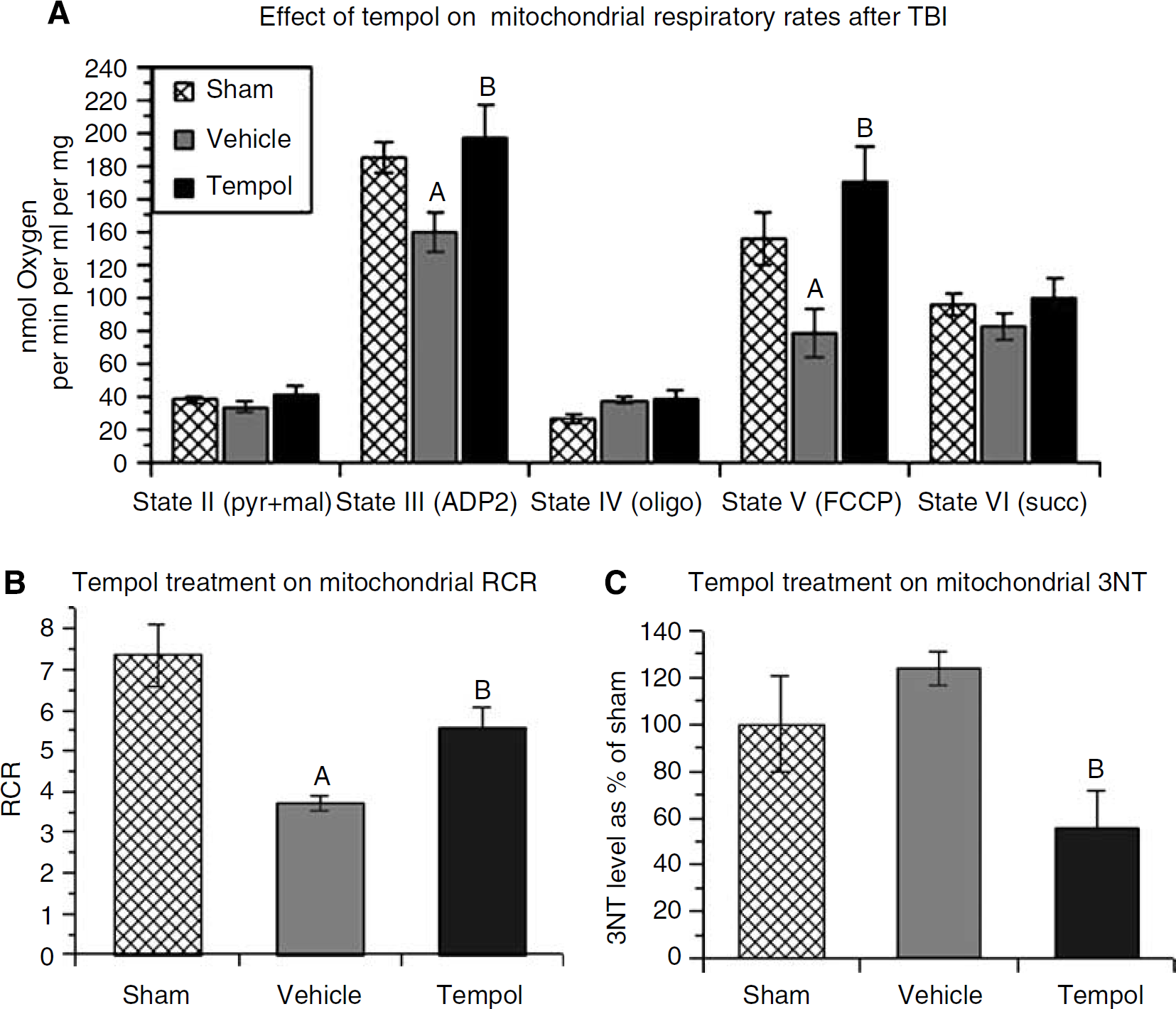
Effect of optimal single-dose tempol treatment on mitochondrial respiration in ipsilateral cortical traumatic brain injury tissues measured by Clarke-type electrode. (
A mitochondrial uncoupler, FCCP, was added to initiate maximal mitochondrial respiration state V by inducing mild proton leak into the mitochondria. This analysis also showed a significant difference across the treatment groups (F(2,15)=6.894; P<0.001). Post hoc testing showed that the injured vehicle-treated group displayed a significantly reduced respiration rate when compared with the sham group, suggesting impairment of the ETC (electron transport chain). However, mitochondria isolated from the tempol-treated mice showed significantly improved state V respiration compared with the vehicle-treated group. Complex-II-driven state VI respiration was tested by adding the complex-II substrate succinate. There was no significant difference observed among the three treatment groups, suggesting that complex-II and the downstream complexes were relatively healthy in contrast to complex-I, which was impaired.
