
Abstract
Cerebral ischaemia usually results in the rapid death of neurons within the immediate territory of the affected artery. Neuronal loss is accompanied by a sequence of events, including brain oedema, blood-brain barrier (BBB) breakdown, and neuroinflammation, all of which contribute to further neuronal death. Although the role of macrophages and mononuclear phagocytes in the expansion of ischaemic injury has been widely studied, the relative contribution of these cells, either of exogenous or intrinsic central nervous system (CNS) origin is still not entirely clear. The purpose of this study, therefore, was to use different durations of transient middle cerebral artery occlusion (tMCAo) in the mouse to investigate fully post-occlusion BBB permeability and cellular changes in the brain during the 72 h post-MCAo period. This was achieved using in vivo magnetic resonance imaging (MRI) and cell labelling techniques. Our results show that BBB breakdown and formation of the primary ischaemic damage after tMCAo is not associated with significant infiltration of neutrophils, although more are observed with longer periods of MCAo. In addition, we observe very few infiltrating exogenous macrophages over a 72 h period after 30 or 60 mins of occlusion, instead a profound increase in proliferating resident microglia cells was observed. Interestingly, the more severe injury associated with 60 mins of MCAo leads to a markedly reduced proliferation of resident microglial cells, suggesting that these cells may play a protective function, possibly through phagocytosis of infiltrating neutrophils. These data further support possible beneficial actions of microglial cells in the injured brain.
Introduction
Experimental paradigms of focal cerebral ischaemia show that tissue damage includes a rapidly and irreversibly damaging core and a surrounding region, known as the ‘penumbra,’ which is potentially salvageable (Ginsberg, 2003). In addition to the early events caused by compromised cerebral blood flow, there is also a delayed inflammatory response that takes place hours to days after ischaemia. Post-ischaemic inflammatory events include activation of brain resident microglia and astrocytes (Clark et al, 1993; Davies et al, 1998; Morioka et al, 1993), as well as the infiltration of exogenous leukocytes such as polymorphonuclear granulocytes (Barone et al, 1991), macrophages (Yamagami et al, 1999), and lymphocytes (Schroeter et al, 1994) to the brain, which can increase neuronal damage (Barone et al, 1991; Dinkel et al, 2004; Jiang et al, 1995). The contribution of activated resident microglia versus infiltrating macrophages to ischaemic brain damage has been difficult to define because of similar immunophenotypes and other histological characteristics for these cells (Campanella et al, 2002; Davis et al, 1994; Zhang et al, 2005). Experiments with bone marrow chimeric mice have indicated significant differences in terms of the ratio and contribution of resident microglia versus exogenous infiltrating macrophages to early (up to 3 days) post-ischaemic inflammatory processes. After middle cerebral artery occlusion (MCAo), massive infiltration of exogenous macrophages was reported 24 to 48 h after the insult as well as a profound dominance of resident microglia over infiltrating blood-borne macrophages during the first 4 days of reperfusion (Kokovay et al., 2006; Schilling et al, 2003, 2005; Tanaka et al, 2003). Unfortunately, the evolution of the ischaemic damage was not correlated with the numbers of microglia/macrophage cells in these studies. It is known that microglia can enter the cell cycle and exert proliferating activity in the brain on various challenges, such as acute brain injury (Gowing et al, 2006; Ladeby et al, 2005) and global cerebral ischaemia (Liu et al, 2001). Microglial proliferation may therefore contribute to increases in the number of mononuclear phagocytes in the brain parenchyma, although the time course and correlation with ischaemic damage of this response has not been studied in detail.
Therefore to investigate cellular changes after MCAo, we assessed the infiltration of mononuclear phagocytes into the brain in vivo and correlated this with intactness of the blood-brain barrier (BBB) (by contrast enhanced magnetic resonance imaging (MRI)), evolution of oedema, and tissue damage (by diffusion and T2 MRI), as well as with histological analysis regarding the number of cells of different origin, and finally, damage formation. As it is likely that the duration of ischaemia and hence extent of neuronal injury will impact on these parameters, we studied two different MCAo periods, namely 30 and 60 mins.
We show that after tMCAo, microglial proliferation is the main factor behind the increase in the number of mononuclear phagocytes seen during the first 3 days and that the number of activated microglial cells negatively correlates with the extent of ischaemic brain damage. These findings suggest an advantageous role of newly produced brain-resident microglia in the post-ischaemic brain possibly owing to phagocytosis of neutrophils.
Materials and methods
Animals
Experiments were carried out in adult male C57Bl/6J mice (Harlan-Olac, UK) aged 12 to 14 weeks weighing 26 to 29 g (n = 62). Animals had free access to food and water and were maintained under temperature, humidity, light-controlled conditions, under 12-h light/12-h dark cycle, with lights on at 0700 hours. All animal procedures were performed under an appropriate Home Office License and adhered to regulations as specified in the Animals (Scientific Procedures) Act (1986).
Transient Middle Cerebral Artery Occlusion
Anesthesia was induced with 1.5% isoflurane in a mixture of 30% oxygen and 70% nitrous oxide. During surgery, core temperature was monitored with a rectal probe and maintained at 37 ± 0.5°C, using a homeothermic blanket (Harvard Apparatus, UK). Animals were exposed to MCAo for 30 or 60 mins as described previously (Wheeler et al, 2003), except that laser-Doppler was not used. Sham-operated animals had the common carotid artery clipped for 30 or 60 mins. In some control animals, the filament was advanced along the ICA until the origin of MCA and was retracted immediately, as a control for endothelial disruption by the filament.
Magnetic Resonance Imaging Scanning and Data Analysis
Mice were re-anaesthetized 4 h after tMCAo and underwent MRI scanning using a horizontal 7 T SMIS system (Guildford, Surrey, UK). A single loop surface coil (3 cm in diameter) was used for both signal reception and transmission. Ten 0.8-mm-thick coronal slices (slice gap 0.8 mm) were selected to cover the forebrain. T1 images were acquired with TR = 700 ms, TE = 10 ms, FOV = 30 mm, 256 × 128 (zero filled to 256 × 256 before FT). For T2 images TR was 1,500 ms and TE either 30 or 60 ms. Diffusion images were obtained with TR = 1,800 ms, TE = 60 ms and b = 1,000 secs/mm2 diffusion weighting applied along the slice selection gradient, data matrix 128 × 128. AMI-227 (Ferumoxtran-10, Sinerem®, Guerbet/France) 160 μmol of Fe/kg together with Magnevist (0.1 mmol/kg) was injected via the tail vein of conscious mice before (n = 8) or immediately after (n = 14) 30 mins of MCAo. Mice were re-injected with AMI-227/Magnevist 24, 48, and 72 h after MCAo occlusion (last AMI injection was injected 2 to 3 h before the last MRI scanning and 4 to 5 h before transcardial perfusion, respectively), anaesthetized with isoflurane, and scanned for MRI as described above.
The images were analysed using the software provided by SMIS. The areas with signal changes were manually delineated and percentage volumes of T1, T2, or diffusion abnormalities of total hemispheric volume determined.
