
Abstract
A new group of proteins, small ubiquitin-like modifier (SUMO) proteins, has recently been identified and protein sumoylation has been shown to play a major role in various signal transduction pathways. Here, we report that transient global cerebral ischemia induces a marked increase in protein sumoylation. Mice were subjected to 10 mins severe forebrain ischemia followed by 3 or 6 h of reperfusion. Transient cerebral ischemia induced a massive increase in protein sumoylation by SUMO2/3 both in the hippocampus and cerebral cortex. SUMO2/3 conjugation was associated with a decrease in levels of free SUMO2/3. After ischemia, protein levels of the SUMO-conjugating enzyme Ubc9 were transiently decreased in the cortex but not in the hippocampus. We also exposed HT22 cells to arsenite, a respiratory poison that impairs cytoplasmic function and induces oxidative stress. Arsenite exposure induced a marked rise in protein sumoylation, implying that impairment of cytoplasmic function and oxidative stress may be involved in the massive post-ischemic activation of SUMO conjugation described here. Sumoylation of transcription factors has been shown to block their activation, with some exceptions such as the heat-shock factor and the hypoxia-responsive factor, where sumoylation blocks their degradation, and the nuclear factor-κB (NF-κB) essential modulator where sumoylation leads to an activation of NF-κB. Because protein sumoylation is known to be involved in the regulation of various biologic processes, the massive post-ischemic increase in protein sumoylation may play a critical role in defining the final outcome of neurons exposed to transient ischemia.
Introduction
Transient cerebral ischemia is a severe form of metabolic stress that interferes with all major biochemical pathways. Ischemia-induced changes in gene expression have been investigated in various studies. Besides genomic and proteomic analyses to elucidate ischemia-induced changes in gene expression, interest has focused on post-translational protein modifications activated by transient cerebral ischemia. A prominent post-ischemic post-translational protein modification is phosphorylation of proteins resulting in activation or inhibition of various signal transduction pathways, including the mitogen-activated protein kinase, extracellular signal-regulated kinase, stress-activated protein kinase, and c-Jun N-terminal kinase pathways (Ferrer et al, 2003). Much attention has been given to phosphorylation of the α-subunit of the eukaryotic transcription factor 2 (eIF2α) because it is believed to be responsible for ischemia-induced shutdown of translation (Burda et al, 1994). Ischemia-induced phosphorylation of eIF2α has been found to be triggered by activation of the double-stranded RNA-dependent protein kinase-like endoplasmic reticulum kinase (Kumar et al, 2001), a protein kinase that is specifically activated under conditions associated with impairment of endoplasmic reticulum function (Harding et al, 1999). This implies that transient ischemia impairs endoplasmic reticulum function (Paschen and Doutheil, 1999), and illustrates that investigating ischemia-induced changes in post-translational modification of proteins could help to elucidate mechanisms underlying the pathologic process triggered by transient ischemia that culminate in cell death. This assumption is supported by the observation that transient ischemia induces a massive ubiquitination of proteins, and that with increasing recovery time poly-ubiquinated proteins form insoluble aggregates that trap proteins that are crucial for cellular functions, including initiation factors and ribosomes (Zhang et al, 2006).
A post-translational protein modification playing a pivotal regulatory role in multiple cellular processes is conjugation by the small ubiquitin-like modifier (SUMO) (Hay, 2005). Four SUMO proteins have been identified so far, SUMO1–4. Like post-translational modification of proteins by ubiquitination, sumoylation is brought about by a sequence of enzymatic reactions involving an activating enzyme (E1), a conjugating (E2), and a ligating enzyme (E3). The C-terminal carboxyl group of SUMO is ligated to the ε-amino group of the lysine residue in the SUMO conjugation motif of the target protein. Small ubiquitin-like modifier can be deconjugated from the target protein by the action of SUMO-specific proteases.
While ubiquitination targets proteins for degradation at the proteasome, sumoylation modifies stability, localization, and activity of transcription factors and other intracellular proteins. For many years, sumoylation of proteins was believed to be predominantly a nuclear process modifying transcription factors, nuclear pore proteins and other nuclear proteins that play a role in genome integrity (for reviews, see Melchior et al, 2003; Ulrich, 2005). Sumoylation of most transcription factors induces transcriptional repression with few exceptions as summarized in more detail in the Discussion. Sumoylation is a rapidly reversible process, but it may produce a long-lasting effect (Hay, 2005). One model to explain this conflicting observation is that after sumoylation SUMO-conjugated transcription factors are incorporated into a repression complex and that the factors are still retained in the repression complex after desumoylation (Hay, 2005). Recently, sumoylation of membrane proteins has been identified, including SUMO conjugation of the leak K2P1 K+ channel, the glucose transporters GLUT1 and GLUT4, and metabotropic glutamate receptors (for a review, see Scheschonka et al (2007)). Sumoylation of K2P1 was found to silence the plasma membrane channel (Rajan et al, 2005), whereas SUMO conjugation of glucose transporter proteins is believed to modulate receptor protein insertion and retrieval from plasma membrane (Giorgino et al, 2000).
Sumoylation of target proteins is activated by various stresses including oxidative stress, heat stress, and metabolic stress (Manza et al, 2004; Shao et al, 2004; Anckar et al, 2005). But no information is available regarding the signal transduction pathways that activate the sumoylation process. In a recent publication, the group of John M Hallenbeck provided evidence that protein sumoylation is massively increased in the hibernation torpor of ground squirrels, and the authors concluded that sumoylation may help to make cells of hibernating animals tolerant to the otherwise lethally low levels of blood flow reduction (Lee et al, 2007). Whether ischemia is associated with a change in the pattern of protein sumoylation has, to the best of our knowledge, never been investigated before. We show here that transient cerebral ischemia is associated with a massive activation of protein sumoylation. Sumoylation was also markedly increased in cell cultures exposed arsenite, a respiratory poison that impairs cytoplasmic function and induces oxidative stress. This implies that impairment of cytoplasmic function and oxidative stress may be involved in the ischemia-induced activation of protein sumoylation described here.
