
Abstract
Tumor necrosis factor-alpha (TNFα) and Fas are induced after traumatic brain injury (TBI); however, their functional roles are incompletely understood. Using controlled cortical impact (CCI) and mice deficient in TNFα, Fas, or both (TNFα/Fas—/—), we hypothesized that TNFα and Fas receptor mediate secondary TBI in a redundant manner. Compared with wild type (WT), TNFα/Fas—/— mice had improved motor performance from 1 to 4 days (P < 0.05), improved spatial memory acquisition at 8 to 14 days (P < 0.05), and decreased brain lesion size at 2 and 6 weeks after CCI (P < 0.05). Protection in TNFα/Fas—/— mice from histopathological and motor deficits was reversed by reconstitution with recombinant TNFα before CCI, and TNFα—/— mice administered anti-Fas ligand antibodies had improved spatial memory acquisition versus similarly treated WT mice (P < 0.05). Tumor necrosis factor-alpha/Fas—/— mice had decreased the numbers of cortical cells with plasmalemma damage at 6h (P < 0.05 versus WT), and reduced matrix metalloproteinase-9 activity in injured brain at 48 and 72 h after CCI. In immature mice subjected to CCI, genetic inhibition of TNFα and Fas conferred beneficial effects on histopathology and spatial memory acquisition in adulthood (both P < 0.05 versus WT), suggesting that the beneficial effects of TNFα/Fas inhibition may be permanent. The data suggest that redundant signaling pathways initiated by TNFα and Fas play pivotal roles in the pathogenesis of TBI, and that biochemical mechanisms downstream of TNFα/Fas may be novel therapeutic targets to limit neurological sequelae in children and adults with severe TBI.
Introduction
Traumatic brain injury (TBI) is a leading cause of morbidity and mortality in young adults and children. Although advances in intensive care management have improved the prognosis for TBI, specific therapy remains lacking, in part owing to incomplete understanding of secondary injury mechanisms that extend primary tissue damage and exacerbate neurological dysfunction in survivors (Marion and Spiegel, 2000).
Tumor necrosis factor receptor (TNFR) family members and their ligands are induced early after acute central nervous system injury and may initiate complex, redundant intracellular signaling pathways that contribute to injury as well as regeneration (Locksley et al, 2001). Tumor necrosis factor-alpha (TNFα) is a pleiotropic cytokine, which transduces death and survival signaling through its cognate receptors TNFR1 and TNFR2, whereas Fas is a death receptor, which was first characterized as a key initiator of programmed cell death in liver and immune cells (Adachi et al, 1995). Fas initiates many of the same signaling pathways as TNFα,including assembly of death-inducing signaling complexes, activation of nuclear factor kappa B, mitogen-activated protein kinases, Jun N-kinase, receptor-interacting protein, and others. Tumor necrosis factor-alpha is induced early after experimental TBI, and assembles a death inducing signaling complex with TNFR1 in brain cell membrane lipid rafts (Lotocki et al, 2004). Tumor necrosis factor-alpha mediates acute cell death and neurological dysfunction after closed head TBI in rodents (Shohami et al, 1999); however, one study reported beneficial roles for TNFα in the chronic phase of TBI using controlled cortical impact (CCI) (Scherbel et al, 1999).
Because its role in experimental brain trauma has notbeendirectlystudied, how Fasinfluencesthe pathogenesis of TBI remains unknown. Fas receptor, and Fas-FADD-caspase-8 DISC, are upregulated in post-traumatic rodent and human brain (Qiu et al, 2002), and Fas inhibition reduces cell death in experimental stroke and traumatic spinal cord injury models (Casha et al, 2005; Demjen et al, 2004; Martin-Villalba et al, 2001, 1999). These studies suggest a detrimental role for Fas, yet Fas signaling can also promote cell survival and proliferation (Lambert et al, 2003). Fas regulates neural branching during development (Desbarats et al, 2003; Zuliani et al, 2006), enhances functional recovery after sciatic nerve and spinal cord injury (Demjen et al, 2004; Desbarats et al, 2003), and protects against neuronal degeneration in a model of Parkinson's disease (Landau et al, 2005). Thus, like TNFα, Fas signaling could exert diverse effects on TBI by co-opting intracellular signaling pathways that influence cell death as well as survival and regeneration.
Previous studies in experimental stroke suggest that in the context of acute brain injury, TNF and Fas signaling exert a net negative influence on outcome. Mice deficient in TNF or Fas receptor had modestly decreased infarct volume versus wild type (WT), but dual antagonism of TNF and Fas ligand together further reduced tissue damage in a middle cerebral artery occlusion model (Martin-Villalba et al, 2001). On the basis of these studies and the known redundancy of TNF receptor signaling, we hypothesized that inhibition of TNFα and Fas together would be required to reduce histopathology and improve motor and cognitive deficits after CCI in mice.
Materials and methods
In all studies, CCI was performed and all data were obtained by experimenters masked to study groups.
Controlled Cortical Impact
The CCI model was used as described previously with minor modifications (Bermpohl et al, 2006). The trauma protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. Mice (20 to 21 days old for immature, 8 to 16 weeks old for adults) were anesthetized with 4% isoflurane (Anaquest, Memphis, TN, USA) in 70% N2O and balance O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, USA) and positioned in a stereotaxic frame. Anesthesia was maintained using 2% to 3% isoflurane. A 5 mm craniotomy was made using a portable drill and trephine over the left parieto-temporal cortex, and the bone flap was removed. Mice were then subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 m/sec, depth of 0.6 mm, and 100 ms impact duration. The bone flap was discarded, and the scalp was sutured closed. The mice were returned to their cages to recover from anesthesia, and were generally able to ambulate within 15 mins.
Knockout Mice
Mice were housed in a pathogen-free facility in 12 h day/night cycles, and treated humanely in accordance with the NIH Guide for Care and Use of Laboratory Animals. Tumor necrosis factor-alpha/Fas double-knockout mice (TNFα/Fas—/—) were generated on a C57Bl/6 background from TNFα—/— mice (stock #3008, Jackson Laboratories, Bar Harbor, ME, USA) (Pasparakis et al, 1996) and Fas—/—mice (stock #3233, Jackson Laboratories) (Adachi et al, 1995). Tumor necrosis factor-alpha—/— and Fas—/— mice were produced by gene targeting and homologous recombination in embryonic stem cells using a gene cassette encoding neomycin resistance. In TNFα—/— mice, all of exon 1 is replaced, resulting in complete loss of functional TNFα. Fas knockout mice lack most of exon 9 and produce no detectable 45 kDa mature Fas protein, but do produce immunoreactive truncated Fas/neomycin-resistance /β-galactosidase fusion protein (approximately 120 kDa on Western blots) in low concentrations (Adachi et al, 1995). Fas— /— mice used to make TNFα/Fas—/— were backcrossed more than 10 generations into C57Bl/6. Tumor necrosis factor-alpha—/— mice are also inbred into the C57 background, but the exact number of backcrosses is not known (Jackson Laboratories). Tumor necrosis factor-alpha—/— and Fas—/— mice were genotyped using polymerase chain reaction primers and protocols recommended by Jackson Laboratories. Tumor necrosis factor-alpha/Fas—/— mice were identified by presence of the neomycin resistance cassette and absence of TNFα and Fas WT alleles. Age-matched male mice were used for all experiments, except adult naïve Morris water maze (MWM) studies, genetic/pharmacological MWM studies, and studies in immature mice, in which equal numbers of male and female mice were used.