In Situ Blood Cell Labelling with CFSE
Circulation-related blood cells were labelled in situ with carboxyfluorescein diacetate succinimidyl ester, injected via the tail vein into conscious, restrained animals. To label almost all blood cells, a slightly modified injection protocol was used (Becker et al, 2004). Carboxy fluoresce in diacetate succinimidyl ester (0.5 μg; Molecular Probes, Eugene, OR, USA) was dissolved in 90 μL of high-quality dimethylsulphoxide according to the manufacturer's instructions. Stock solution (19 μL) was mixed with 80 μL of 100% ethyl alcohol, 300 μL of phosphate-buffered saline and with 0.5 μL of Pluronic F127 (Molecular Probes, Eugene, OR, USA), immediately before administration. After thorough mixing, 9 μL/g body weight of the labelling solution was injected into the tail vein at an average rate of 50 μL/min. Carboxyfluorescein succinimidyl ester (CFSE) was administered 4h before 30 (n =17) or 60 (n = 12) mins of MCAo followed by 4, 24, 48, 72 h after MCAo, and 4, 24, 48 h after 30 (n = 14) or 60 (n = 12) mins of MCAo.
Blood samples were routinely collected to 3.8% sodium citrate (Sigma-Aldrich, UK) 30 mins, 4, 8, and 24 h after tracer administration. Cellular labelling with CFSE was evaluated by fluorescent microscope on native blood and bone marrow samples, as well as on leukocytes prepared from blood samples by hypoosmotic lysis with ACK solution (8.4 g/L ammonium chloride, 1 g/L potassium bicarbonate, and 1 mmol sodium ethylenediaminete-traacetic acid).
Administration of Bromodeoxyuridine
Bromodeoxyuridine (BrdU) (Sigma-Aldrich) was dissolved in 0.9% NaCl and 50 mg/kg was administered intraperitoneally twice daily after 30 or 60 mins of MCAo (n = 5 to 7/group). Mice were killed 24, 48, and 72 h after MCAo. In a separate set of experiments, mice (n = 8) received a single BrdU injection 22, 46, or 70 h after MCAo (2 h before transcardial perfusion). These experiments were performed to reveal the in situ dividing cells in the brain, testing the theoretical possibility that exogenous macrophages, which have incorporated BrdU migrate into the brain and might escape identification because of dilution of CFSE by cellular division, thereby appearing falsely as dividing microglia.
Tissue Processing
Under terminal anesthesia, mice were transcardially perfused with 10 mL saline followed by 40 mL 4% paraformaldehyde (PFA). Brain, spleen, femoral, and tibial bones (to sample bone marrow) were removed and post-fixed in 4% paraformaldehyde at 4°C for 24 h. After cryoprotection of brains and spleens in 20% sucrose/phosphate-buffered saline for 24 h, five alternate sets of 20 μm coronal brain sections and two sets of spleen sections were cut on a sliding microtome. All sections were collected into an antifreeze solution (containing 30% ethylene glycol and 20% glycerol in phosphate-buffered saline) and stored at 20°C until processing.
Measurement of Infarct Volume using Histology
The volume of ischaemic damage was measured using a modification of the method described previously (McColl et al, 2004). Briefly, areas of ischaemic damage were identified on cresyl violet-stained sections at eight neuroanatomically defined coronal levels. Digitized images were created and areas of damage measured using ImageJ software. The volume of damage was calculated by integration of areas of damage with the distance between coronal levels. The end points for integration were 2.9 mm (rostral limit) and −4.9 mm (caudal limit) regarding bregma. Volumes are expressed as a percentage of total hemispheric volume.
Immunofluorescence
Double- or triple-labelling immunofluorescence was performed on free-floating sections. After blocking in 2% normal donkey or goat sera (Vector Laboratories, Burlingame, CA, USA) sections were incubated overnight at 4°C in various mixtures of the following primary antibodies: monoclonal rat anti-mouse F4/80 1:400 (Serotec, UK), rat anti-mouse CD45 1:250 (Serotec, UK), polyclonal rabbit anti-Iba1 1:2,000 (Wako Chemicals, Germany), rat anti-mouse NIMP-R14 1:100 (Hycult Biotechnology, Uden, The Netherlands), rat anti-mouse BrdU 1:400 (Serotec, UK), goat anti-BrdU 1:1000 (Abcam, UK), rat anti-mouse CD34 1:400 (Pharmingen, San Diego, CA, USA). For detection of BrdU-labelled cells, sections were pretreated for 30 mins in 1 N HCl at 37°C.
The antigens were visualized with the adequate fluorochrome-conjugated secondary antisera (donkey anti-rat Alexa 594, goat anti-rabbit Alexa 594, donkey anti-goat Alexa 594, or donkey anti-rabbit Alexa 350, Molecular Probes) used in 1:500 dilutions for 2 h at room temperature. Sections were mounted onto gelatin-coated slides and cover-slipped with Vectashield mounting medium (Vector Laboratories) or with Vectashield mounting medium containing diamidinophenylindole.
To localize microglial cells and brain macrophages, sections were incubated with 6 μg/mL biotinylated tomato lectin (Sigma, UK) for 2 h at room temperature according to Acarin et al (1994) and visualized in streptavidin-conjugated Alexa-350, 1:500 (Molecular Probes).
Mounted sections and stained cell cultures were analysed and images were viewed using an Olympus microscope. Double- or triple-fluorescent images were generated using MetaView software (Universal Imaging Corp., Downingtown, PA, USA). Quantification was performed on fluorescent images using the ImageJ (NIH, Bethesda, MD, USA) software. With the exception of phasing the brightness of the images and mild contrasting of pictures, no other modifications of presented images were done.
Histological Detection of AMI-227
Iron oxide particles were detected with Prussian Blue staining (2% HCl mixed with 2% potassium hexacyanoferrat Fe2 + (1:1), freshly prepared) on bone marrow and blood smears, blood leukocyte preparations, spleen, and on brain sections. On two sets of brain and spleen sections, after Prussian Blue staining diaminobenzidine enhancement was performed combined with subsequent silver/gold intensification as described recently (Schroeter et al, 2004). One set of sections was mounted and the histochemical iron staining was performed as originally described. The other set of free-floating sections was incubated in Prussian Blue solution and developed with diaminobenzidine, and then mounted and silver/gold intensification was performed. After the staining procedure sections were dehydrated and coverslipped with Depex mounting medium.
Statistical Analysis
Data are expressed as mean ± s.d. One-way analysis of variance followed by post hoc Bonferroni's comparison was used to assess statistical differences in the cell numbers and proliferation among the different groups.
Results
Magnetic Resonance Imaging shows Blood-Brain Barrier Breakdown with no Significant Infiltration of AMI-227 Labelled Cells after Middle Cerebral Artery Occlusion
T1-weighted MRI after Magnevist injection revealed no signs of contrast agent leakage 4 h after 30 mins of MCAo, although BBB leakage in all mice at 24 h after MCAo was evident (Figure 1A). T2-weighted MRI showed the ischaemic lesion and oedema were complete by 24 h after MCAo, whereas diffusion MRI images revealed consistent size of abnormal brain volume with those determined by T2-weighted MRI (Figures 1A and 1B).