Materials and methods
Animal Experiments
This study was approved by the Duke University Animal Care and Use Committee. Transient severe forebrain ischemia was induced in male mice as described elsewhere (Sheng et al, 2000). In short, mice were fasted overnight but allowed free access to water. Mice were anesthetized with 3% isoflurane and the trachea was intubated with a 20-gauge intravenous catheter (Insyte-W, Becton-Dickinson, Sandy, UT). The isoflurane concentration was reduced to 1.8% of the inspired air, and the lungs were mechanically ventilated at a rate of 130 breaths/min with a delivered tidal volume of 0.7 mL. A needle thermistor was placed adjacent to the skull to monitor pericranial temperature. During ischemia and for 30 mins after onset of reperfusion pericranial temperature was servocontrolled at 37°C by surface cooling or heating. The right femoral artery was cannulated (PE 10, Becton-Dickson, Sparks, MD) to allow measurement of mean arterial blood pressure and blood gases, and the right internal jugular vein was cannulated to allow withdrawal of blood. Heparin (6 IU) was given intraarterially. A surgical incision was made to identify the common carotid arteries, which were encircled with sutures. To induce ischemia, blood was withdrawn from the venous catheter, reducing mean arterial blood pressure to 30 mm Hg, and the common carotid arteries were occluded. After 10 mins of ischemia, the carotid arteries were deoccluded and withdrawn blood was reinfused. Fifteen-microliter NaHCO3 solution (8.4%) was given intravenously, and catheters were removed. The wounds were infiltrated with bupivicaine and closed with sutures. At the end of the experiments, animals were reanesthetized with 5% isoflurane and decapitated. Brains were quickly removed, the hippocampus and cortex dissected out, and samples were immediately frozen and stored at −80°C until being used for analyses. Control mice were anesthetized and decapitated in the same way as experimental animals.
Cell Culture Experiments
HT22 cells, a stable murine hippocampal cell line (Davis and Maher, 1987), was used for the experiments. Cells were cultured in Dulbecco's minimal essential medium, supplemented with fetal calf serum (5%), and penicillin/streptomycin and amphotericin. Cells were plated on polyethelenimin-coated dishes and were used for the experiments at a density of approximately 70%. Cells were exposed to arsenite (100 μmol/L) for 3 h, a respiratory poison that impairs cytoplasmic function and induces oxidative stress. At the end of the experiments, cells were rapidly trypsinized, centrifuged at 4°C, and stored at −80°C.
Sample Preparation and Western Blot Analysis
Proteins were extracted with lysis buffer composed of β-glycerophosphate (50 mmol/L; pH 7.4), 1 mmol/L EDTA, 1 mmol/L EGTA, 0.5 mmol/L Na3VO4, 1% Triton X-100, 2% sodium dodecyl sulphate (SDS), and the protease inhibitor cocktail I (Sigma-Aldrich, St Louis, MO; 1%). Frozen brain samples were weighed in the cold, and the same volume of extraction buffer was used per mg of tissue. Samples were homogenized by a short sonication at 4°C for 10 secs, immediately followed by heating to 99°C for 5 mins to heat-inactivate enzymes. Homogenates were centrifuged at 14,000 × g for 10 mins at 4°C. Supernatant samples were mixed with 4 × loading buffer (0.25 mol/L Tris/Hcl (pH 6.8), 4% SDS, 40% glycerol, and 10% mercaptoethanol) and heated to 99°C for 10 mins. Discontinuous SDS-polyacrylamide electrophoresis (16%, 12%, 8%, and 4%) was used to separate free SUMO and SUMO conjugates, and 12% SDS-polyacrylamide electrophoresis was used to analyze ischemia-induced changes in Ubc9 protein levels. Ubc9 is the only SUMO conjugation enzyme identified so far. After electrophoresis, protein bands were transferred to nitrocellulose membranes (Hybond, Amersham Biosciences, Freiburg, Germany) at 4°C for 2 h. Membranes were blocked for 1 h in Tris/HCl-buffered saline solution (pH 7.6), supplemented with 0.1% Tween 20 (TBST) and 5% skim milk powder, and incubated with the first antibody for 16 h at 4°C. The following primary antibodies were used: rabbit polyclonal antibody against SUMO1 (by courtesy of Dr John M Hallenbeck), dilution 1:1000; rabbit polyclonal antibody against SUMO2/3 (by courtesy of Dr John M Hallenbeck), dilution 1:1000; mouse monoclonal antibody against Ubc9, dilution 1:250 (BD Transduction Laboratories, Franklin Lakes, NJ). After extensive washing with TBST, membranes were incubated with donkey anti-rabbit or donkey anti-mouse horseradish peroxidase conjugates (Affinity Bioreagents, Golden, CO; dilution 1:2000) for 1 h at room temperature. Bands were visualized using the ECL Western blot analysis system (Amersham Biosciences). After development of films, optical densities of bands were evaluated by image analysis. For analysis of ischemia- or arsenite-induced changes in levels of SUMO conjugates, the higher molecular weight area in each lane was cropped and analyzed, as indicated in the legends to the respective figures. To take into account any heterogeneity in film background for quantitative analysis of SUMO conjugation, the film background optical density of each lane was measured and subtracted from the respective SUMO conjugation optical density. A monoclonal antibody against β-actin (Sigma-Aldrich; dilution 1:5000) was used as loading control. Membranes were stripped and incubated with the monoclonal β-actin antibody.
Quantitative Polymerase Chain Reaction
Ischemia-induced changes in mRNA levels of SUMO2, SUMO3, and Ubc9 were evaluated by quantitative polymerase chain reaction (PCR). To illustrate that the brains used for analyses of mRNA levels responded in the expected way to transient cerebral ischemia, we also measured changes in hsp70 and heme oxygenase-1 (HO1) mRNA levels. For quantification, changes in individual mRNA levels were related to β-actin mRNA levels. Total RNA was extracted from hippocampal and cortical samples using the Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction. Two micrograms of total RNA was reverse-transcribed into cDNA using the reverse transcriptase reaction (Invitrogen) and a mixture of random hexamers (50 ng) and oligo(dT) (500 ng) as primers. Polymerase chain reactions were run in the LightCycler 2.0 (Roche Diagnostics GmbH, Mannheim, Germany). Quantification was achieved by using a standard curve created with serial dilutions of PCR products produced with the same sets of primers and run in the GenAmp PCR System 2700 (Applied Biosystems, Foster City, CA). The following sets of primers were used: for Ubc9, upper strand primer: 5′-GGTTTCAGGTG GAACTAAGGG-3¶ime;, lower strand primer: 5′-TGGTGG TGAGGACGGATAGTC-3¶ime;; SUMO2: upper strand primer: 5′-GGCAGGGTTTGTCAATGAGGC-3′, lower strand primer: 5′-CTGGAGTAAAGTAGCAGGCTC-3′; SUMO3: upper strand primer: 5′-GAGGCAGGGCTTGTCAATGAG-3′, lower strand primer: 5′-GGTCAGGACAACGGTTGGGTG-3′; hsp70: upper strand primer: 5′-AGGAGCGTACCCACGAGTGT, lower strand primer: 5′-GGAGATGACCTCCTGGCACTT; HO1: upper strand primer: 5-GCACAGGGTGACAGAA GAGGC-3′, lower strand primer: 5′-CACAGGGAGTGGGC TAGGGAC-3′; β-actin: upper strand primer: 5′-CTATTGG CAACGAGCGGTTCC-3′, lower strand primer: 5′-TTGGCA TAGAGGTCTTTACGG-3′.