Figure 3B displays the RCRs of isolated mitochondria from the three different treatment groups, which ANOVA showed were significantly different (F(2, 15)=11.910; P<0.001). The RCR indicates how well oxygen consumption is coupled to ATP generation and is often used as the index of mitochondrial function (healthy: RCR>5). In Figure 3B, mitochondria from injured mice without drug treatment show a mean RCR of 3.7, which was significantly lower than the sham group (RCR=7.3). The tempol-treated group showed a mean RCR of 5.6, suggesting that the mitochondria remain on an average within the healthy range (i.e., >5.0). The mean from the tempol-treated group was significantly higher than the vehicle-treated group. However, although tempol treatment was able to improve mitochondrial function to a healthy level, the RCR in the injured tempol-treated group was significantly lower than that in the sham group, suggesting that mitochondrial function was not fully protected.
Figure 3C shows mitochondrial 3NT level at 12 h after injury subjected to different treatments (n=4). The ANOVA revealed a significant difference across the treatment groups (F(2, 9)=4.843; P<0.05). Post hoc analysis showed that even though 3NT level in the injured vehicle-treated group was increased when compared with the sham group, this increase was not significant. Consistent with this observation, we had previously found that although a significant increase in 3NT level in mitochondrial proteins is seen as early as 30 mins after injury, and up to at least 3 h, the persistent mean increase in 3NT at 12 h after TBI loses significance in comparison with sham (Singh et al, 2006). Nevertheless, in the injured tempol-treated group, 3NT level was significantly lower than the injured vehicle-treated group by 55%. Moreover, the mean 3NT level in the injured tempol-treated group was also lower than that seen in the sham group, but this difference was not significant.
Effect of a Single Dose of Tempol on Calpain-Mediated α-Spectrin Breakdown
Posttraumatic cytoskeletal degradation was measured by western-blotting detection of α-spectrin breakdown products (SBDPs) in cortical tissues ipsilateral to the injury. Mice were treated with a single dose of vehicle or tempol (300 mg/kg) 15 mins after TBI and were killed at 1 or 6 h after injury (n=8 per group). Figure 4A shows that there was a statistically significant difference across the three groups for the calpain-specific SBDP145 analysis (F(2, 21)=5.590; P<0.05), but not in the case of SBDP150, which is generated by both calpain and caspase 3 (Wang, 2000). Post hoc testing revealed that there was a significant increase in SBDP145 at 1 h after injury in the injured vehicle-treated group compared with the sham vehicle-treated group. However, tempol treatment significantly reduced SBDP145 compared with vehicle treatment by 45%.
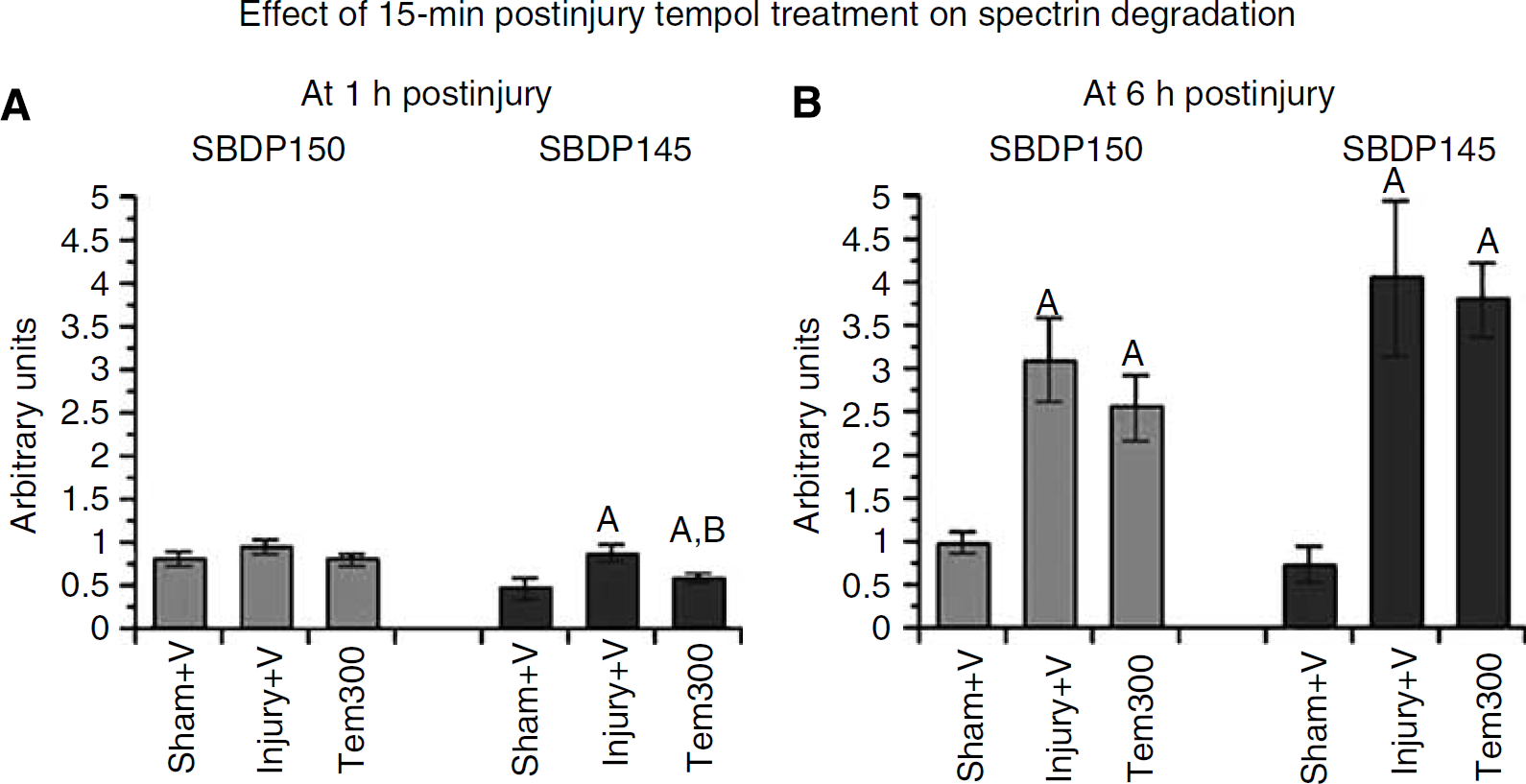
Effect of optimal single-dose tempol treatment on calpain-mediated α-spectrin breakdown in ipsilateral cortical traumatic brain injury tissues measured by quantitative western blotting. (