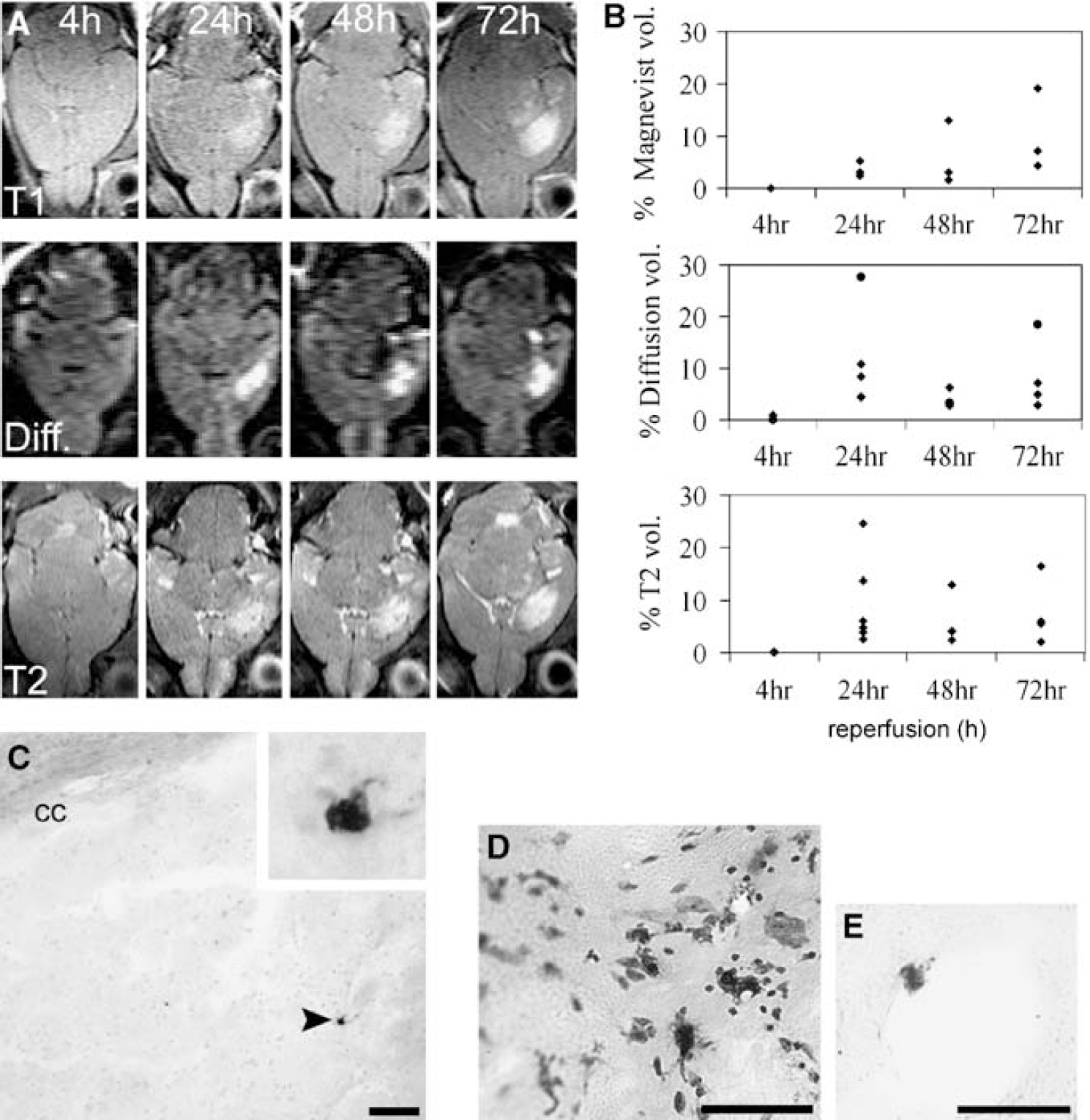
MR images showing damage evolution and histological verification of infiltration of iron-laden macrophages. (
AMI-227 is taken up by reticuloendothelial cells in vivo, including circulating macrophages. Owing to its superparamagnetic property intracellular iron oxide nanoparticles cause strong T*2 (Rausch et al, 2001) and T2 (Kavec et al, 2004) signal decrease as well as T1-weighted signal (Rausch et al, 2001) decrease. Interestingly, T2-weighted MRI showed no signal void areas in or surrounding the ischaemic lesion in the mice injected repeatedly with AMI-227 (data not shown). This finding suggests that no leakage of this intravascular contrast agent through the BBB had occurred and/or that no significant number of AMI-227 containing cells were present in the post-ischaemic brain.
Iron-laden cells were detectable with Prussian Blue staining (with or without diaminobenzidine and silver/gold intensification) in bone marrow, blood smears, and in spleen sections up to 72 h after injection of AMI-227 (data not shown). However, although animals undergoing MCAo showed many more iron-laden cells in blood smears than sham-operated mice, in the brain parenchyma the average number of iron-laden cells was surprisingly low, with only a few single cells being seen in the ischaemic striatum (Figure 1C). Rarely, exogenous AMI-containing cells were recruited into the ischaemic striatum in small clusters, but the maximal number of these cells never exceeded 6 to 8 cells/section in any of the examined brain sections (Figure 1D). Scattered iron-laden macrophages were also sometimes found attached to the wall of blood vessels (Figure 1E). Therefore, the absence of T2-weighted MRI or Prussian Blue staining evidence for AMI-227 labelled cells in the brain parenchyma indicates that there is virtually no exogenous macrophage infiltration into the brain after MCAo.
In vivo Cell Labelling Confirms the Lack of Significant Exogenous Macrophage Infiltration in the Ischaemic Brain
To clarify further whether exogenous circulation-related cells enter the brain, CFSE leakage into the brain independent from cellular infiltration and BBB damage was determined in the brains of sham-operated animals 4 and 24 h after carboxyfluorescein diacetate succinimidyl ester injection. Cellular CFSE labelling was never observed in the parenchyma and only occasional labelling was present in the wall of blood vessels. Strong CFSE uptake was observed in certain cells of the circum-ventricular organs (eminentia mediana and area postrema) and in the choroid plexus, although resident ependymal cells in these structures were not positive to the CD45 (common leukocyte) antigen (Figure 2A).
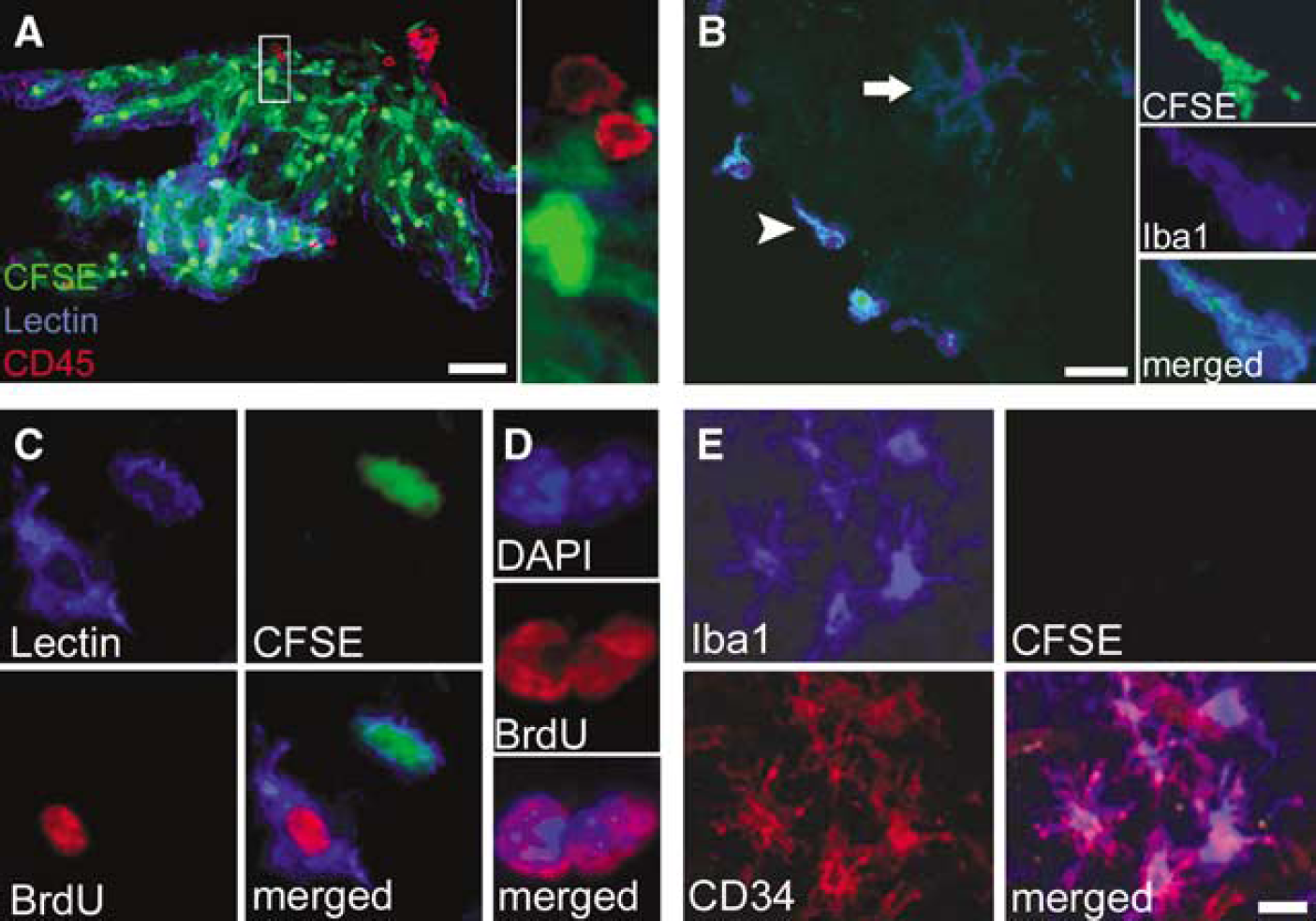
Proliferation, but not infiltration of exogenous macrophages, is the main mechanism underlying early increase in phagocyte numbers in the ischaemic brain. CFSE staining reveals qualitative differences between infiltrating macrophages and resident microglia in the brain parenchyma after MCAo. (
Twenty-four hours after 30 or 60 mins of MCAo, CFSE-positive macrophages appeared at the meninges, only very few being found infiltrated into the brain parenchyma (Figure 2B), which is in support of the AMI-227 findings. After MCAo (48 to 72 h), the number of CFSE-positive macrophages slightly increased in the ipsilateral hemisphere, but never exceeded 2% to 3% of the Iba1-positive cells.