Statistical Analysis
Data are presented as means ± s.d., with five independent brain samples and three independent tissue culture samples per group. Statistically significant differences between groups were evaluated by analysis of variance (ANOVA), followed by Fisher's protected least-significant differences test. A probability of 95% was taken to indicate significant differences between groups.
Results
Protein Modification with SUMO1
The pattern of protein modifications with SUMO1 in control samples and experimental samples derived from mouse brains subjected to 10 mins forebrain ischemia and 3 or 6 h of reperfusion is shown in Figure 1. In control samples from the hippocampus and cerebral cortex, several sumoylated proteins were identified with the SUMO1 antibody (Figure 1). We were, however, not able to visualize free SUMO1, possibly because levels were too low to be detected by the antibody. Transient forebrain ischemia did not induce major changes in the pattern of SUMO1 conjugation. When we cropped in each lane the higher molecular weight area (above 70 kDa) for analysis of ischemia-induced changes in SUMO1 conjugation, we observed a small but significant post-ischemic increase in protein sumoylation in the cerebral cortex (Figure 1C).
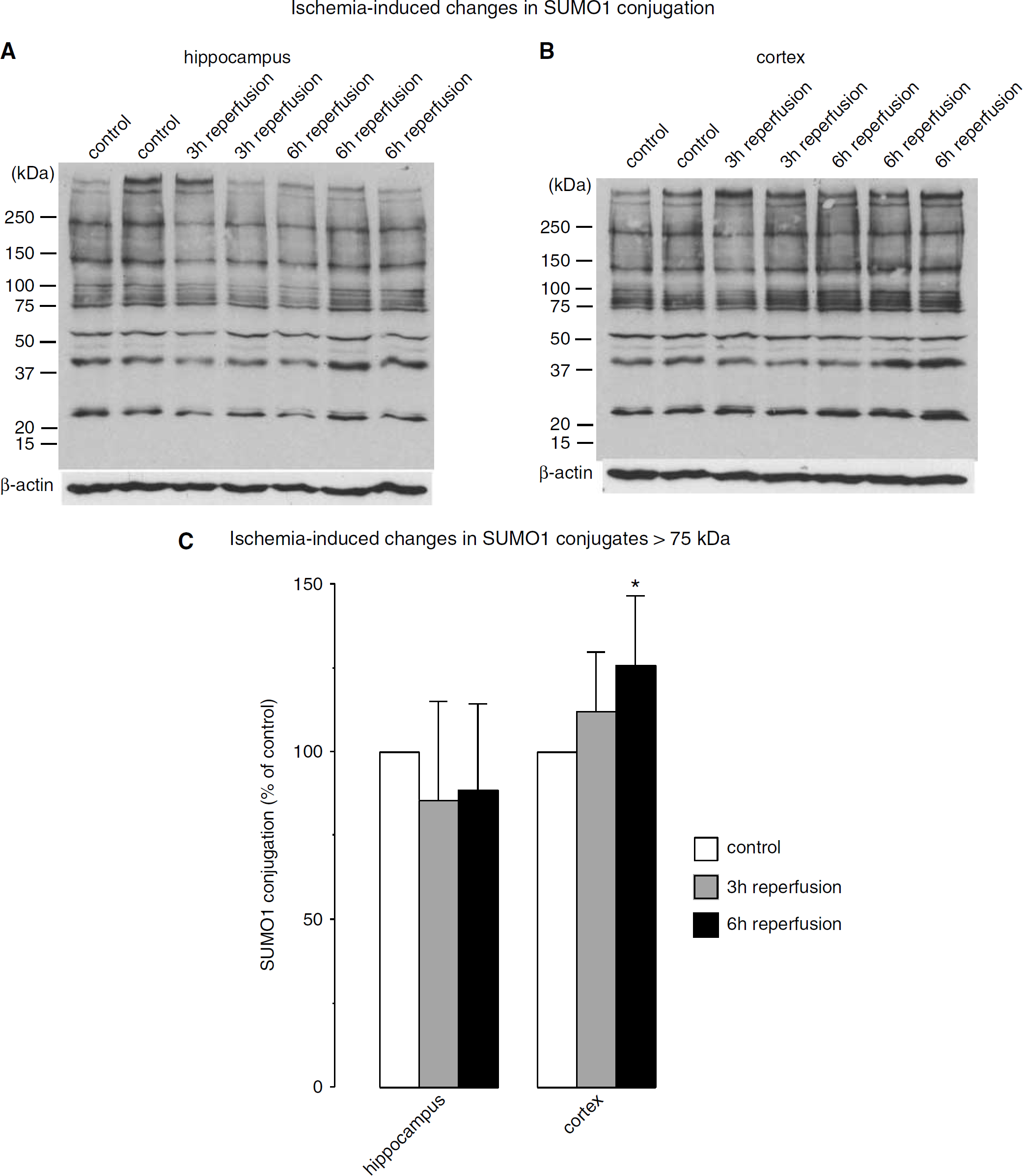
Effect of transient forebrain ischemia on protein modification with SUMO1. Mice were subjected to 10 mins transient forebrain ischemia and 3 or 6 h of reperfusion. The pattern of protein sumoylation was evaluated by Western blot analysis with samples taken from (
Protein Modification with SUMO2/3
The pattern of SUMO2/3 conjugation in control brains is illustrated in Figure 2A and summarized in Figure 2B. The extent of protein sumoylation differed significantly in the cortex and hippocampus. Although the optical densities of SUMO2/3 bands at approximately 45 and 35 kDa and free SUMO2/3 (at approximately 18 kDa) were significantly higher in the cortex than in the hippocampus implying larger levels of sumoylated proteins, the extent of the smear of sumoylated proteins above 170 kDa was larger in the hippocampus compared with the cortex. Ischemia-induced changes in SUMO2/3 conjugation are shown in Figures 3A and 3B and summarized in Figures 3E and 3F. For clarity of band identification, we used short and long exposure times to reveal SUMO2/3 conjugation and levels of free SUMO2/3, respectively. In samples from control brains, three prominent bands of SUMO2/3-conjugated proteins were identified exhibiting molecular weights of approximately 35, 45, and 170 kDa (Figures 3A and 3B). Free SUMO2/3 was detectable in samples from both the hippocampus and cerebral cortex. Free SUMO2/3 levels decrease markedly after ischemia, to 46.1 ± 12.0% and 18.8 ± 16.1% of control in the hippocampus, and to 16.3 ± 2.3% and 26.9 ± 19.6% of control in the cortex after 3 and 6 h of reperfusion, respectively (Figures 3C and 3D).