Genotyping Protocols
Primers oIMR0013, oIMR0014, oIMR0745, and oIMR0746 were used to determine the presence of neomycin cassette (Neo) and TNFα WT allele. The sequences of the primers are oIMR0013: 5′-CTTgggTggAgAggCTATTC-3′; oIMR0014: 5′-AggTgAgATgACAggAgATC-3′; oIMR0745: 5′-gCACAgA AAgCATgATCCg-3′; oIMR0746: 5′-TAgACAgAAgAgCgTgg Tgg-3′. oIMR0013 and oIMR0014 amplify a 280bp DNA fragment on the neomycin cassette, whereas oIMR0745 and oIMR0746 amplify a 146 bp DNA fragment from the TNFα gene. Primers oIMR1360, oIMR1361, oIMR158, and oIMR159 were used to identify the Fas WT allele and Neo cassette, respectively. The sequences of the primers are oIMR1360: 5′-TgTCCATCAAtggAAggTCT-3′; oIMR1361: 5′-gCTTgAgTAAATACATCCCg-3′; oIMR158: 5′-CTGAATGAACTGCAGGACGA-3′; oIMR159: 5′-ATACT TTCTCGGCAGGAGCA-3′. oIMR1360 and oIMR1361 amplify a 700 bp DNA product from the Fas WT allele, whereas oIMR158 and oIMR159 amplify a 172 bp DNA fragment on the neomycin cassette.
Administration of Anti-Fas Ligand Antibodies and Recombinant Tumor Necrosis Factor-Alpha
For genetic/pharmacological inhibition of TNFα/Fas, TNFα—/— mice or WT controls (n = 10/group) were administered anti-mouse Fas ligand antibody (BD Pharmingen, San Diego, CA, USA) 2 mg/kg intraperitoneally 1 to 2 h before CCI and 2 μg intracerebroventricularly at the time of CCI. Mice were then subjected to CCI. These routes of administration were based on increased blood—brain barrier permeability early after CCI, and previous studies showing that anti-TNF and anti-Fas ligand antibodies effectively reduce stroke volume when administered intraperitoneally (Martin-Villalba et al, 2001). We chose to administer anti-Fas ligand antibodies intra-cerebroventricularly as well as intraperitoneally before CCI to avoid potential delay of central nervous system penetration related to the time course of blood—brain barrier damage. For TNFα reconstitution experiments, TNFα/Fas—/— or WT mice were anesthetized and administered 0.2 μg of recombinant mouse TNFα (R&D Systems, Minneapolis, MN, USA) in 2 μL of phosphate-buffered saline (PBS) intracerebroventricularly just before CCI.
Assessment of Injury after Tissue Damage
Morphometric image analysis (MCID, Imaging Research Inc., St Catherines, ON, Canada) was used to determine lesion volume and brain tissue loss after CCI. Mice were deeply anesthetized with isoflurane, decapitated, and brains were removed and frozen in nitrogen vapor. Coronal brain sections (12 μm) were cut on a cryostat at 0.5 mm intervals from anterior to posterior and mounted on poly-
Assessment of Brain Edema
Brain edema was assessed by measuring brain water content using the (wet—dry)/wet brain weight method. Mice subjected to CCI were killed at 24 h and brains removed and bisected at the midline to separate right and left hemispheres. The cerebellum and brain stem were removed. Uninjured brain tissue anterior and posterior to the contusion was removed by sectioning in the coronal plane. The wet weight of the remaining injured and uninjured hemispheres was obtained. Brain tissue was dried at 99°C for 48 h and dry hemispheric weights were obtained. Brain water content was expressed as (wet—dry weight)/wet weight × 100%.
Evaluation of Motor Function
Vestibulomotor function was assessed using a wire-grip test (Bermpohl et al, 2006). Mice were placed on a metal wire (45 cm long) suspended 45 cm above protective padding and were allowed to traverse the wire for 60 secs. The latency that a mouse remained on the wire within a 60 secs interval was measured, and a wire grip score was quantitated using a 5-point scale. Testing was performed in triplicate and an average value calculated for each mouse on each test day.
Assessment of Spatial Memory Function
The MWM was used to evaluate spatial memory performance as described previously (Bermpohl et al, 2006). The apparatus consisted of a white pool (90 cm diameter and 60 cm deep) filled with water to 29 cm depth with several highly visible cues located on the walls of each of the four quadrants. Water temperature was maintained at 21 to 25°C. A clear Plexiglass goal platform 5 cm in diameter was positioned 0.5 cm below the water's surface approximately 15 cm from the Southwest wall. Each mouse was subjected to a series of 4 to 8 trials per day. For each trial, mice were randomized to one of four starting locations (north, south, east, and west) and placed in the pool facing the wall. Mice were given a maximum of 60 or 90 secs to find the submerged platform. If the mouse failed to reach the platform by the allotted time, it was placed on the platform by the experimenter and allowed to remain there for 10 secs. Mice were placed in a warming chamber for at least 4 mins between trials. To control possible differences in visual acuity or sensorimotor function between groups, two trials were performed using a visible platform raised 0.5 cm above the surface of the water. Performance in the MWM was quantitated by latency to find the platform.
Administration of Propidium Iodide and Preparation of Tissue Sections for Cell Counting
Propidium iodide (PI; 10 mg/mL, Sigma, St Louis, MO, USA) diluted in 0.9% NaCl was administered by intraperitoneal injection (1 mg/kg) to mice in a total volume of not more than 100 μL at 5 h after CCI. Mice were killed 1 h after PI injection (6 h after CCI) and brains were frozen in nitrogen vapor. Cryostat brain sections (12 μm) were cut at 150 to 200 μm intervals from anterior to posterior hippocampus and placed on poly-
Assessment of Propidium Iodide-Positive Cell Counts
We chose regions of interest for cell counts in contused cortex based on a previous report showing robust cell death and plasmalemma permeability of cells in perilesional cortex in our CCI model (Bermpohl et al, 2006). Cortical regions of interest were × 200 microscopic fields (1,100 × 1,100 μm) at the medial and lateral edges of the contusion, and one cortical field directly under the impact site. Brain sections were photographed on a Nikon Eclipse T300 fluorescence microscope (Tokyo, Japan), using excitation/emission filters 568/585. Propidium iodide-positive cells were counted in a total of three brain sections located within the center of the contusion (bregma −1.90 to −2.70) and separated by at least 150 μm, for a total of nine × 200 cortical fields assessed per animal. Cell count data for each mouse were the average of the nine cortical brain fields.
Immunohistochemistry for Neutrophils, Astrocytes, and Microglia
At 48 h after CCI mice were anesthetized with isoflurane and the brain was rapidly removed intact or transcardially perfused with 4% paraformaldehyde, frozen in nitrogen vapour, and stored at −80°C. Coronal brain sections (12 μm) were placed on poly-
Microglia staining was performed using paraformaldehyde-fixed brain tissue. Cryostat-fixed brain sections were treated with 1% hydrogen peroxide in PBS. The slides were blocked in PBS with 3% normal goat serum for 1 h and incubated overnight at 4°C with rabbit anti-Iba-1 antibody (1:200; Wako Pure Chemical Industries Ltd., Osaka, Japan). The sections were then washed in PBS with 1% normal goat serum and incubated with biotinylated goat anti-rabbit immunoglobulin G (1:200; Vector Laboratories, Burlingame, CA, USA) antibody for 1 h. After washing, the signal was detected using an ABC kit (Vector Labs) and diaminobenzidine (Vector Labs).
Quantitation of Neutrophils, Astrocytes, and Activated Microglia in Mouse Cortex after CCI
For the estimation of neutrophils in injured cortex, cortical brain fields (× 200; 1,100 × 1100 μm) from three brain sections separated by at least 150 μm and within the approximate center of the contusion were selected for analysis. Neutrophils were photographed and quantitated in four × 200 cortical fields randomly chosen from each brain section. Thus, a total of 12 × 200 fields were analyzed per mouse. From these data, an average neutrophil count per × 200 field was obtained for each animal.