Microglial Proliferation rather than Infiltration of Exogenous Macrophages Increases the Population of Mononuclear Phagocytes in the Brain Parenchyma after Middle Cerebral Artery Occlusion
Resident microglia exhibited intense proliferating activity at 48 and 72 h, as revealed by BrdU uptake (Figure 2C). CFSE-positive infiltrating macrophages were void of BrdU labelling both 48 and 72 h after 30 and 60 mins of MCAo. Processes of cellular division and BrdU uptake were observed in several diamidinophenylindole-stained microglial nuclei in the ischaemic hemisphere (Figure 2D), indicating that proliferation takes place locally in the parenchyma. Occasionally, dividing microglia contained CFSE-positive particles in their cytoplasm, as a result of phagocytosis of infiltrating neutrophils (see below), but this labelling never extended to the whole cytoplasm with either 30 or 60 mins of MCAo.
Iba1-positive microglia exhibited increased staining over time to the stem cell antigen, CD34 (Figure 2E) in the ischaemic striatum. BrdU-labelled microglia located under the corpus callosum and near the left ventricle were CD34-positive 24 h after 30 and 60 min of MCAo, the remaining dividing microglial cells in the ipsilateral hemisphere being positive by 72 h after MCAo.
Reduced Numbers of Microglia after Middle Cerebral Artery Occlusion are Associated with more Severe Injury
There was no difference in the number of Iba1-positive cells during the 72 h examination period on the contralateral hemisphere and Iba1 numbers were similar between cases of 30 and 60 mins of MCAo (207 ± 14 cells/mm2 in the striatum, 223 ± 17 cells/mm2 in the cortex after 30 mins of MCAo and 202 ± 12 cells/mm2 in the striatum, 218 ± 12 cells/mm2 in the cortex after 60 mins of MCAo at 72 h reperfusion). The number of Iba1-positive cells in the contralateral hemisphere was similar to that observed in sham animals (Figure 3D).
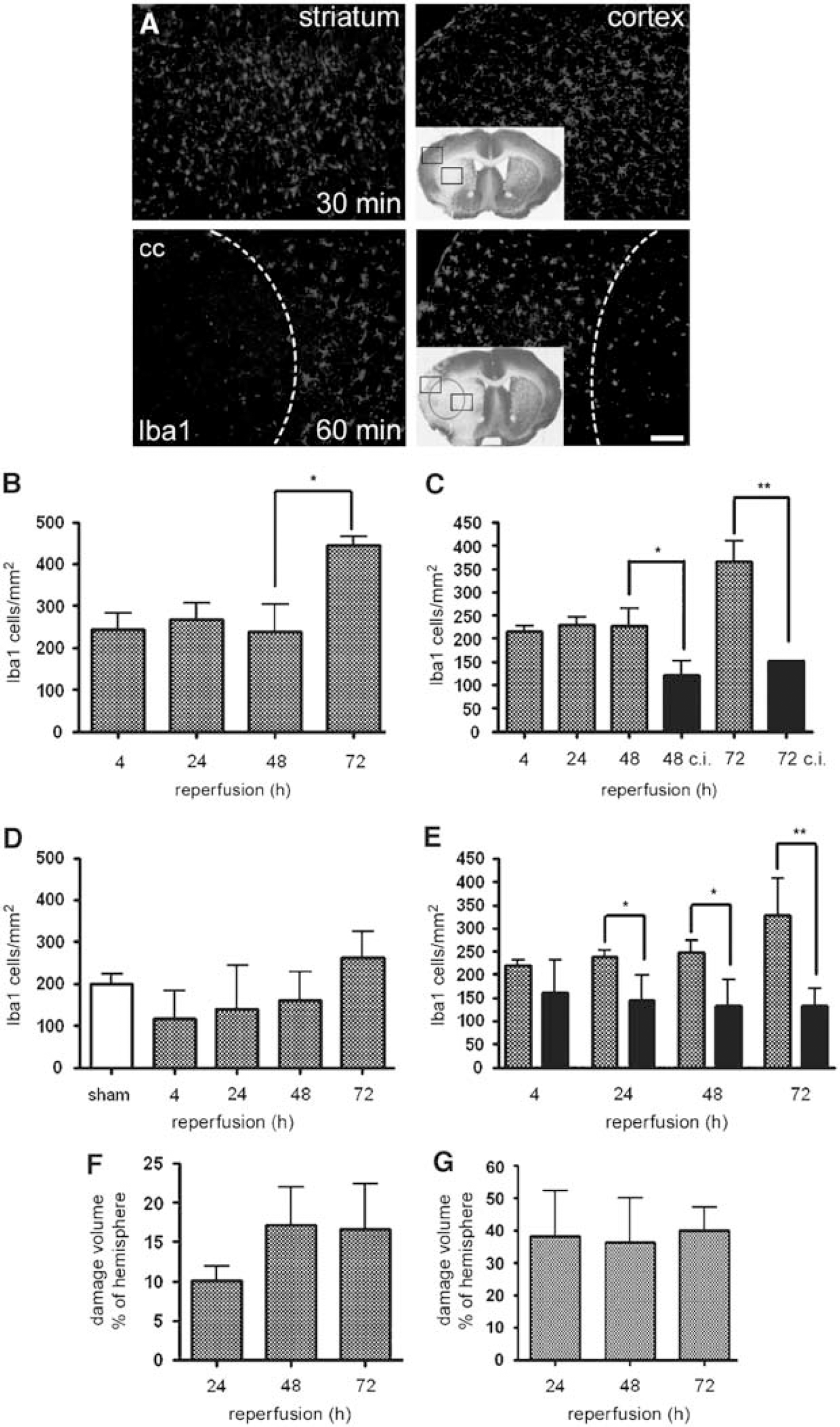
Effect of tMCAo of different duration on resident microglial population. (
At 4 h after 30 mins of MCAo, no obvious cell death was apparent on cresyl violet-stained brain sections in the ipsilateral hemisphere (not shown) and there was no change in Iba1 cell numbers in the striatum or cortex compared with the contralateral hemisphere and sham animals. In contrast, 60 mins of MCAo caused significant loss of Iba1-stained microglia in the inner part of the ipsilateral striatum near the corpus callosum, and to a lesser extent in the deeper ipsilateral cortex layers (Figure 3A). Twenty-four hours after 60 mins of MCAo Iba1-positive cells were found throughout the injured striatum, but only occasionally were these cells positive to CFSE (0 to 2cells/mm2). Microglial