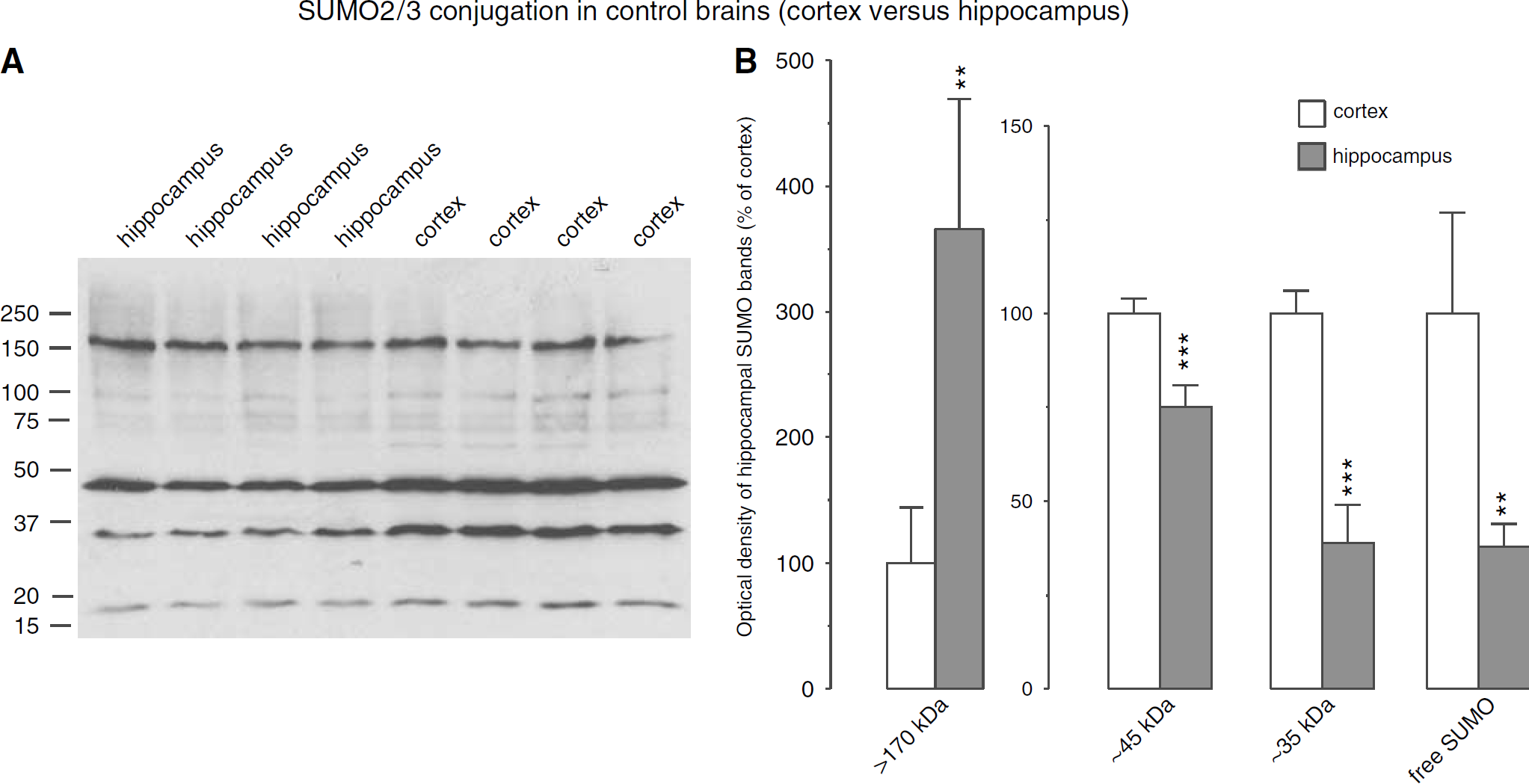
SUMO2/3 conjugation in control brains. (
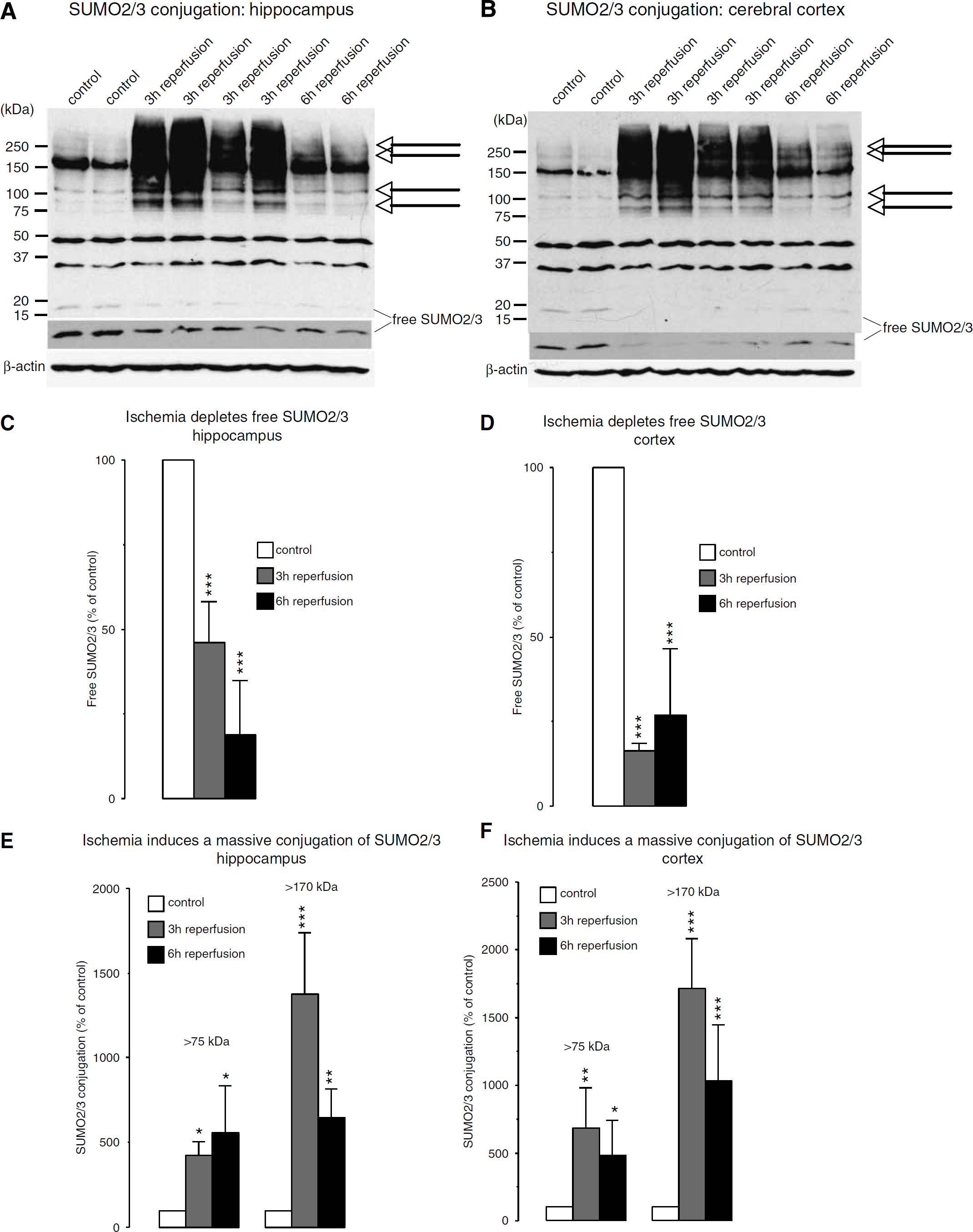
Effect of transient forebrain ischemia on protein modification with SUMO2/3. Mice were subjected to 10 mins transient forebrain ischemia and 3 or 6 h of reperfusion. The pattern of protein sumoylation was evaluated by Western blot analysis in samples taken from the hippocampus and cerebral cortex, as described in Materials and methods. (
Transient cerebral ischemia induced a massive increase in SUMO2/3 conjugation (Figures 3A and 3B). For evaluation of ischemia-induced changes in SUMO2/3 conjugation, we performed two analyses. We cropped the high molecular weight area above 75 or 170 kDa, respectively, from each lane and displayed results as percentage change relative to control. The ischemia-induced rise in SUMO2/3 conjugation was particularly pronounced when we cropped the high molecular weight area above 170 kDa from each lane, because at this high molecular weight there was very little SUMO2/3 conjugation in the control samples (Figures 3A and 3B and 3E and 3F). After 6 h of reperfusion, we observed a heterogeneous pattern of SUMO2/3 conjugation, with two animals displaying a moderate, and three animals exhibiting marked conjugation. After 3 h of reperfusion, we identified individual bands with markedly increased SUMO2/3 conjugation (Figures 3A and 3B, arrows) and a smear of bands. This suggests that SUMO2/3 conjugates are formed for many proteins under these conditions.
Arsenite Exposure Activates SUMO1 and SUMO2/3 Conjugation
We exposed HT22 cells, a stable cell line derived from murine hippocampal neurons, to arsenite, a respiratory poison that has secondary effects such as inducing oxidative stress and disturbing cytoplasmic function. In control cultures, we observed two prominent SUMO1 bands at approximately 75 and 85 kDa, and a smear of bands above 150 kDa (Figure 4A). SUMO2/3 conjugation, in contrast, was confined to the high molecular weight area above 100 kDa (Figure 4B). Arsenite induced a significant increase in SUMO1 and SUMO2/3 conjugation (Figure 4C). Free SUMO1 protein levels were not changed by arsenite exposure, whereas free SUMO2/3 protein levels were significantly reduced to 3 ± 3% of control (Figure 4D).