For the analysis of glial fibrillary acidic protein-positive astrocytes or Iba-1-positive microglia, photomicrographs of a × 200 field from the medial and lateral edges of the contusion were analyzed from three brain sections chosen from the approximate center of the contusion. Using MCID image analysis software, activated glial fibrillary acidic protein-positive astrocytes were quantified by measuring the average density values of all the pixels of each image. Because the density of activated microglia in injured cortex was less than that of astrocytes, appropriate morphological and density criteria were chosen and MCID software was used to estimate the total area occupied by microglia, and the average density value for Iba-1 immunopositivity, per × 200 field. The product of the total area multiplied by the average density value of this area was calculated for each × 200 field, and an average value obtained from the six × 200 fields was obtained for each mouse.
Gelatin Gel Zymography
Tissue samples were homogenized in ‘working buffer’ (50 mmol/L Tris—HCl, pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, and 0.02% NaN3). Proteins (25 μg) were separated by electrophoresis on 10% polyacrylamide zymogram gels containing gelatin (Invitrogen, Carlsbad, CA, USA). After washes in 2.5% Triton X-100 to remove sodium dodecyl sulfate and then in 50 mmol/L Tris—HCl, pH 7.5, the gels were incubated overnight in developing buffer (50 mmol/L Tris—HCl, pH 7.5, 10 mmol/L CaCl2, 1 μm ZnCl2, and 0.05% BRIJ-35) at 37°C. Gels were stained with 0.25% Coomassie Blue G-250 in 20% methanol and 10% acetic acid, then were washed in 20% methanol/10% acetic acid and photographed. Samples of recombinant matrix metalloproteinase-9 (MMP-9) were included in the zymograms to identify MMP-9 in tissue homogenates. To ensure equal protein loading, aliquots of brain tissue homogenates used for zymography were also analyzed by Western blot for β-actin content.
Immunoblots
Brain tissue dissected from contused cortex within the injured hemisphere or from the left hemisphere of uninjured control mice was homogenized on ice in a modified radioimmunoprecipitation buffer (50 mmol/L Tris—HCl, pH 7.5, 50 mmol/L NaCl, 4 mol/L urea, 0.5% SDS, 0.5% NP-40, 0.5% Na-deoxycholate, 5 mmol/L phenylmethylsulfonylfluoride, 1 mmol/L ethylenediami-netetraacetic acid, 5 mmol/L ethyleneglycoltetraacetic acid, 10 mmol/L dithiothreitol, and protease inhibitor cocktail (1:100, Sigma Aldrich, St Louis, MO, USA). Homogenates were cleared by centrifugation at 14,000 r.p.m. for 15 mins at 4°C. Protein content of the supernatant was assayed (Bio-Rad Laboratories, Hercules, CA, USA) and aliquots of protein were boiled in denaturing sample buffer (62.5 mmol/L Tris pH 6.8, 2% sodium dodecyl sulfate, 5 mmol/L ethylenediaminetetraacetic acid, 10% glycerol, 0.25% 2-mercaptoethanol, 0.01% Bromophenol Blue). Brain samples were loaded 20 μg/lane. Denatured protein was size fractionated on 10% sodium dodecyl sulfate—polyacrylamide gels (Invitrogen) and blotted onto Immobilon 0.45 μm polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Membranes were blocked for 1 h in 5% milk in Tris-buffered saline (pH 7.4) containing 0.1% Tween 20, then incubated overnight at 4°C with anti-mouse β-actin (Sigma Aldrich, 1:1,000). Membranes were washed in Tris-buffered saline Tween 20 then incubated for 1 h with an appropriate horseradish peroxidase-conjugated secondary antibody (1:20,000 in Tris-buffered saline Tween 20, 5% milk) at room temperature. Proteins of interest were detected using the enhanced chemiluminescence Western blotting detection system kit (ECL Plus, Amersham, Buckinghamshire, UK) and Hyperfilm (Amersham, Oakville, ON, Canada).
Statistical Analyses
Data are mean ± s.e.m. Lesion volume, PI-positive cell count data, edema data, neutrophil counts, astrocytosis, and microgliosis data were analyzed by rank-sum test. Motor and MWM test data in each mutant mouse strain were compared with WT controls using two factor repeated measures analysis of variance (group and time). For immature mice, lesion size analysis for Fas—/—, TNFα/ Fas—/—, and WT groups was assessed by analysis of variance on ranks followed by Dunn's test, as these three groups were included in one experiment. For all comparisons, P < 0.05 was regarded as significant.
Results
Generation and Characterization of TNFα/Fas—/— Mice
Similar to findings previously reported in TNFα/gld double-knockout mice (Martin-Villalba et al, 2001; Pasparakis et al, 1996), TNFα/Fas—/— mice were viable, fertile, and remained healthy for at least 6 months. Polymerase chain reaction analysis of TNFα/Fas—/— mice yielded the expected results based on analysis of TNFα—/— and Fas—/— single knockout mice, and TNFα/Fas—/— mice lacked obvious brain anatomical or behavioral abnormalities (Figures 1 to 3). At 3 mins after the start of general anesthesia with body temperature maintained at 37°C, TNFα/Fas—/— mice had baseline blood pressure and blood gases similar to WT mice (P = NS for all pairwise comparisons; Table 1). The observed values for arterial PaCO2 were subphysiological in both mutant and WT mice, likely owing to the effects of general anesthesia on minute ventilation in the first 3 mins. All mice had no response to tail pinch at the time the blood gases were drawn, ruling out inadequate anesthesia as the cause of hyperventilation. Compared with WT, TNFα/ Fas—/— mice recovered from CCI without any obvious increased incidence of infectious or other complications.
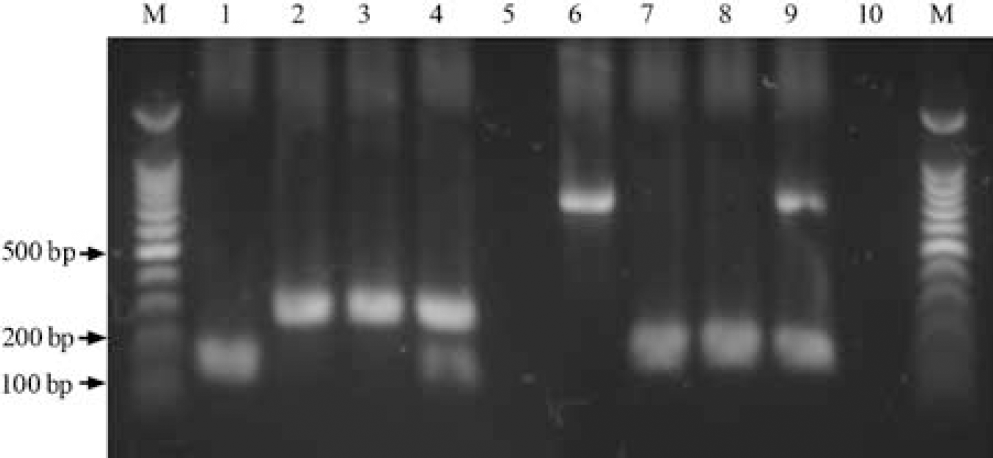
Genotyping of TNFα/Fas—/— mice analyzed by polymerase chain reaction of tail deoxyribonucleic acid. M, molecular weight markers (100 bp). Lanes 1 to 5: wild type (WT) TNFα allele (146 bp) and Neomycin resistance cassette (280 bp) DNA sequences amplified from tail DNA of mice using Jackson Laboratories protocol #3008. Lane 1, WT; lane 2, TNFα—/—; lane 3, TNFα/Fas—/—; lane 4, TNFα+/—/Fas+/—double heterozygote; lane 5, water control. Lanes 6 to 10: wildtype Fas allele (700 bp) and Neomycin resistance cassette (172 bp) DNA sequences amplified from tail DNA of mice using Jackson Laboratories protocol #3233. Lane 6, WT; lane 7, Fas—/—; lane 8, TNFα/Fas—/—; lane 9, TNFα + /—/Fas+/—double heterozygote; lane 10, water control.