numbers tended to increase in the striatum over time during the 72 h after 60 mins of MCAo (Figure 3D), whereas Iba1 cell numbers remained low in the ischaemic cortex but continuously increased over time in the peri-infarct cortex (Figure 3E). The ratio of CFSE-positive Iba1 cells to total Iba1 cell population never exceeded 0.05 in the ischaemic hemisphere. The average volume of the ischaemic damage did not increase after 24 h of reperfusion (Figure 3G). After 30 mins of MCAo, the number of Iba1 cells was similar both at 4 and 24h (Figures 3B and 3C). At 24h, the ischaemic damage was confined to the striatum, but in two of six animals there was subsequent evolution of injury to the cortex by 48 h (Figure 3F). In these animals, the number of Iba1-positive microglia was lower both in the ipsilateral striatum (194 ± 11 cells/mm2 at 48 h and 399 ± 29 cells/mm2 at 72 h) and ipsilateral cortex (120 ± 21 cells/mm2 at 48 h and 150 ± 2 cells/mm2 at 72 h) than in the animals without visible cortical damage (282 ± 47 cells/mm2 at 48 h and 475 ± 13 cells/mm2 at 72 h in the striatum, 221 ± 26 cells/mm2 at 48 h, P < 0.05 and 369 ± 28 cells/mm2 at 72 h, P < 0.01 in the ipsilateral cortex). Overall, the average number of Iba1-positive cells showed negative correlation with damage size both at 48 and 72 h after 30 mins of MCAo (Table 1). This correlation was not observed after 60 mins of MCAo, when neither the average size of the ischaemic damage nor the average number of microglia increased after 24 h in the ischaemic cortex.
Correlation between damage size and average microglial cell numbers in the left cerebral cortex after 30 mins of MCAo
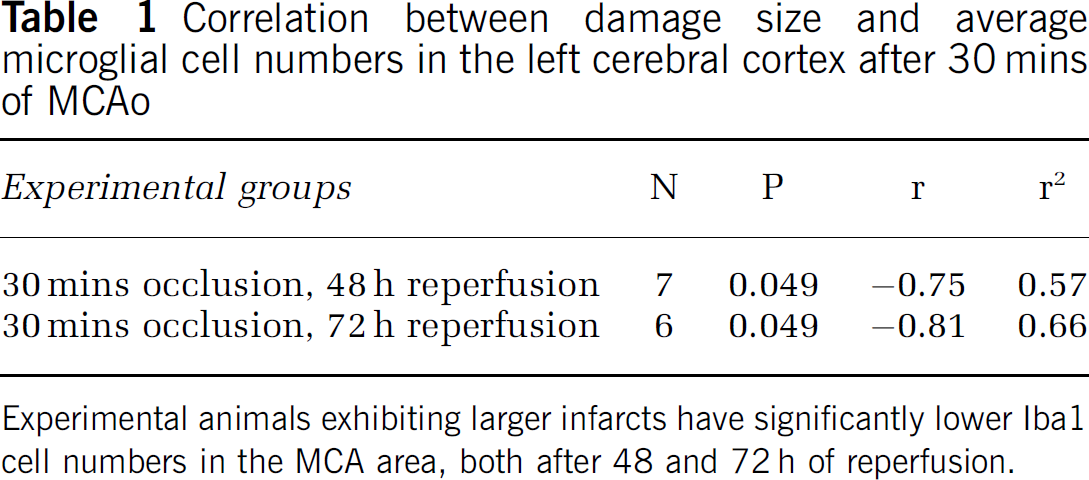
Experimental animals exhibiting larger infarcts have significantly lower Iba1 cell numbers in the MCA area, both after 48 and 72 h of reperfusion.
Cortical Microglial Proliferation is Reduced by Prolonged Middle Cerebral Artery Occlusion
Only scattered BrdU-positive cells were present in the ipsilateral striatum and cortex at 24 h after 30 and 60 mins of MCAo (Figure 4A). In contrast, by 48 h reperfusion after MCAo, intensive microglial proliferation was observed in some regions of the ipsilateral hemisphere dividing cells being apparent in the inner part of the ipsilateral striatum near the subventricular zone and under the corpus callosum (Figures 4A and 4B). In animals displaying no cortical damage after 30 mins of MCAo, the ipsilateral cortex contained proliferating microglia (Figure 4B). In contrast, in the two of six animals where damage was observed in the ipsilateral cortex, only scattered dividing microglial cells, similar to the cases with 60 mins of MCAo, were seen (Figures 4B and 4C).