Figure 4B displays the level of SBDPs at 6 h after TBI with or without tempol treatment. Both SBDPs showed significant increase compared with the sham group. Although there was a slight decline of both SBDPs in the injured tempol-treated group compared with the vehicle-treated group, the decrease was not significant (P<0.01). The magnitude of both SBDPs in injury groups at 6 h after injury was higher than that at 1 h after injury, which indicated a progressive increase of calpain activity over time after TBI until it reaches its peak at 24 h in the CCI model (Deng et al, 2007).
Effect of Multiple Doses of Tempol on Calpain-Mediated α-Spectrin Breakdown
The short-term effect of tempol in reducing SBDP145 at 1 h but not 6 h after injury caused us to next explore a tempol treatment regimen involving repeated doses administered over the full course of the oxidative damage (i.e., 12 h). Accordingly, multiple doses of vehicle or tempol (300 mg/kg) were administered intraperitoneally to mice at 15 mins, 3, 6, 9, and 12 h after injury followed by euthanasia at 24 h when calpain-mediated SBDPs reach their peak in the CCI model (Deng et al, 2007). As indicated previously (Deng et al, 2007), at 24 h after injury, the magnitude of calpain-specific SBDP145 is more than twice as much as that of calpain/caspase-3-produced SBDP150, suggesting a the dominant role of calpain in posttraumatic proteolysis. However, at 24 h after injury, the difference across groups for both SBDPs is significant, for SBDP145: F(2, 20)=16.513 and P<0.0001; for SBDP150: F(2, 20)=5.241 and P<0.05. In Figure 5A, tempol treatment decreased both SBDPs significantly compared with vehicle treatment (P<0.05). The effect on SBDP150 was essentially complete as the magnitude of nonspecific SBDP150 is much lower than that of calpain-specific SBDP145. However, although tempol treatment also reduced SBDP145 by 45% compared with the injured vehicle-treated group, the SBDP145 level in the injured tempol-treated group remained significantly elevated compared with the sham level, suggesting that proteolytic damage induced by calpain was not fully inhibited even with this repeated-dose treatment regimen.
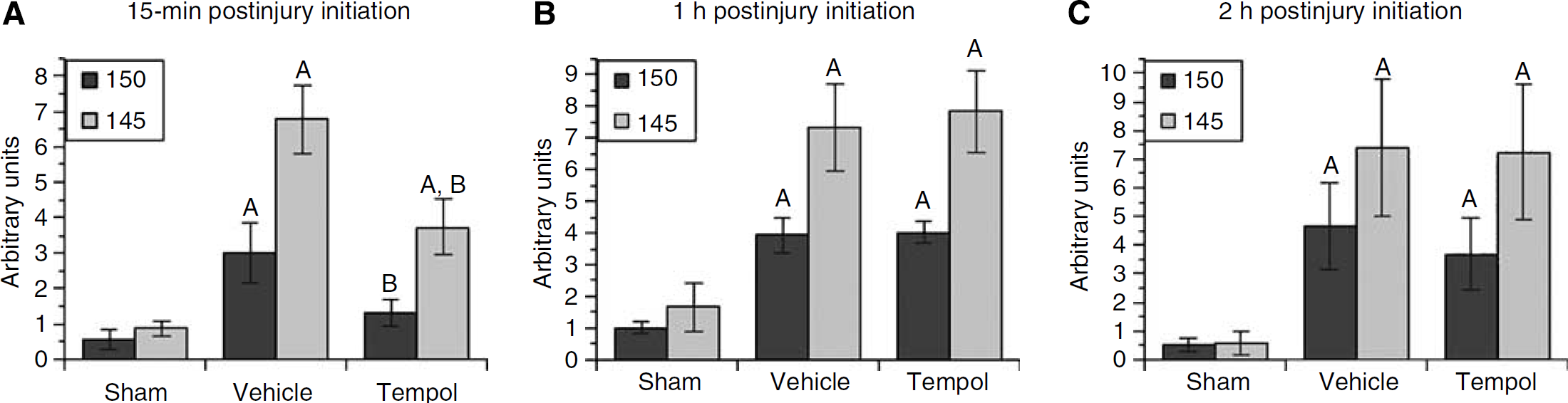
Effect of multiple-dose tempol treatment on calpain-mediated α-spectrin breakdown in ipsilateral cortical traumatic brain injury tissues measured by quantitative western blotting. (
As part of this experiment, a therapeutic window study was conducted in which the onset of tempol multiple-dose treatment was delayed from 15 mins after injury to either 1 or 2 h. We administered multiple intraperitoneal-doses treatment starting at 1 or 2 h after injury, with four additional bolus intraperitoneal injections every 3 h. Animals were then studied at 24 h after the first injection. As shown, a 1 h (Figure 5B) or a 2 h (Figure 5C) delay in treatment with tempol showed no effect on either SBDP145 or SBDP150. Both SBDPs remained at similar levels as the injured vehicle-treated group.