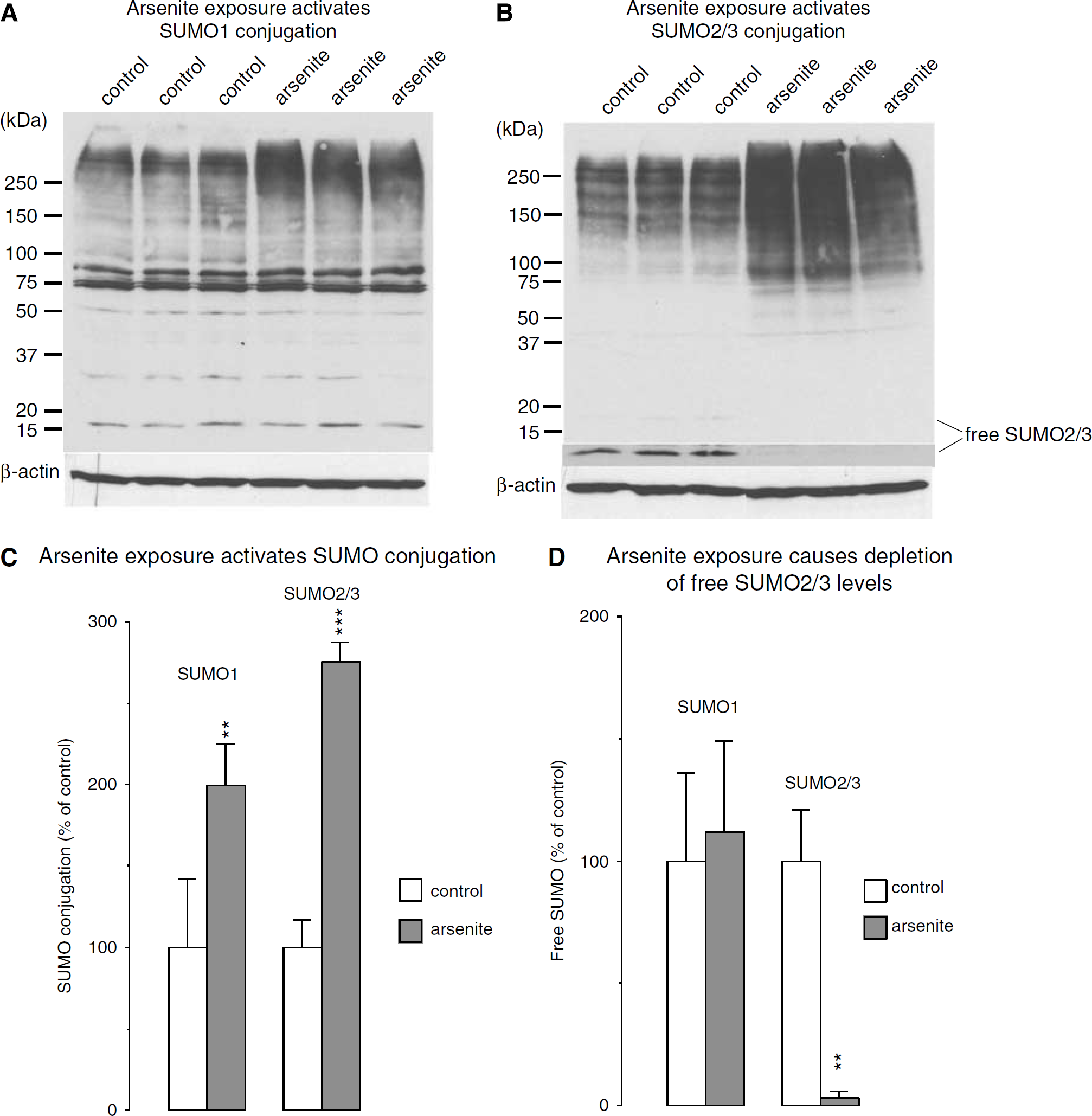
Changes in the pattern of protein sumoylation induced by exposing HT22 cells to the oxidative stressor arsenite. HT22 cells were exposed to 100 μmol/L arsenite for 3 h. Proteins were extracted and loaded on discontinuous SDS-polyacrylamide electrophoresis as described in Materials and methods. (
Transient Forebrain Ischemia Induces a Transient Reduction in Ubc9 Protein Levels in the Cortex
Ubc9 is the only SUMO-conjugating enzyme identified so far. To investigate whether transient forebrain ischemia causes any change in Ubc9 protein levels, we measured Ubc9 levels by Western blot analysis using the same samples as for SUMO conjugation analysis. We observed a marked transient decrease in Ubc9 protein levels in the cortex (to 26.9 ± 17.5% of control; P < 0.001) but not in the hippocampus (Figures 5A to 5C). Arsenite exposure, in contrast, induced a moderate increase in Ubc9 protein levels to 140 ± 12.9% of control (P < 0.05).
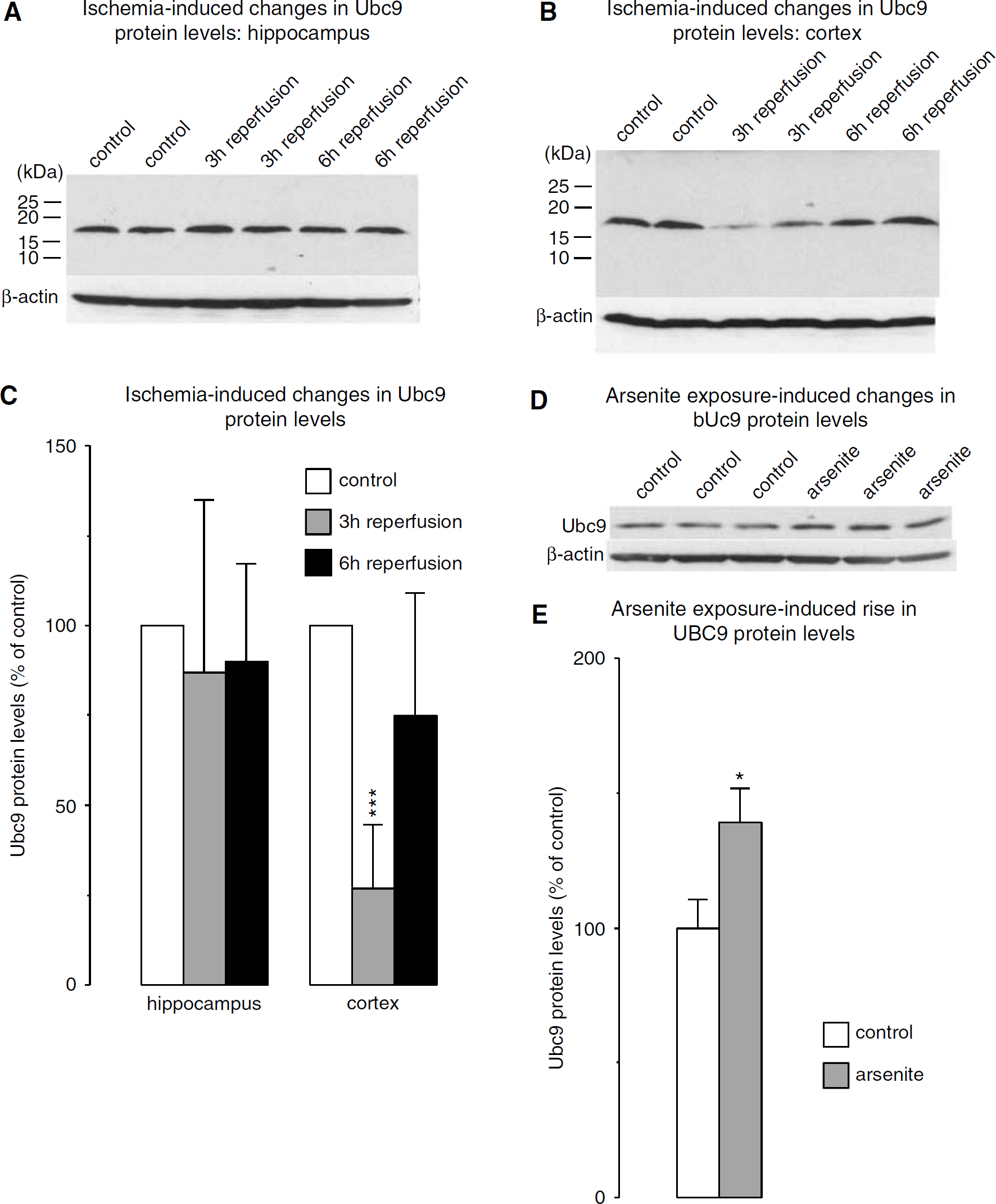
Stress-induced changes in protein levels of the SUMO-conjugating enzyme Ubc9. (
Transient Forebrain Ischemia did not Change mRNA Levels of SUMO2, SUMO3, or Ubc9
To investigate whether the severe form of metabolic stress induced by transient forebrain ischemia activates transcription of SUMO2, SUMO3, and Ubc9 genes, we evaluated ischemia-induced changes in the respective mRNA levels by quantitative PCR (Figure 6). We did not find any significant change in SUMO2, SUMO3, or Ubc9 mRNA levels in the hippocampus or cortex of animals subjected to 10 mins forebrain ischemia and 6 h of reperfusion (Figure 6A). To confirm that transient ischemia induced the expected genetic response, we also analyzed changes in hsp70 and HO1 mRNA levels in the same samples used for SUMO2/3 and Ubc9 quantitative PCR. We did indeed observe the expected post-ischemic rise in hsp70 and HO1 mRNA levels (Figure 6B). In the hippocampus and cerebral cortex, hsp 70 mRNA levels rose 67.3 ± 10.7- and 129.0 ± 30.7-fold and HO1 mRNA levels 14.1 ± 3.7- and 10.3 ± 4.8-fold, respectively.
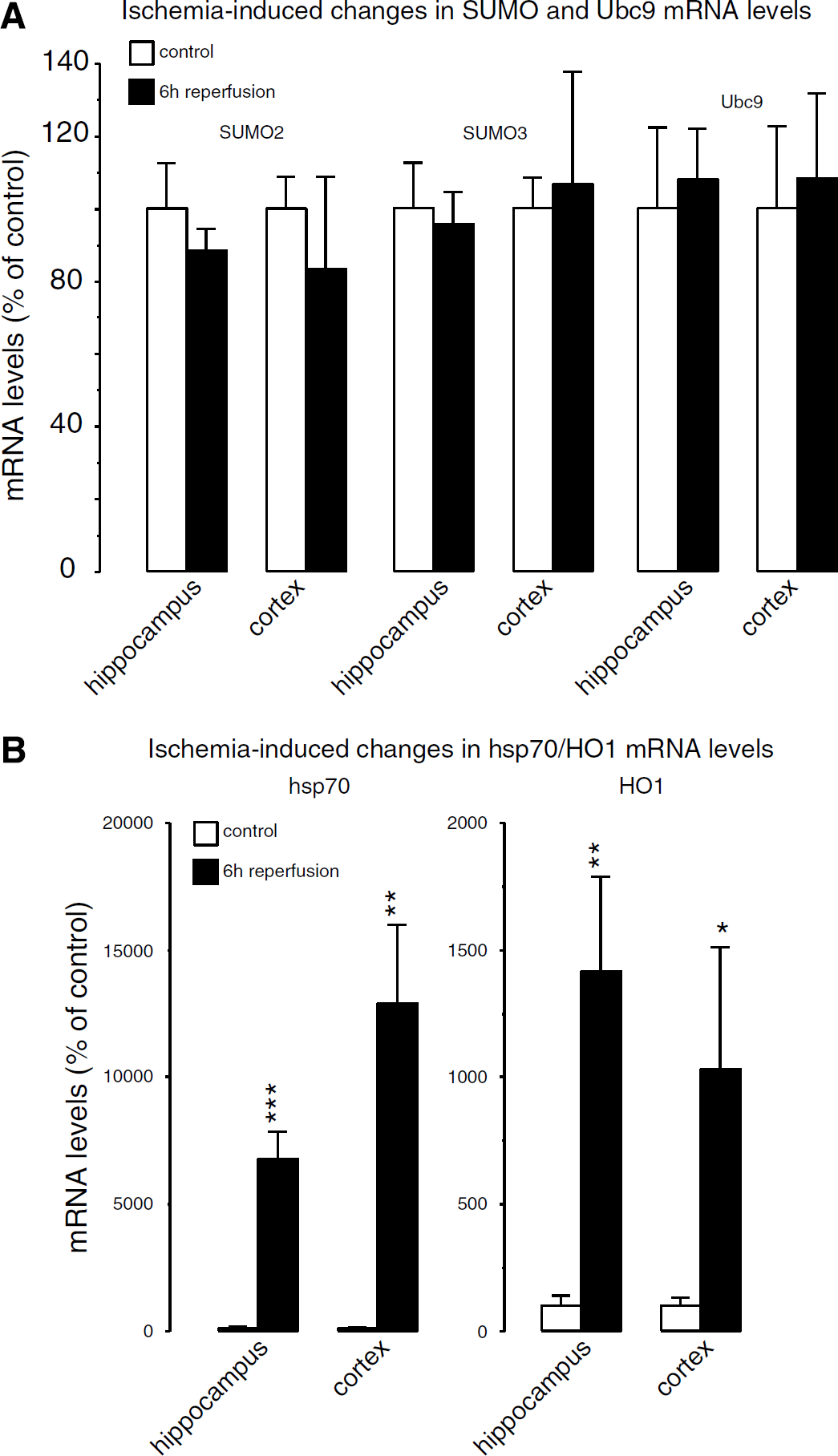