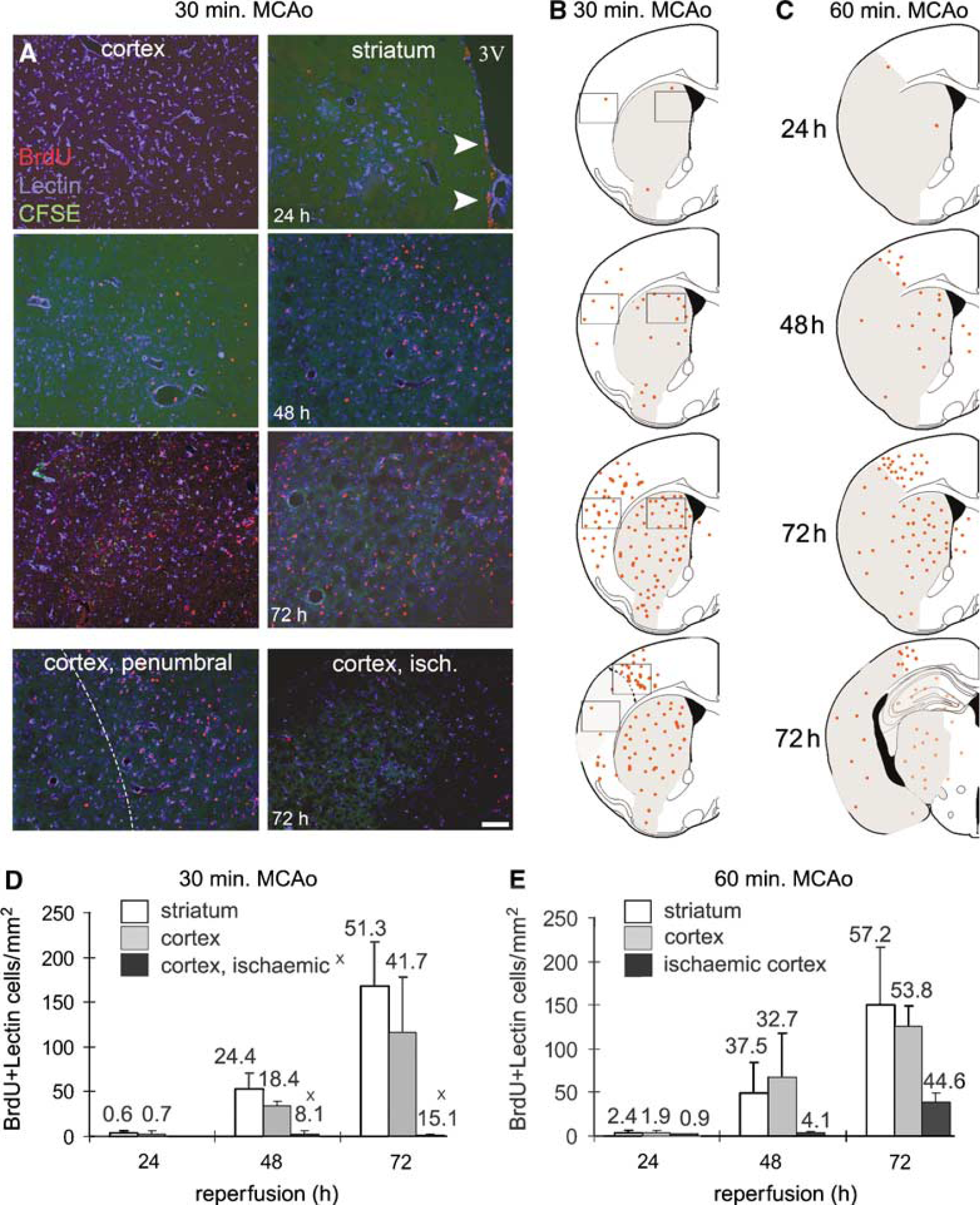
Cortical microglial proliferation is reduced by prolonged MCAo. (
Seventy-two hours after 30 mins of MCAo, the entire striatum as well as the undamaged cortical MCA territory contained very high numbers of BrdU-positive microglia in four of six animals. The two animals with cortical damage after 30 mins of MCAo still only showed a few scattered dividing cells, lower in number than found in the animals exposed to 60 mins of occlusion, where numerous proliferating microglia were present in the ischaemic striatum but relatively few in the ischaemic cortex (Figures 4D and 4E).
Neutrophil Numbers are Controlled by Microglial Phagocytosis in the Ischaemic Brain
In animals undergoing 30 mins of MCAo, the number of infiltrating neutrophils were low during the first 24 h. Nevertheless, in animals with delayed cortical damage after 30 mins of MCAo, the number of neutrophils were very high in the striatum (Figure 5A) as well as the cortex compared with the ones with only striatal damage (23.4 ± 9.33 versus 2.9 ± 2.1 cells/mm2 after 48 h and 11.0 ± 3.4 versus 2.5 ± 0.7 cells/mm2 after 72 h of reperfusion in the left striatum; 20.2 ± 5.1 versus 1.3 ± 1 cells/mm2 after 48 h and 13.6 ± 2.9 versus 2.4 ± 1.1 cells/mm2 after 72 h in the left cerebral cortex, respectively; P < 0.001). In animals undergoing 60 mins of MCAo, the numbers of infiltrating neutrophils were low during the first 24 h, similar to those seen after 30 mins of occlusion. In contrast increasing numbers of neutrophils were seen 48 and 72 h after the insult (Figure 5B).
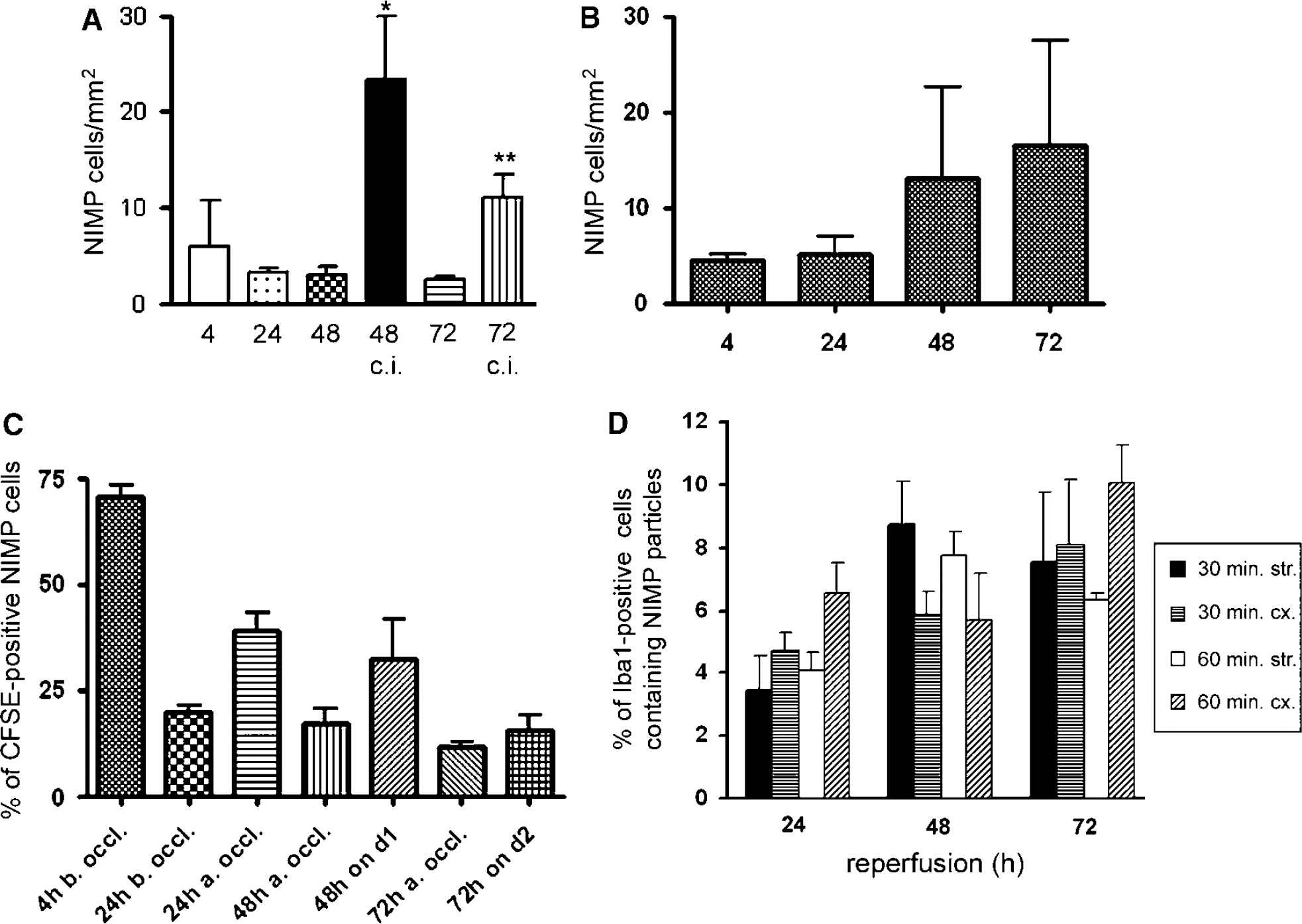
Microglial phagocytosis controls neutrophil granulocyte number in the ischaemic brain. (
NIMP staining was visualized together with CFSE to evaluate the approximate ratio of in situ-labelled versus all blood-derived cells infiltrated into the brain after MCAo at various time points (Figure 5C). Carboxyfluorescein succinimidyl ester-labelled cells were clearly detectable in the peripheral blood and brain neutrophils were positive to CFSE up to 72 h (Figure 6A). The ratio of labelled neutrophils (used either to evaluate the efficiency of CFSE staining or to confirm phagocytosis, see below) was dependent on the time point of in situ labelling and survival after MCAo (Figure 5C). The percentage of neutrophils labelled with CFSE was the highest at 4 h of reperfusion (approximately 70% of total neutrophils). Administration of the tracer 4, 24, or 48 h after the MCAo resulted in higher ratio of CFSE-labelled neutrophils in the brain compared with when given 4 h before the occlusion. This indicates that a new population of neutrophils were formed and/or released into the circulation. By 72 h of reperfusion, the percentage of labelled neutrophils was around 15% to 20% of the total infiltrated population in the animals receiving one single injection of the tracer.