Behavioral Outcome Measured by Neuroscore Motor Test after Tempol Treatment
Five intraperitoneal doses of tempol (300 mg/kg each; last dose at 12 h after injury) were administered to the animals at 3-h intervals beginning at 15 mins after the CCI injury. Behavioral outcome was assessed at 48 h and 7 days after TBI, respectively measured by neuroscore. We divided the scores into two categories: fore limb and hind limb. At 48 h after injury (Figure 6), ANOVA revealed a significant difference across the treatment groups in both fore limb (F(2, 33)=3.610; P<0.05) and hind limb (F(2, 33)=3.927; P<0.05) motor function. Post hoc analysis showed that motor function in the injured vehicle-treated group was significantly impaired compared with the scores of the sham group. Although the 48 h fore limb (Figure 6A) and hind limb (Figure 6B) scores for the injured tempol-treated group were not significantly different from the injured vehicle-treated group, the scores of the tempol-treated group were also not significantly lower than the sham group, implying some improvement in the 48 h neurologic status as a result of the tempol treatment started at 15 mins after injury. At 7 days after injury, both fore limb (F(2, 33)=2.668; P=0.0843) (Figure 6C) and hind limb (F(2, 33)=2.797; P=0.08) (Figure 6D) motor function showed no significant difference among treatment groups. Although injured vehicle-treated animals showed decreased scores compared with the sham group, this slight deficit was not statistically significant.
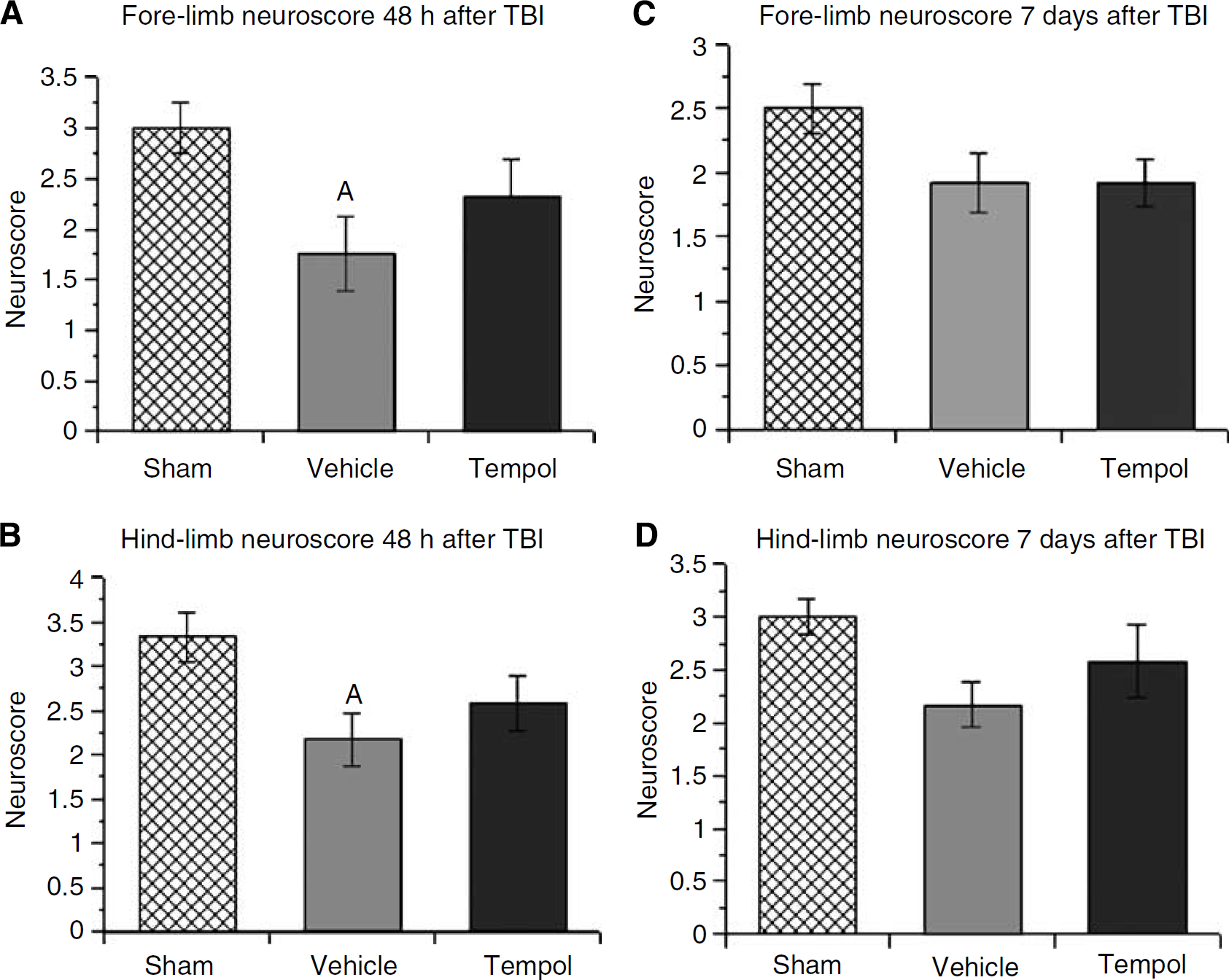
Behavioral outcome of multiple-dose tempol treatment measured by neuroscore motor test after TBI. Neuroscore was accessed at 48 h and 7 days after injury. N=12 animals per treatment group; values=mean+s.e.; one-way ANOVA and Student-Newman—Keuls post hoc test: A=P<0.05 versus sham.
Histologic Evaluation of Hemispheric Neurodegeneration Measured by de Olmos Aminocupric Silver Staining and Cortical Lesion Volume After Tempol Treatment