Tumor Necrosis Factor-Alpha and Fas Mediate Brain Tissue Damage in a Redundant Manner After Controlled Cortical Impact
We first examined the contribution of TNFα and Fas to brain tissue damage after CCI. At 2 weeks after CCI, brain lesion size was similar between TNFα—/—and WT and between Fas—/— and WT (Figure 2A), but was reduced by 30% in TNFα/Fas—/— versus WT mice (P < 0.05, Figure 2B). To provide further proof for redundant function of TNFα and Fas, and rule out the possibility that genetic background influenced tissue sparing in TNFα/Fas—/— mice, TNFα/Fas—/— and WT mice were administered recombinant mouse TNFα or vehicle (see Materials and methods) and subjected to CCI. Brain lesion size was increased by 28% in TNFα/Fas—/— mice administered TNFα (P < 0.05 versus vehicle; Figure 2B). However, brain lesion size did not differ in WT mice administered TNFα versus vehicle (Figure 2B), arguing against a nonspecific toxic effect of exogenously administered TNFα on histopathology.
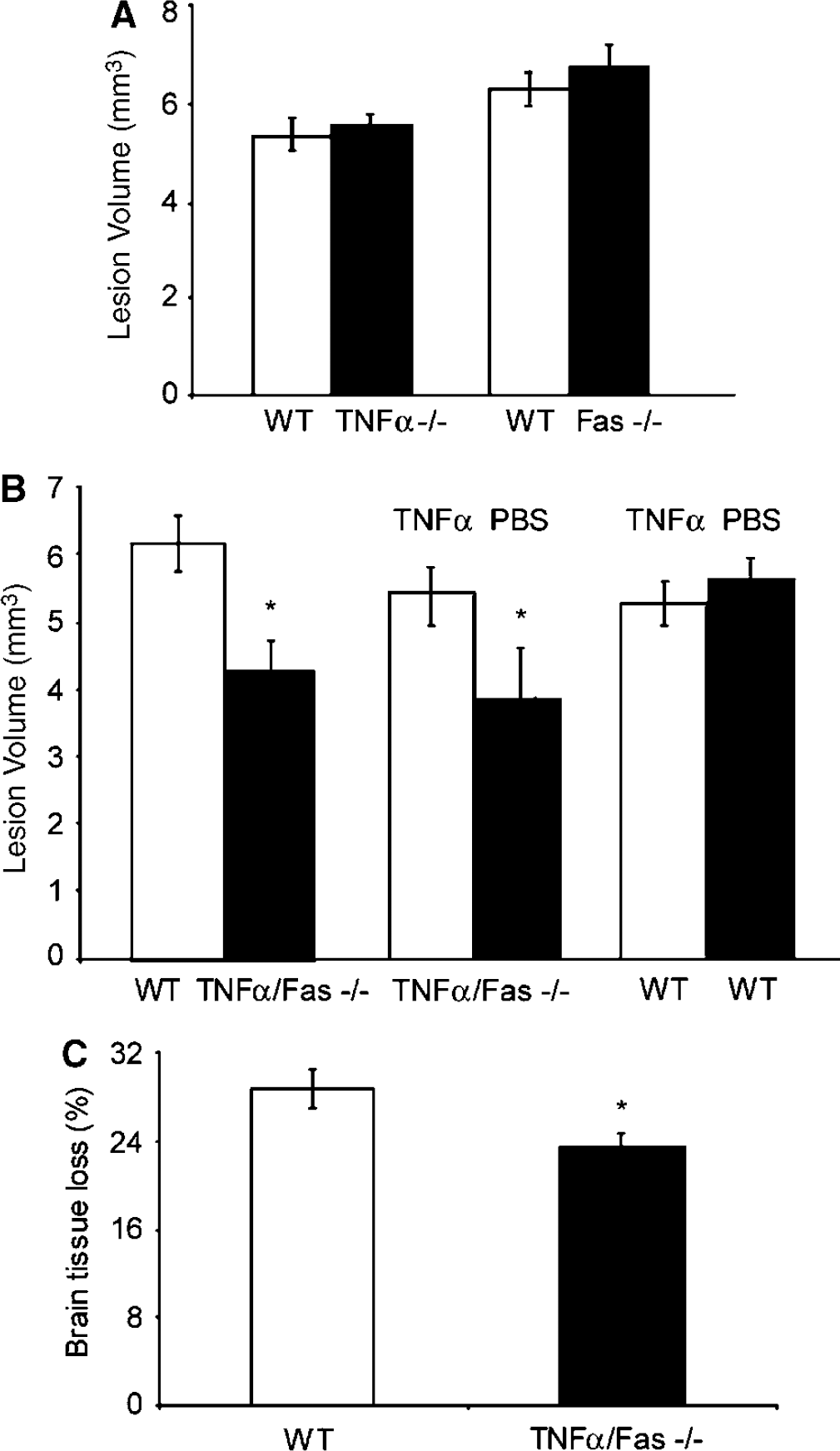
Lesion volume and brain tissue loss are reduced in TNFα/Fas—/— mice after controlled cortical impact (CCI). (
To determine whether the observed protection in TNFα/Fas—/— mice was transient or persistent, we performed CCI in adult TNFα/Fas—/— and WT mice and measured hemispheric brain tissue loss at 35 days after injury. Hemispheric brain tissue loss in TNFα/Fas—/— mice was decreased by 18% compared with WT (Figure 2C; P < 0.05). Thus, the protective effects of TNFα/Fas—/— on tissue damage persisted in the chronic phase of TBI.
Tumor Necrosis Factor-Alpha/Fas Knockout Improves Recovery of Motor Performance after Controlled Cortical Impact
No difference in baseline motor function was observed between groups of uninjured knockout and WT animals (Figures 3A to 3C). After CCI, recovery of motor function was similar between WT and TNFα—/— (Figure 3A) and WT and Fas—/—(Figure 3B). Although TNFα—/— mice showed transient improvement on the first post injury day versus WT, the data were not significantly different when all days were included in the repeated measures analysis of variance. In contrast to results in single knockout mice, recovery of motor function was significantly improved in TNFα/Fas—/— mice compared with WT (P < 0.01 for group effect, Figure 3C). Reconstitution of TNFα/Fas—/— mice with recombinant TNFα reversed the beneficial effect of TNFα/Fas—/— on post injury motor performance (P < 0.05 for group effect, Figure 3D), however administration of TNFα to WT mice did not affect recovery of motor function compared with administration of vehicle (Figure 3E). Thus, TNFα and Fas contribute to motor dysfunction after CCI in a redundant manner.