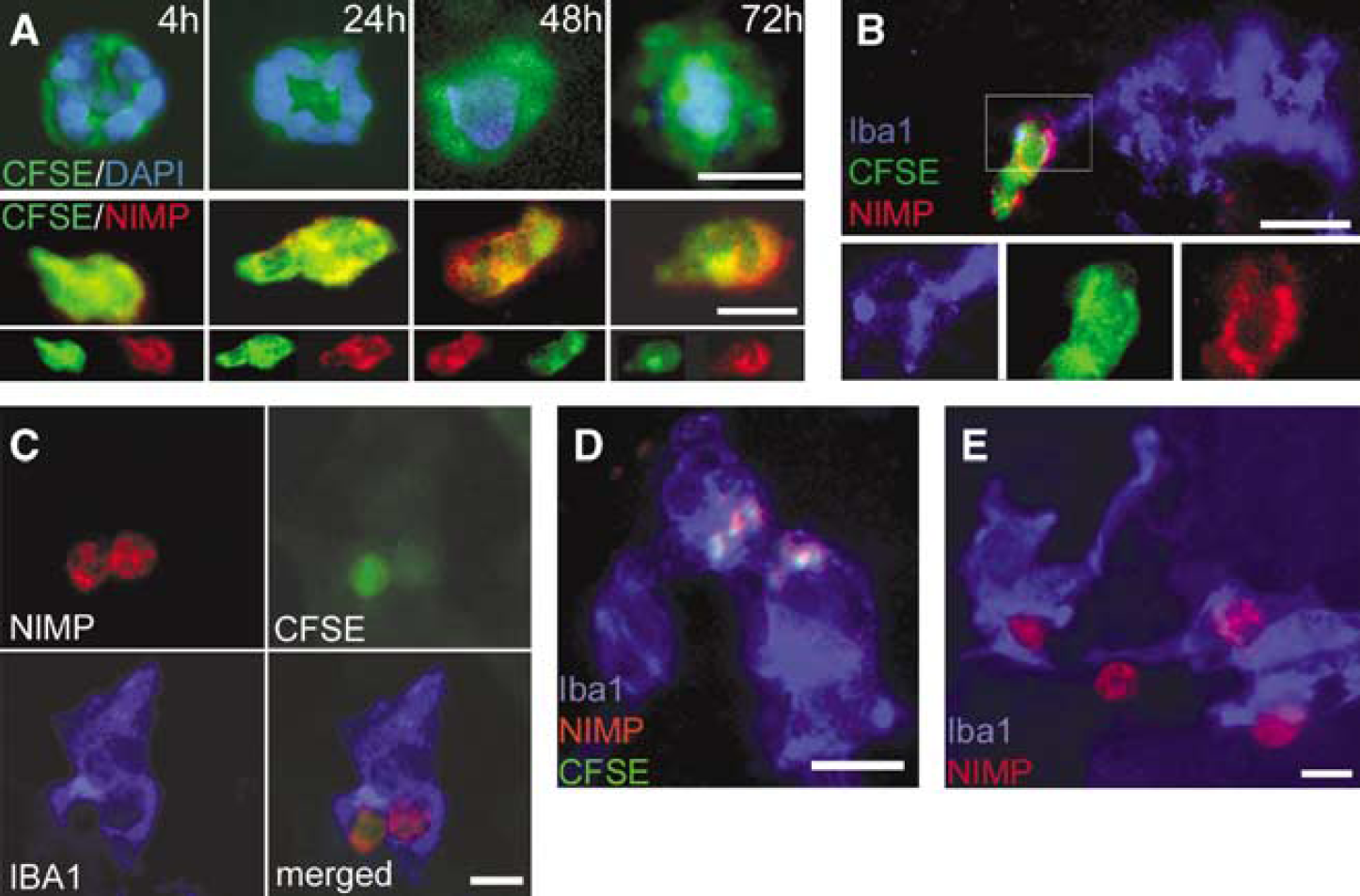
Cellular processes of neutrophil phagocytosis by microglia. (
Given the long half-life of CFSE in labelled leukocytes and decreasing ratio of CFSE-positive neutrophils in the brain parenchyma over time, we examined the possibility that these cells die and/or are being phagocytosed in the brain. Simultaneous visualization of Iba1, NIMP, and CFSE staining revealed phagocytosis of neutrophils by microglial cells in the ischaemic striatum and cortex (Figures 6B to E). The cytoplasm of mononuclear brain phagocytes contained NIMP-positive cells labelled with CFSE (Figure 6C) or CFSE/NIMP particles (Figure 6D). At the meninges near the ischaemic areas similar processes were seen, where infiltrating neutrophils as well as mononuclear brain phagocytes appeared in large numbers (Figure 6E). The meningeal regions likely contained both exogenous macrophages as well as resident brain phagocytes, since CFSE staining often covered the whole cytoplasm (exogenous macrophages), although sometimes staining was confined only to the phagosomes (phagocytosing microglia, data not shown). The ratio of microglia containing NIMP particles was 3% to 4% of the total microglial population at 24 h and 6% to 8% at 48 as well as 72 h of reperfusion time after both 30 and 60 mins of MCAo (Figure 5D). Processes of cellular phagocytosis (i.e., whole NIMP-positive cells attached to microglia or being internalized in the cytoplasm of microglia) were observed mainly after 48 and 72 h of reperfusion in 0.6% to 1.8% of microglia in the ischaemic hemisphere.
Discussion
We did not observe significant invasion of blood-borne macrophages into the brain after 30 or 60 mins of MCAo, despite prolonged disruption of the BBB. This suggests that these cells are not directly involved in the infarct maturation process in the present mouse model. In contrast, we show extensive proliferation and activation of resident microglial cells in the ischaemic brain. A key function of activated microglia is, among others, to phagocytose neutrophils, and we believe that this could represent a potential protective effect, phagocytosis preventing spread, and induction of a more severe inflammatory response in the post-ischaemic brain.
Blood-brain barrier damage after MCAo was revealed with Gd-DTPA MRI both after 30 and 60 mins of occlusion, yet no evidence for leakage of AMI-227 was detected in either animal group within 3 days, suggesting that a degree of selectivity to the permeability was retained at the BBB. In the rat, 90 mins of MCAo leads to permeation of the BBB with a few AMI-227-positive macrophages (Kavec et al, 2004). We administered AMI-227 daily and, given the long plasma half-life (hours in rodents) of the compound (Payen et al, 1998), conditions were favourable for direct penetration to the brain. However, neither AMI-227 deposits nor a large number of iron-containing macrophages in the brain parenchyma were observed. Therefore, leakage of AMI-227 through the BBB was insignificant in mice and all the iron-positive cells detected histologically were likely to be blood-borne (microglia may phagocytose leaked AMI-227 as well). Overall, the number of infiltrating leukocytes was relatively low in the ischaemic striatum throughout the study and the majority of these cells were neutrophils. An explanation may be that iron-laden macrophages in vivo have reduced capacity to migrate into the brain on ischaemic challenge. Internalization of small superparamagnetic iron oxide/ultrasmall superparamagnetic iron oxide particles was shown to shift macrophages towards an anti-inflammatory, less responsive phenotype by enhancing interleukin (IL)-10 and inhibiting tumor necrosis factor-α production in vitro (Siglienti et al, 2006). However, we observed some CFSE-labelled exogenous macrophages in the brain, which suggests the labelling procedure did not inhibit the ability of macrophages to infiltrate into the brain.