After the 7-day neuroscore test, brains were harvested for histopathological analysis of neurodegeneration using the de Olmos aminocupric silver staining method. Figure 7A displays the volume of hemispheric neurodegeneration in injured vehicle-treated and injured tempol-treated animals. Both injured groups displayed a significant increase in neurodegeneration silver staining volume compared with the sham group. However, the injured tempol-treated group showed significantly reduced neurodegeneration compared with the injured vehicle-treated group (F(2, 33)=125.864; P<0.05; SNK comparing vehicle- and tempol-treated groups P<0.05). Tempol reduced the silver staining volume by 17.4%. Selected coronal brain sections at the level of the injury epicenter illustrate the degree of neurodegeneration with tempol treatment versus vehicle treatment.
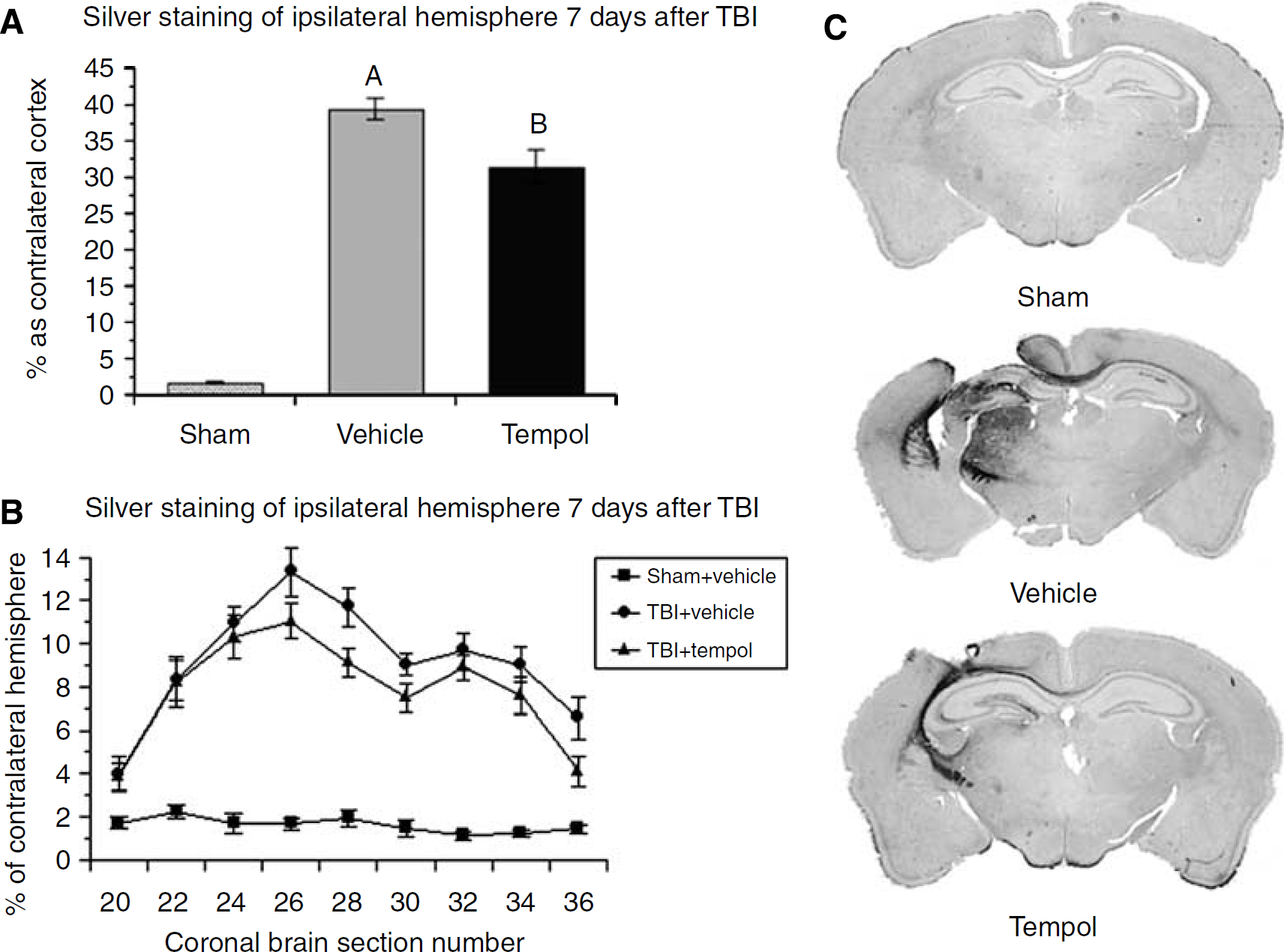
Histologic evaluation of multiple-dose tempol treatment on silver staining in ipsilateral hemisphere after TBI. (
Figure 7B shows the percentage of silver staining compared with nonstained coronal brain sections from the anterior hemispheric boundary of the posttraumatic neurodegeneration to the posterior boundary. Injured groups at all sections showed a significant increase of silver staining compared with the sham group. The injured tempol-treated group displayed significantly less silver staining than injured vehicle-treated group at section 28, which is slightly posterior to the epicenter of the injury. Typical examples of silver staining at 7 days after injury are shown for each group.
The effect of tempol on the cortical lesion volume was also assessed. In vehicle-treated mice, the cortical lesion volume at 7 days after injury was 13.2%+1.2%. Tempol treatment reduced the lesion volume to 9.0%+2.1% (F(2, 33)=24.664; P<0.0001; SNK comparing vehicle- and tempol-treated P<0.03). The percentage reduction in lesion volume measured by this more traditional method was 32% (9.0/13.2). Thus, the effect of tempol on posttraumatic cortical neurodegeneration is greater than the effect on hemispheric neurodegeneration as assessed by silver staining.
Discussion
The current study employed a pharmacological approach to help elucidate the pathophysiological relationship of PN-induced oxidative damage, mitochondrial dysfunction, calcium-activated, calpain-mediated cytoskeletal degradation, and neurodegeneration (Deng et al, 2007; Singh et al, 2006) by testing the ability of the multimechanistic nitroxide antioxidant tempol, to inhibit these secondary injury events in the CCI-TBI mouse model.