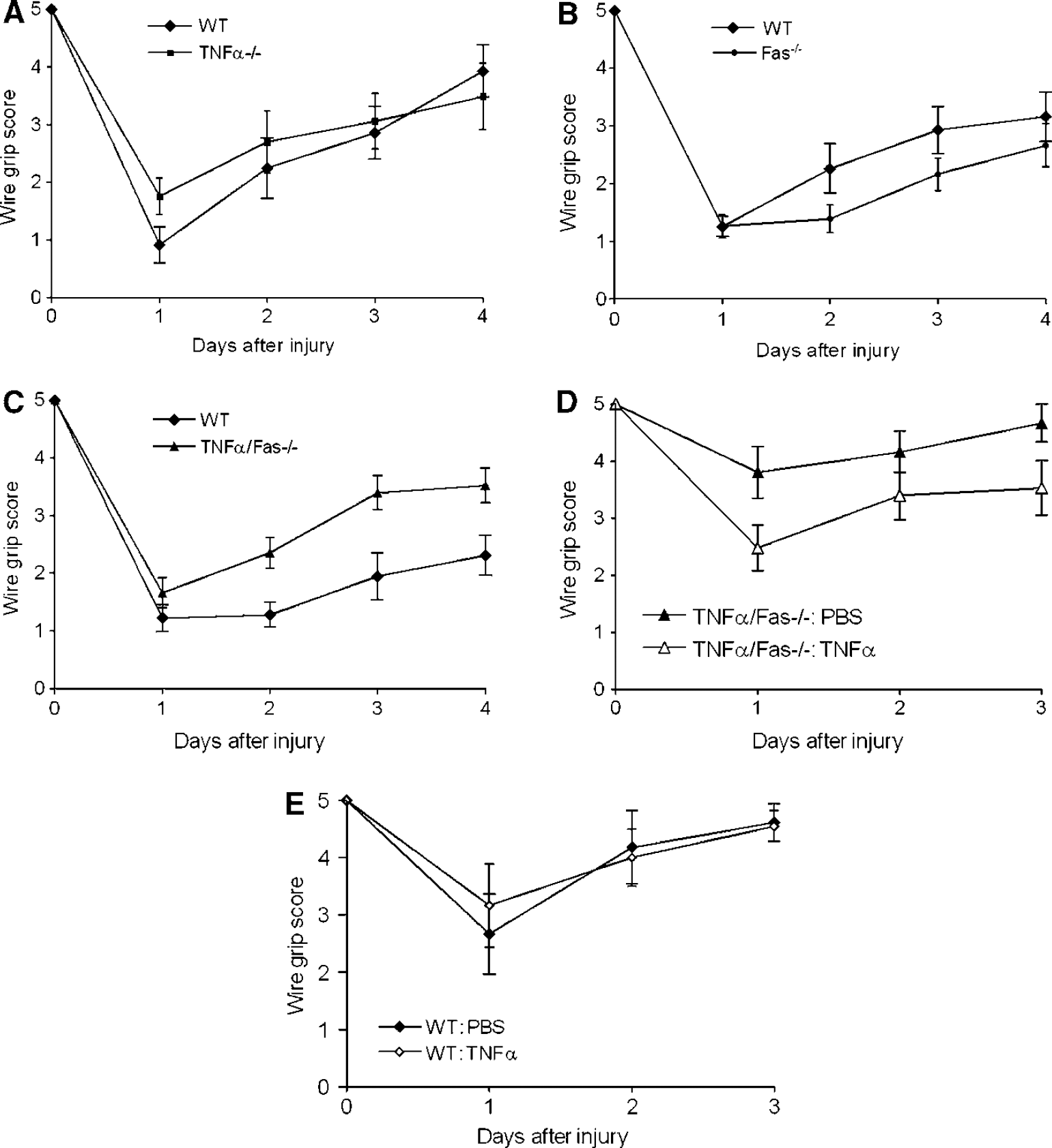
Reduced motor deficits after controlled cortical impact (CCI) in TNFα/Fas—/— mice. Vestibulo-motor function was assessed by wire grip test before CCI and up to 4 days after CCI in (
Tumor Necrosis Factor-Alpha/Fas Knockout Improves Spatial Memory Acquisition after Controlled Cortical Impact
To examine the contribution of TNFα and Fas to spatial memory acquisition, we first assessed naïve (uninjured) WT, TNFα—/—, Fas—/—, and TNFα/ Fas—/— mice using a MWM paradigm. Performance in the MWM did not differ among groups of uninjured animals (Figure 4A). We next asked whether antagonism of TNFα or Fas alone, or concomitant antagonism of TNFα/Fas together, influences spatial memory acquisition after CCI. Compared with WT, mice deficient in TNFα or Fas alone (single knockouts) had slightly worse performance, whereas performance in TNFα/Fas—/— mice was significantly improved versus WT (P < 0.05 for group effect, Figures 4B to 4D).
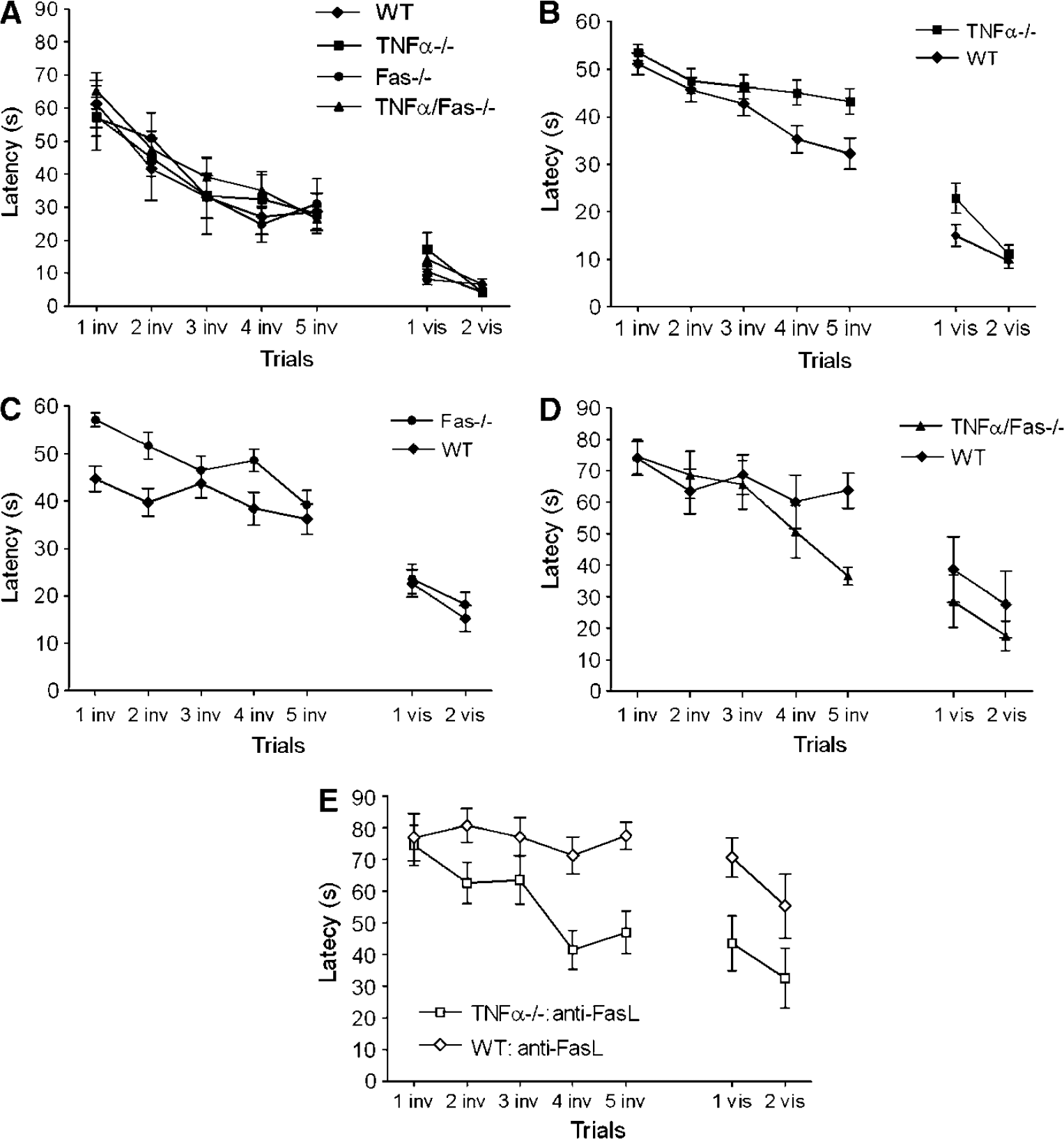
Inhibition of TNFα/Fas improves Morris water maze (MWM) performance after CCI. (
To confirm that the effect of TNFα/Fas knockout on MWM performance is not due to effects of genetic background, we used a pharmacological/genetic approach. Adult WT or TNFα—/— mice were administered anti-Fas ligand antibodies before CCI (see Materials and methods) and subjected to MWM testing at 1 to 2 weeks after injury. Similar to results in TNFα/Fas—/— mice, post injury MWM performance was significantly improved in TNFα-/- mice administered anti-Fas ligand antibody (P < 0.05 for group effect versus similarly treated WT mice, n = 10/group, Figure 4E). Contusion volume at 2 weeks did not differ between a subgroup of WT (6.2 ± 0.4 mm3, n = 5) and TNFα—/— (5.8+ 0.7 mm3, n = 4) mice administered anti-Fas ligand antibodies before CCI. These data strongly suggest that TNFα and Fas contribute redundantly to spatial learning deficits after CCI, and that differences in genetic background or development do not account for improved posttraumatic MWM performance in TNFα/Fas—/—versus WT mice.