Previous studies using AMI-227 to label peripheral macrophages in the rat in vivo showed that Prussian Blue-positive cells accumulate in the ischaemic core and peri-infarct zone. ED-1-positive macrophages were shown to appear in the infarct and peri-infarct areas (Rausch et al, 2001; Schroeter et al, 2004). However, direct visualization of iron-burdened macrophages by immunohistochemistry was only possible later than day 3 after transient MCAo (Rausch et al, 2001) or only minor iron-positive infiltrates were seen histologically, or by T*2-weighted MRI (Schroeter et al, 2006). In addition, activated phagocytic microglia express ED-1 (Rinner et al, 1995) and other phenotypic markers similar to macrophages (Stoll and Jander, 1999), so in these studies the possibility that phagocytosing microglia might take up iron oxide particles through the leaky BBB has not been completely excluded.
One plausible mechanism behind the beneficial action of resident microglia and perivascular macrophages might be that we observed phagocytosis of neutrophils by these cells. Polymorphonuclear granulocytes are known to accumulate in the brain after stroke and contribute to the ischaemic inflammatory damage, chiefly by producing free radicals and inflammatory molecules (Akopov et al, 1996; Davies et al, 1998; Dinkel et al, 2004; Matsuo et al, 1995). Resident microglial cells, and in lesser part, infiltrating haematogenous macrophages were shown to exert active phagocytosis of neuronal cell debris in the brain exposed to 30 mins of focal ischaemia (Schilling et al, 2005). In accordance with our findings, very recent studies also reported the engulfment of neutrophils and myeloperoxidase by macrophages after endothelin-1 induced MCAo. In addition, correlation of myeloperoxidase activity with temporal changes in brain injury and modulation of neurtophil infiltration as well as neutrophil phagocytosis by AM-36 (a Na+ channel blocker antioxidant) were observed (Weston et al, 2006, 2007). Macrophages acquire granules by phagocytosing apoptotic neutrophils, which increases antimicrobial activity against intracellular pathogens (Tan et al, 2006). In the striatum after 30 mins of MCAo, the resident microglial population showed no signs of damage and these, together with meningeal and perivascular macrophages, might contribute to limiting of the effects of infiltrated neutrophils by phagocytosis. It is not clear how microglia can recognize infiltrating neutrophils in the brain parenchyma and what type of signals can trigger the phagocytotic process. It was reported that proteolytic enzymes divert the recognition and clearance of polymorphonuclear leukocytes by macrophages. Certain proteinases (of bacterial origin) can sensitize healthy neutrophils for uptake by macrophages due to proteolytic cleavage of an antiphagocytic signal (CD31) and the generation of a novel ‘eat-me’ signal on the neutrophil surface (Guzik et al, 2007). Similar processes might be involved after MCAo, elicited by activated brain proteases. Interestingly, our results show that a low number of microglial cells and/or a profound reduction in dividing cells strongly correlated with the number of neutrophils present in the parenchyma and, ultimately, with the extent of neuronal injury. It is possible, therefore, that increase of microglial proliferation is a direct response to the increasing neutrophil numbers in the brain parenchyma. Because degeneration of microglia was observed after prolonged MCAo it seems that proliferating microglia is protective at certain degrees of ischaemia but can be overcome by increased ischaemia, possibly as a result of increased neutrophil infiltration in these animals. These observations support the hypothesis that resident microglial activation may serve as an endogenous mechanism to limit the inflammation-mediated worsening of ischaemic brain damage. In addition, the authors emphasize that phagocytosis of neutrophils is not the only potentially protective role of microglia after stroke.
We identified activation of microglia by means of increased expression of activation markers (Iba1, CD45, F4/80) and microglial proliferation, which profoundly increased by 72 h of reperfusion. Microglial proliferation is initially observed under the corpus callosum as well as adjacent to the lateral ventricle. Dividing microglia-expressed CD34, which is a hallmark of dividing, self-renewing microglia, as described previously after acute neuronal injury (Ladeby et al, 2005).
Proliferation resulted in an increase of microglial cell numbers, which was not accompanied with larger ischaemic damage. In contrast, the more extensive ischaemic injury observed after 60 mins of MCAo was associated with low microglial cell numbers and an impaired proliferative response in the cerebral cortex. Degeneration of microglia in the ischaemic core after prolonged MCAo was also reported by other studies either in a permanent model of rat (Davies et al, 1998) or mouse MCAo (Tanaka et al, 2003). Infiltrating macrophages were not positive to BrdU, which indicates that these cells may differ from resident microglia regarding their ability to divide after entering the brain. Blood-brain barrier damage and infiltrating blood-borne substances can trigger microglia to enter the cell cycle. Evidence supporting these ideas was recently provided by studies showing that pure albumin is a potent trigger of calcium signaling and proliferation in microglial cells, but not of macrophages or astrocytes (Hooper et al, 2005).
The relative contribution of exogenous macrophages to post-ischaemic processes is controversial. In a permanent model of MCAo (Tanaka et al, 2003) as well as in a transient model (Kokovay et al., 2006), several Iba1-positive exogenous macrophages were found infiltrated into the brain 24 to 48 h after the insult. In contrast, others found a minor contribution of blood-borne macrophages to the inflammatory infiltrates in the 4 days after transient MCAo (Schilling et al, 2003, 2005). It was reported that head irradiation dose-dependently increased the number of infiltrating Iba1-positive cells in the MCA territory and the olfactory bulb in naive animals 6 weeks after bone marrow transplantation (Tanaka et al, 2003). Therefore, it may be difficult to compare the number and ratio of infiltrating cells between different studies using either naive or bone marrow chimeric animals.
In our experiments, the population of peripheral leukocytes labelled with CFSE or AMI-227 was in accordance with previous data (Becker et al, 2004; Ristevski et al, 2003; Weissleder et al, 1990), but relatively few exogenous macrophages infiltrated into brain parenchyma over the first 72 h after tMCAo, parallel to progression of the ischaemic damage. In this process, the number of microglial cells that survive the ischaemic challenge might be of importance in controlling the number of blood-borne macrophages and other proinflammatory cells. The fact that the ratio of labelled brain neutrophils in post-ischaemic brain was different depending on the time point of carboxyfluorescein diacetate succinimidyl ester administration relative to exposure to MCAo suggests that MCAo either triggers formation or release of leukocytes from the bone marrow or from peripheral pools (or both these effects), which were differently available for CFSE injected into the circulation. The gene expression profile of neutrophils are reported to undergo rapid changes and early mobilization of these cells is observed shortly after stroke in human patients, followed by a marked reduction 5 to 7 days after the insult (Emsley et al, 2003; Tang et al, 2006).
Taken together, our data show no significant exogenous macrophage infiltration early (4 to 72 h) after MCAo, but rather microglial proliferation and repopulation. The microglia can exert a phagocytic role that may be advantageous in stroke in maintaining the brain microenvironment in spite of BBB breakdown and in the prevention of ischaemic exacerbation of the damage in the penumbral region, although this remains to be demonstrated.