Effect of Tempol on PN-Mediated Oxidative Damage
The neuroprotective effect of tempol was previously explored in a rat closed TBI model. Tempol showed cerebroprotective effects in terms of limiting edema formation, ameliorating blood—brain barrier disruption, and improving functional recovery (Beit-Yannai et al, 1996; Zhang et al, 1998). In these studies, it was concluded that the beneficial effects of tempol were because of its catalytic scavenging of superoxide radicals. However, oxidative damage markers were not examined, and more recent experiments have suggested that an important antioxidant property that may contribute significantly to the inhibition of posttraumatic oxidative damage in brain tissue involves catalytic scavenging of the PN-derived radicals •NO2 and •CO3 (Carroll et al, 2000).
A useful biomarker of PN oxidative damage to proteins involves the nitration of protein tyrosine residues, which produces the product 3NT. Although it was previously suggested that nitric oxide (NO•) could give rise to tyrosine nitration, subsequent analyses have shown that this is probably caused by PN-derived •NO2. Alternatively, PN is able to react with superoxide dismutase to form nitronium ion (NO2+)-like intermediate, which may also cause tyrosine residue nitration (Ischiropoulos et al, 1992). In the current study, we used quantitative immunoslot-blotting detection of the PN-specific marker 3NT to investigate the effect of tempol on PN-induced oxidative damage. Animals were administered with a single dose of tempol intraperitoneally at 15 mins and killed at 1 h after injury when PN-induced oxidative damage was at its peak (Deng et al, 2007). A significant increase in cortical 3NT level in vehicle-treated group compared with sham was observed. Compared with the vehicle-treated group, tempol treatment at 30, 100, and 300 mg/kg significantly decreased 3NT level in the cortical tissue ipsilateral to the injury, showing that tempol has a wide range of antioxidant potency, and that scavenging of PN-derived •NO2 may be a key aspect of tempol's antioxidant mechanism in vivo. The highest dose of tempol caused complete inhibition of posttraumatic oxidative damage.