Physiological variables in TNFα/Fas—/— and wild-type mice
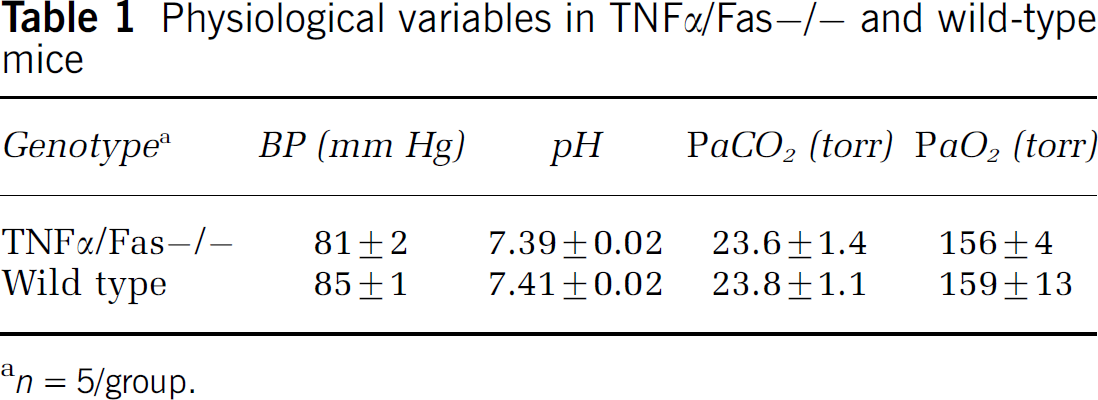
n = 5/group.
Effect of Tumor Necrosis Factor-Alpha/Fas knockout on Brain Edema and Plasmalemma Permeability in Injured Cells after Controlled Cortical Impact
To begin to address possible mechanisms governing the observed improvements in histopathology and functional outcome in TNFα/Fas—/— mice, we assessed the relationship between TNFα/Fas—/—and post-traumatic brain water content at 24 h after injury, the time of peak brain edema in the CCI model (Kochanek et al, 1995). The change in percent brain water content (injured—uninjured hemispheres) did not differ between injured TNFα/ Fas—/— (2.8 ± 0.4) and WT mice (2.6 ± 0.2, P = NS, 5 to 9/group), suggesting that neither TNFα nor Fas plays a significant role in development of brain edema in our CCI model.
We next assessed the effect of TNFα/Fas—/— on loss of plasmalemma integrity, an early marker of cellular injury and death after ischemic and traumatic neuronal injury, in cortical brain regions (Figure 5A) using in vivo PI labeling. Compared with WT, TNFα/Fas—/— mice had decreased numbers of PI-positive cortical cells at 6 h after CCI (P < 0.05, Figures 5B and 5C). In a separate experiment, no differences were observed in PI-positive cell counts among TNFα—/— (244 ± 29), Fas—/— (207 ± 33), and WT mice (184 ± 26 cells/ × 200 field) (P = 0.45 analysis of variance). These data suggest that early loss of plasmalemma integrity after CCI is mediated in part by redundant signaling between TNFα and Fas, and that inhibition of early loss of plasmalemma integrity, and hence acute cell death, may account for some of the reduced histopathology in TNFα/Fas—/— mice.
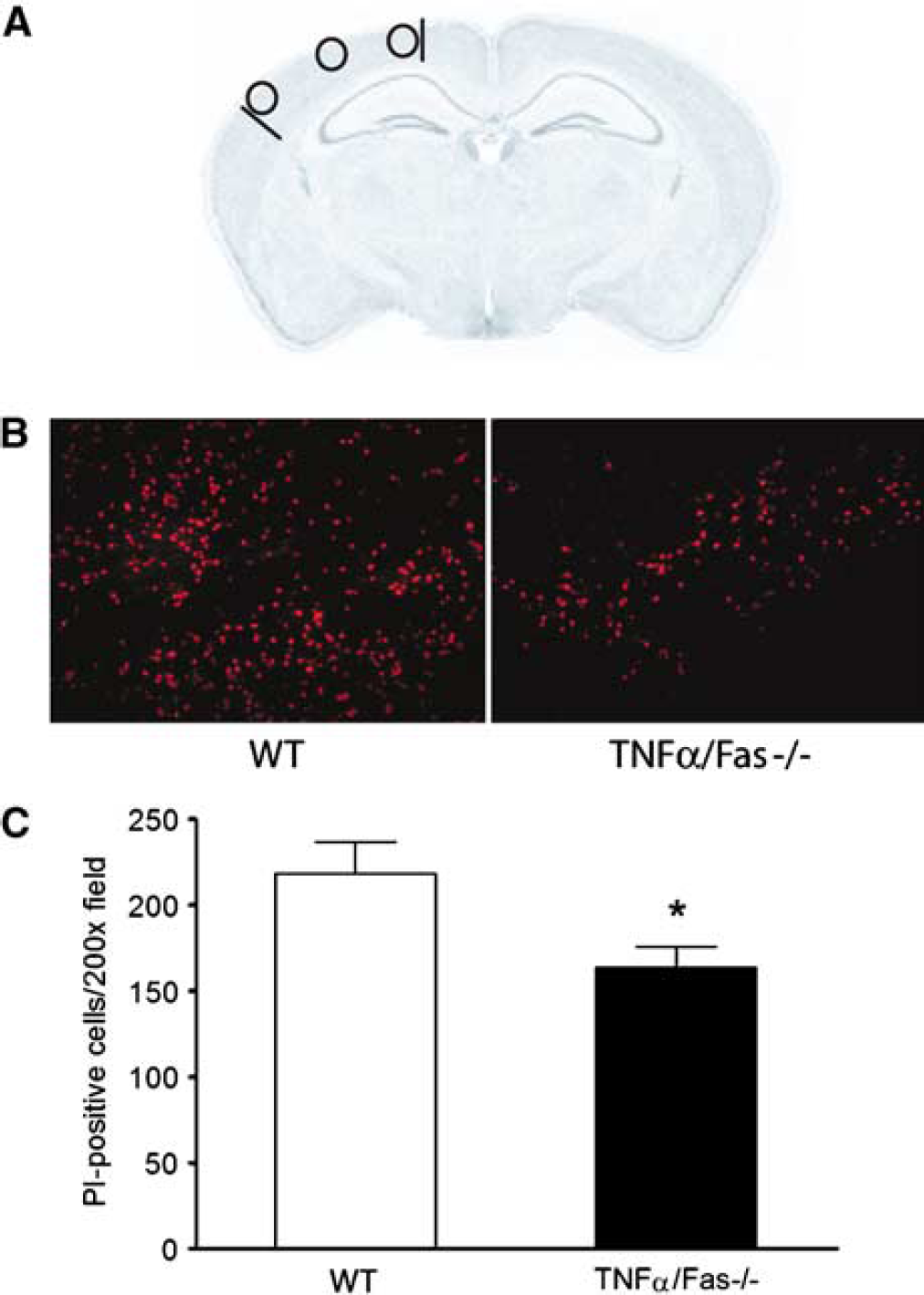
Genetic inhibition of TNFα/Fas decreases numbers of cells with loss of plasmalemma integrity after CCI. (
Effect of Tumor Necrosis Factor-Alpha/Fas Knockout on Inflammatory Markers in Mouse Brain after Controlled Cortical Impact
Because TNF and Fas are key initiators of the brain's inflammatory response to injury, we predicted that a general reduction in neuroinflammation would underlie the decreased histopathology observed after CCI in TNFα/Fas—/— mice. To assess neuroinflammation we examined brain neutrophil counts, astrocytosis, and the microglial response to CCI in injured mouse cortex at 48 h after injury. This time point was chosen because all three of these markers of cellular inflammation are robust in injured brain at 48 h. Figure 6 shows the quantitative assessment of cellular neuroinflammation after CCI. Neutrophil counts in injured cortical brain regions did not differ between TNFα/Fas—/— and WT mice (Figure 6A). Similarly, number and immunohistochemical density analysis of astrocytes and microglia showed no differences between groups (Figures 6B and 6C). These data suggest that the cellular neuroinflammatory response to CCI does not differ significantly between TNFα/Fas—/— and WT mice.