Ischemia-induced changes in mRNA levels of SUMO2, SUMO3, and Ubc9. Mice were subjected to 10 mins forebrain ischemia and 6 h of reperfusion, and mRNA levels of SUMO2, SUMO3, Ubc9 were analyzed by quantitative PCR and compared with ischemia-induced changes in mRNA levels of hsp70 and HO1. Transient ischemia did not induce changes in SUMO2, SUMO3, or Ubc9 mRNA levels (
Discussion
We describe here the results of experimental studies showing that 10 mins transient forebrain ischemia causes a huge increase in proteins sumoylation. This is, to the best of our knowledge, the first study investigating ischemia-induced changes in the pattern of protein modification by SUMO. The pattern of proteins modified by SUMO1 and SUMO2/3 differed considerably in the control state and was even more pronounced after transient forebrain ischemia. In samples from control animals, we found many SUMO1-conjugated proteins but only very few prominent SUMO2/3-conjugated proteins. This implies that in the nonstressed brain SUMO1 conjugation plays a more important role than SUMO2/3 conjugation. This sumoylation pattern changed significantly after ischemia. We did not find any major change in the SUMO1 conjugation pattern. SUMO2/3 modification, however, was massively increased after ischemia, suggesting that SUMO2/3 conjugation plays an important role in the cellular response to the metabolic stress induced by transient interruption of the blood supply. One plausible explanation for the different response of SUMO1 and SUMO2/3 conjugation to transient forebrain ischemia is that there are already many SUMO1-conjugated proteins in the nonstressed state and there is therefore not enough free SUMO1 to further activate SUMO1 conjugation after ischemia. We were indeed unable to identify free SUMO1 in our brain samples. Free SUMO2/3, in contrast, was present in our control brain samples and it markedlydecreased after ischemia when SUMO2/3 conjugation was massively increased. This assumption is supported by the observation that in samples from HT22 cells we could identify both free SUMO1 and SUMO2/3, and oxidative stress was associated with a marked increase in SUMO1 and SUMO2/3 conjugation.