Effect of Tempol on Mitochondrial Oxidative Damage and Respiratory Dysfunction
In our previous study, quantitative slot-blotting measurement of postinjury mitochondrial lipid peroxidation and protein nitration levels showed immediate increase of PN-induced oxidative damage, which persisted for hours after injury and correlated with a time-related depression of mitochondrial complex-I-driven respiratory function (Singh et al, 2006). Although the level of 3NT in injured brain mitochondria remains slightly elevated, it loses significance by 12 h after injury due perhaps to its decreased production and/or its increased proteolytic removal (Grune et al, 1997). Moreover, proteomic identification of specific rat mitochondrial fraction proteins showed that key components of the ETC are highly oxidized as early as 3 h after CCI-TBI (Opii et al, 2007). In the current study, we showed that the early (15 mins after injury) tempol treatment completely suppressed the level of 3NT in whole-tissue homogenates taken from the injured cortical tissue at 1 h after injury and in mitochondria harvested from the injured cortex at 12 h. Together with the inhibition of mitochondrial oxidative damage, mitochondrial respiration was almost completely preserved by early tempol administration. Specifically, state III respiration in the tempol-treated group showed full preservation compared with the sham group, and the RCR (state III/state IV) showed maintenance to nearly normal limits.
The effects of TBI on mitochondrial oxidative damage and respiratory dysfunction have been replicated in in vitro studies in which isolated brain mitochondria have been exposed to PN. The first such study found that exposure of isolated mitochondria to either PN or high levels of Ca2+ induced membrane protein thiol cross-linking together with mitochondrial swelling (Gadelha et al, 1997). A second investigation showed that PN exposure can increase mitochondrial proton leak and electron transport uncoupling (Echtay et al, 2003). We have recently shown that the exposure of isolated mouse brain mitochondria to the PN-generating compound SIN-1 can elicit a rapid depression in complex-I-driven respiratory function (Singh et al, 2007) together with an increase in 3NT and lipid peroxidative damage. In contrast, pretreatment of the mitochondria with tempol before SIN-1 application significantly reduced 3NT levels along with a partial preservation of complex-I-driven respiration. Thus, the similar effects of in vivo postinjury tempol treatment in the present study are no doubt because of a direct protective effect of tempol on the injured cortical mitochondria.
It should also be noted that mitochondria in the injured brain that are severely stressed by elevations in intracellular Ca2+ are probably the source of the PN that in turn contributes to their posttraumatic secondary insult. In short, mitochondria have long been known to be a main resource for reactive oxygen species. The mono-electron transport through the mitochondrial ETC spins-off oxidants (mainly O2−) as by-products during metabolism (Shigenaga et al, 1994). It has been proposed that the complex-I flavin mononucleotide group is the major site of mitochondrial O2− leak (Liu et al, 2002), although others hypothesize that the leak may preferably occur at the level of coenzyme Q (Kowaltowski et al, 1995). More recently, a novel Ca2+-dependent isoform of nitric oxide synthase has been identified within mitochondria, which is activated on mitochondrial Ca2+ uptake (Giulivi, 2003). Although NO• by itself has been suggested to be toxic, most investigators now believe that the damaging effects of NO• are mediated by PN, which is formed by the diffusion rate-limited reaction of NO• with O2−. Thus, the discovery of a mitochondrial isoform of nitric oxide synthase together with the well-known mitochondrial O2− leak provides an intramitochondrial source of PN that can result in posttraumatic PN-mediated oxidative damage.
Effect of Tempol on Calpain-Mediated Cytoskeletal Degradation
Calpain, a cysteine protease, is activated in response to cytosolic Ca2+ elevation. It has been suggested that calpain plays a role in dendritic remodeling, in membrane repair and resealing after injury (Faddis et al, 1997). However, the disruption of neuronal Ca2+ homeostasis may result in excessive activation of calpain and proteolytic degradation of a number of neuronal proteins, which has been suggested to be a final common pathway of neuronal cell death after various acute brain insults and in chronic neurodegenerative diseases (Bartus, 1997). In particular, evidence for an important role of calpain-mediated cytoskeletal proteolyses in posttraumatic secondary injury has been obtained in multiple TBI models that progress for as much as 24 h after injury (Buki et al, 1999; Deng et al, 2007; Povlishock et al, 1999; Ringger et al, 2004; Saatman et al, 1996) Our previous work (Deng et al, 2007) showed that posttraumatic calpain activation and cytoskeletal proteolysis is preceded by PN-mediated oxidative damage, mitochondrial dysfunction, impairment of Ca2+ homeostasis, and presumably intracellular Ca2+ overload, and is then followed by neurodegeneration. If these processes are indeed linked as a series of events, then tempol treatment, which is known to interfere with the initiating PN-induced oxidative damage and mitochondrial dysfunction, should also attenuate the cytoskeletal damage.