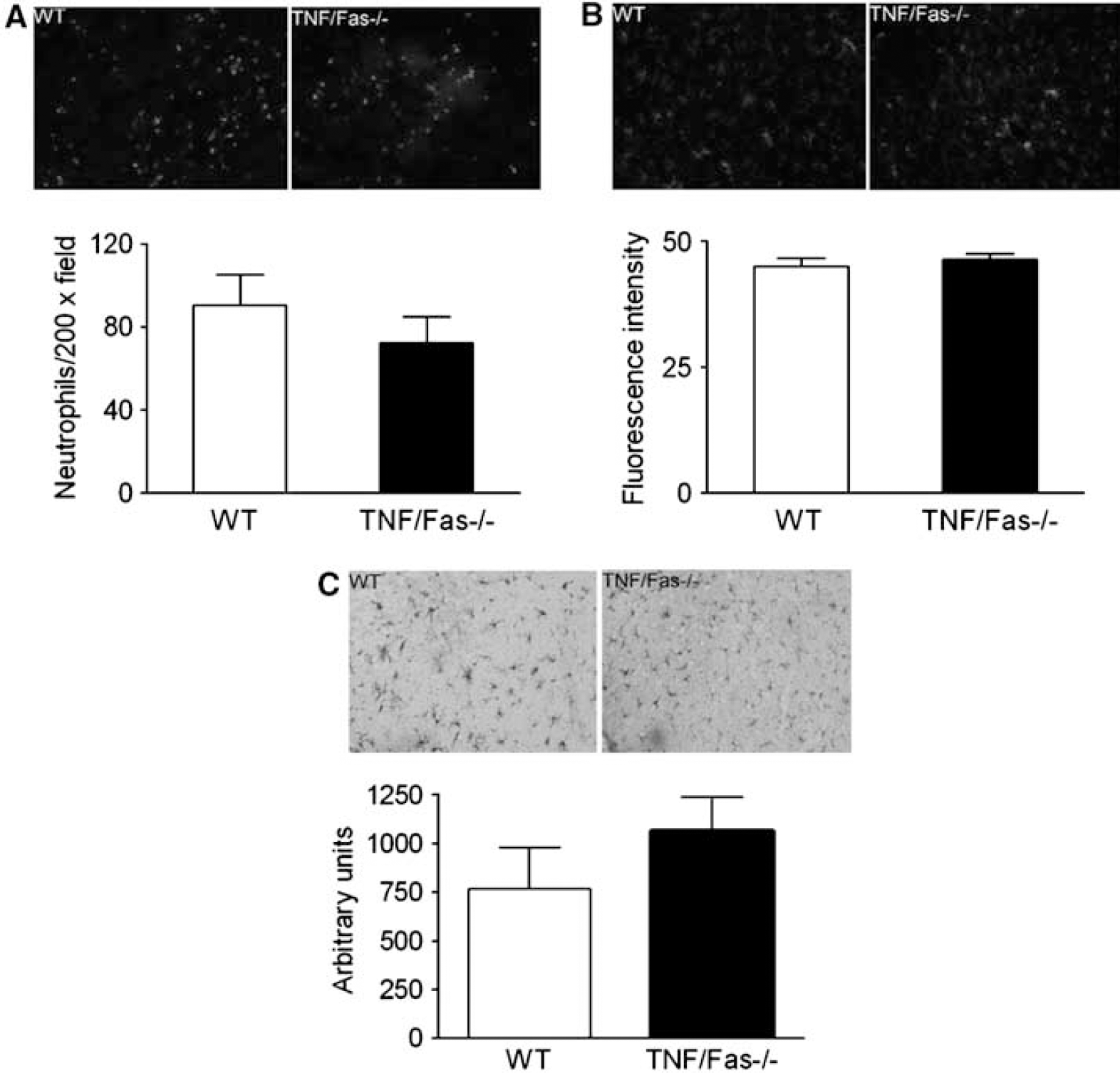
Assessment of brain neutrophils, astrocytosis, and microglial activation in injured cortex at 48 h after CCI. (
We next examined activity of a specific inflammatory component, MMP-9, an extracellular protease under the control of TNFα and Fas signaling which has been implicated in the pathogenesis of edema, tissue damage, and motor deficits in the CCI model (Wang et al, 2000). Matrix metalloproteinase-9 activity peaks at 24 h after CCI in mice and remains increased in contused brain for at least 7 days (Wang et al, 2000). Figure 7 shows the results of gel zymography in naïve and injured mice at 24 to 72 h after CCI. At 24 h, MMP-9 activity was robust in all groups of mice (WT, TNFα—/—, Fas—/—, TNFα/ Fas—/—). However, by 48 h and especially 72 h, MMP-9 activity was strikingly reduced in TNFα/ Fas—/— compared to WT, TNFα—/—, and Fas—/—mice (Figure 7 and (48 h) data not shown). These data suggest that decreased MMP-9 activity may play a role in protection from tissue and functional deficits after CCI observed in TNFα/Fas—/— mice.