After transient ischemia, Ubc9 protein levels did not change in the hippocampus but were transiently decreased in the cortex. This was an unexpected finding, because Ubc9 is the only SUMO-conjugating enzyme identified so far, and it is therefore difficult to understand why SUMO2/3 conjugation should be massively increased in the cortex after ischemia at a reperfusion time when levels of the SUMO-conjugating enzyme were reduced. This pattern is different to the pattern observed inHT22 cells exposed to oxidative stress where a marked increase in SUMO conjugation was associated with a moderate increase in Ubc9 protein levels. Protein sumoylation is a complex reversible process involving SUMO conjugation and desumoylation (Di Bacco and Gill, 2006; Il Kim and Baek, 2006). A plausible explanation for the massive increase in SUMO2/3 conjugation coinciding with a decreased in Ubc9 protein levels in the cortex is that transient ischemia may suppress cleavage of SUMO2/3 from sumoylated proteins, thereby inducing a net rise in levels of SUMO2/3-conjugated proteins. An alternative plausible explanation is that Ubc9 is not a limiting factor in the sumoylation process.
It is well established that oxidative stress plays a role in the pathologic process induced by transient ischemia and culminating in neuronal cells death(Chan et al, 1998). We show here that arsenite exposure induced a marked increase in SUMO1 and SUMO2/3 conjugation. Arsenite is a respiratory poison that has been shown to induce oxidative stress, to suppress protein synthesis by phosphorylation of eIF2α, and to activate the heat-shock response (Bernstam and Nriagu, 2000; Mengesdorf et al, 2002; Roybal et al, 2005), a stress-response pattern similar to that found after transient ischemia. The observation that arsenite exposure induces a marked activation of sumoylation implies that impairment of cytoplasmic function and oxidative stress may be involved in ischemia-induced activation of SUMO2/3 conjugation.
What are the possible consequences of the massive increase in SUMO2/3 conjugation observed after transient forebrain ischemia? It has been shown that sumoylation changes the stability, localization, and activity of proteins. Many sumoylated proteins have been identified, and it has been demonstrated that a major fraction of SUMO-conjugated proteins are transcription factors. It is widely believed that the activity of most transcription factors is suppressed by sumoylation. For example, desumoylation of SUMO-conjugated proteins by SUMO-specific proteases have been shown to play a role in the development of prostate cancer through the activation of androgen receptors, c-Jun and cyclin D1 (Cheng et al, 2006). Furthermore, SUMO conjugation of c-Jun and c-Fos has been shown to downregulate the transcriptional AP-1 complex (Bossis et al, 2005). There are, however, some exceptions, including the heat-shock factor HSF-1α where sumoylation has been shown to increase its stability and transcriptional activity (Bae et al, 2004). A more complex example is the transcription factor nuclear factor-κB (NF-κB), which is activated after ischemia (for a review, see Mattson and Meffert, 2006) and associated with inflammation, cell survival, and apoptosis (Karin and Ben-Neriah, 2000). In the physiologic state, NF-κB activation is blocked by the inhibitory protein IκB. Stress associated with NF-κB activation results in phosphorylation of IκB, which is then ubiquitinated and degraded at the proteasome. Small ubiquitin-like modifier competes with ubiquitin for the same modification site. Sumoylation of IκB therefore suppresses ubiquitination and degradation (Desterro et al, 1998). However, sumoylation of the NF-κB essential modifier results in activation of NF-κB (Huang et al, 2003). As a consequence, SUMO conjugation may result in both NF-κB activation and suppression of NF-κB activation.
The results of cell culture experiments suggest that activation of sumoylation enhances protection from the stress induced by transient oxygen/glucose deprivation, an experimental paradigm modeling the metabolic stress induced by transient cerebral ischemia in vivo (Lee et al, 2007). Whether the massive activation of SUMO conjugation observed here after transient forebrain ischemia helps cells to recover from ischemia remains to be elucidated in further experiments. Because, however, sumoylation has a major influence on the stability, localization, and activity of intracellular proteins, the outcome of ischemia is unlikely to be wholly unrelated to the post-ischemic activation of protein sumoylation. To elucidate the significance of this process it will be of key importance to answer the following questions: (1) What is the temporal profile of post-ischemic SUMO conjugation? (2) Does preconditioning activate sumoylation and how does it affect SUMO conjugation induced by ischemia? (3) Which proteins are sumoylated after ischemia and what are the possible consequences for the respective proteins? (5) Can the sumoylation process be modified in such a way as to make the brain more tolerant to the severe form of metabolic stress induced by transient ischemia? Experiments are under way to address these questions.
Footnotes
Acknowledgements
The authors are grateful to Dr John M Hallenbeck, Laboratory of Gene Regulation and Development, National Institute of Neurological Disorders and Stroke, National Institutes of Health, for providing SUMO1 and SUMQ2/3 polyclonal antibodies.