We first tested this hypothesis through a single intraperitoneal-dose tempol treatment administered immediately after CCI, which showed a complete inhibitory effect on calpain activity at 1 h after injury. This suggests that the membrane permeable tempol is rapidly taken up by mitochondria where it prevents their oxidative damage and loss of Ca2+-buffering capacity leading to an attenuation of intracellular Ca2+ overload and prevention of calpain overactivation. However, when we waited until 6 h after injury, this protective effect observed at 1 h was no longer apparent in the case of early single-dose tempol treatment. This is probably because tempol has a relatively short half-life in mice (Kamataria et al, 2002). Accordingly, we administered repeated doses of tempol at 3 h intervals up to 12 h after injury and measured its effect at 24 h after injury. This experiment showed that this more aggressive multiple-dose tempol treatment had a partial inhibitory effect on calpain-mediated spectrin proteolysis. However, the therapeutic window for the achievement of this effect is disappointingly short as delayed initiation of tempol treatment until 1 or 2 h after injury failed to attenuate the 24 h peak of calpain-mediated α-spectrin degradation. This result may indicate that even though PN-induced oxidative damage is a critical early event in post-TBI injury, the potential therapeutic practicality of its inhibition is limited by a short therapeutic time window. Similarly, a short therapeutic window has also been reported for the antioxidant compound LY341122 in the rat fluid percussion TBI model (Wada et al, 1999).
Effect of Tempol on Motor Dysfunction and Neurodegeneration
The neuroprotective potential of a pharmacological compound that targets a particular secondary injury mechanism ultimately requires it to act to improve posttraumatic behavioral function and decrease neurodegeneration in injured brain areas. In the current study, we assessed motor functional status of brain-injured mice at 48 h and 7 days using the neuroscore test (Raghupathi et al, 1998). A significant forelimb and hindlimb motor deficit compared with sham noninjured mice was detectable at 48 h after injury in mice that received only the saline vehicle. Although the tempol group did not manifest a significant difference compared with the vehicle-treated group, neither the fore-limb nor the hind-limb functional scores in the tempol-treated mice were significantly differently from the sham group. This is consistent with the work of others who observed a beneficial effect of tempol on behavioral recovery in a rat TBI model (Beit-Yannai et al, 1996; Zhang et al, 1998).
After the 7-day motor functional assessment, we used the de Olmos aminocupric silver staining method to measure posttraumatic neurodegeneration (de Olmos et al, 1994; Switzer, 2000). Our previous studies using this method have shown that posttraumatic neurodegeneration peaks at 48 h in the CCI-TBI model and remains ongoing up to at least 7 days (Hall et al, 2005). Widespread silver staining in the cortex and subcortical tissues, including the hippocampus and thalamus, is observed. Using this method, we were able to show a significant attenuation of posttraumatic neurodegeneration in the injured hemisphere after tempol treatment. However, the effects of tempol on the volume of overall hemispheric neurodegeneration were modest (17.4% lower than that seen in the vehicle-treated mice). In the case of cortical neurodegeneration alone, tempol reduced cortical lesion volume by 32%, although still less than the neuroprotective effect that would have been predicted by the much larger decrease in protein oxidative damage and mitochondrial dysfunction.
There may be multiple reasons for the modest neuroprotective effect of tempol. Firstly, the testing of tempol for its ability to reduce calpain-mediated cytoskeletal degradation, which was assessed at 24 h after injury, only showed a 45% reduction in calpain-mediated cytoskeletal degradation. We suspect that the partial inhibition of calpain-mediated proteolysis is part of the reason why tempol did not have a more pronounced effect in preserving neuronal tissue. Secondly, in the present experiments, neurodegeneration was not examined until 7 days after injury rather than at its peak, which is 48 h after injury (Deng et al, 2007). This was done so that we could assess neurologic status up to 7 days before performing histopathology. However, the 7-day time point where less silver staining is seen compared with the 48 h peak may make it more difficult to observe a more robust neuroprotective effect. Thirdly, the half-life of tempol in mice is fairly short (i.e., <30 mins) (Kamataria et al, 2002) and therefore even repeated dosing with the compound is unable to maintain therapeutic levels adequately during the first 12 h after injury. Longer acting PN-directed antioxidants may be more effective because of the ability to maintain a steady, nonfluctuating radical scavenging effect.
Conclusion
In conclusion, despite the possibly limited therapeutic potential of tempol by itself, the protective efficacy of the compound is at least adequate to support our original hypothesis that PN-mediated oxidative damage is an upstream posttraumatic event that contributes to mitochondrial dysfunction, intracellular Ca2+ overload, calpain-mediated cytoskeletal degradation, and neurodegeneration, as illustrated in Figure 1. However, the observation that even though early tempol administration was able to almost completely attenuate early oxidative damage and mitochondrial dysfunction up to 12 h after injury, but only partially block cytoskeletal degradation and, even less so, posttraumatic neuronal loss, suggests that antioxidant treatment may need to be combined with other agents that perhaps target mitochondrial failure and/or calpain activation more directly to achieve a more pronounced and translatable neuroprotective strategy. However, this possibility will have to be carefully tested in future studies.