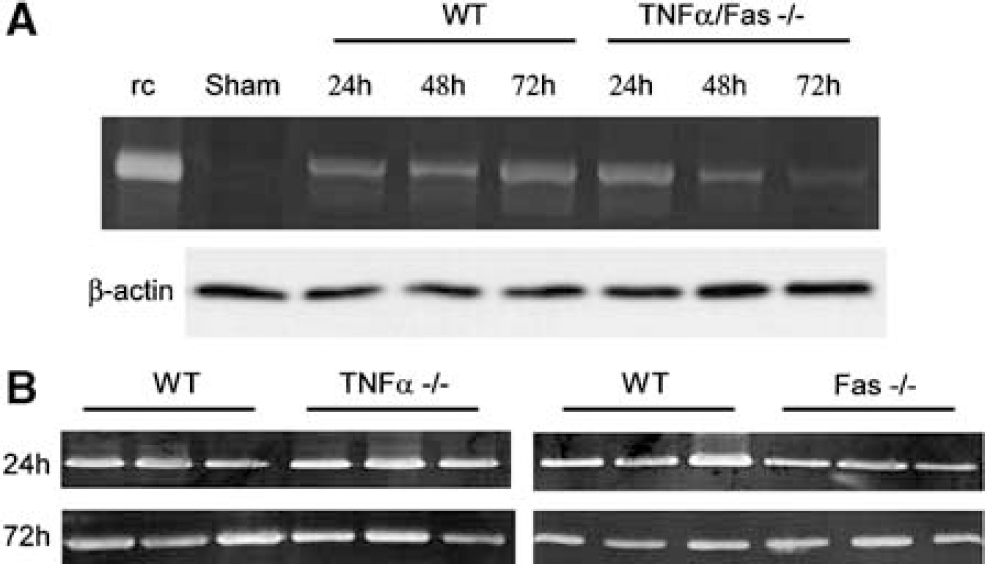
Matrix metalloproteinase-9 (MMP-9) activity is reduced after controlled cortical impact (CCI) in TNFα/Fas—/—mice. (
Tumor Necrosis Factor-Alpha/Fas Knockout Preserves Brain Development after Controlled Cortical Impact in Immature Mice
To assess further the importance of the TNFα/Fas pathway in TBI, we examined the response to CCI in immature mice (Tong et al, 2002). Immature (20 to 21 day old) mice were subjected to CCI and tested in the MWM from 28 to 35 days after injury. Brain lesion size was assessed at 35 days. At 4 to 5 weeks after CCI, MWM performance was significantly improved in TNFα/Fas—/— mice compared with WT, (P < 0.05 for group effect, Figure 8A), whereas performance in immature Fas—/— mice did not differ from WT. These long-lasting effects on brain development were further confirmed by lesion size analysis, as brain lesion size in TNFα/Fas—/— mice was reduced by 33% compared with WT at 35 days after injury (P < 0.001, Figure 8B). In contrast, brain lesion size did not differ between immature TNFα—/— (9.6 ± 0.9 mm3, n = 5) and WT (8.8 ± 0.6mm3, n = 9) or immature Fas—/— (9.3 ± 0.4mm3, n = 5) and WT (10.8 ± 0.5 mm3, n = 22) mice. In addition, no gender differences were observed in 35-day lesion size between immature male TNFα/Fas—/— (7.5 ± 0.9 mm3, n= 8) and female TNFα/ Fas—/— (7.2 ± 0.9 mm3, n = 10) mice, or between immature WT male (10.9 ± 0.6, n = 12) and female (10.7 ± 0.9 mm3, n = 10) mice.
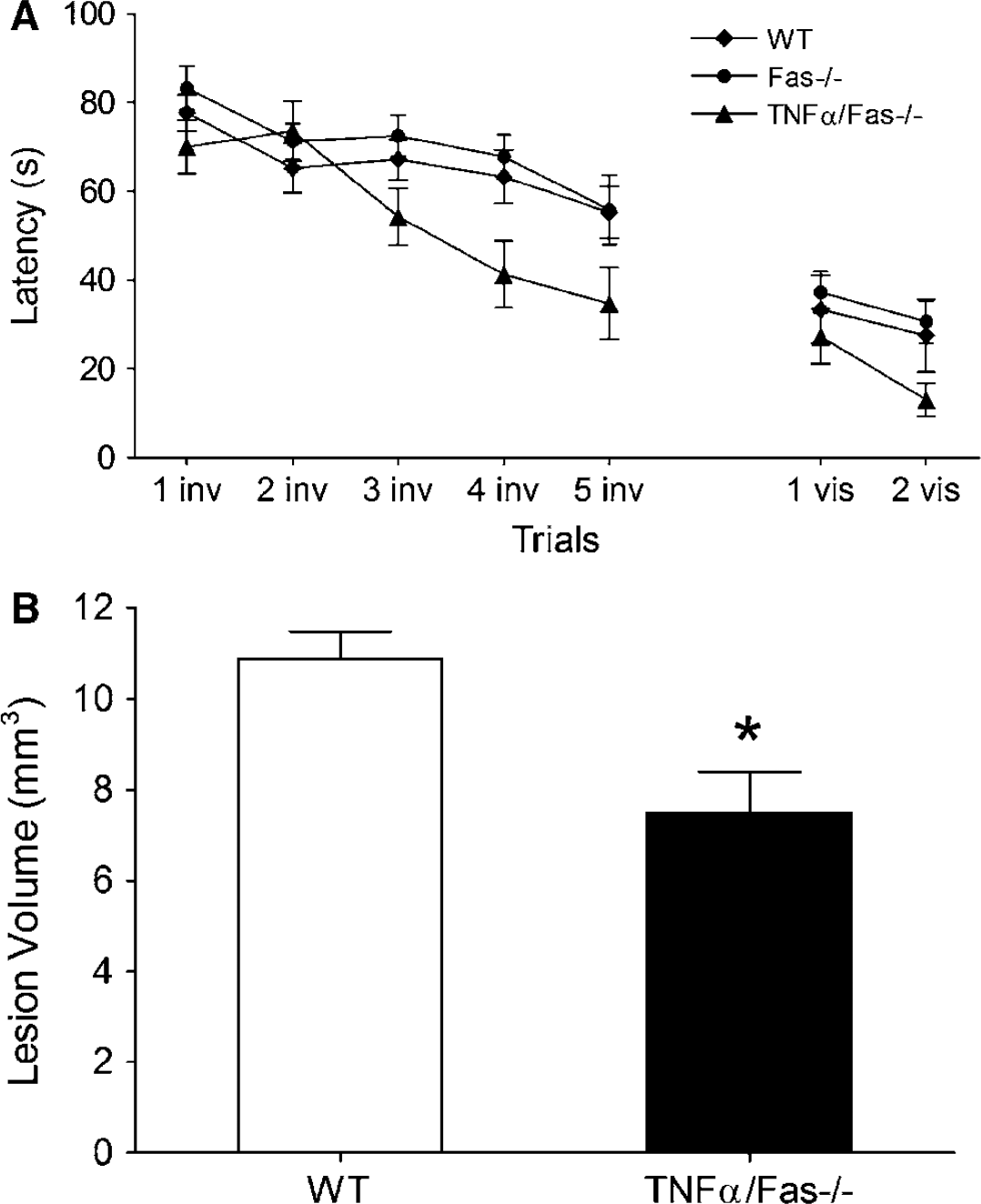
Genetic inhibition of TNFα/Fas improves Morris water maze (MWM) performance and reduces brain tissue damage after controlled cortical impact (CCI) in immature mice. Immature TNFα/Fas—/— and WT mice were subjected to CCI. Morris water maze performance was assessed during adulthood at 4 to 5 weeks after injury, and lesion size was determined at 5 weeks after injury. (
Discussion
The major findings of this study are that dual inhibition of TNFα and Fas reduced brain tissue damage, motor dysfunction, and spatial learning deficits after CCI in mice. Importantly, studies employing pharmacologic/genetic inhibition of TNFα/Fas, and reconstitution of TNFα/Fas—/— mice with recombinant TNFα, provided additional proof that redundant effects of TNFα and Fas mediate outcome after CCI. Perhaps most importantly, immature mice deficient in TNFα and Fas had long-lasting beneficial effects on post-traumatic cognitive and histopathological outcome assessed in adulthood. These novel findings suggest a fundamental role for TNFα and Fas in the pathogenesis of TBI.
Dual inhibition of both TNFα and Fas was required to reduce brain tissue damage after CCI. In contrast, inhibition of TNFα or Fas alone reduced ischemic infarct size in neonatal and adult mice, but concomitant antagonism of TNFα and Fas ligand together afforded the most protection against focal stroke (Graham et al, 2004; Martin-Villalba et al, 1999, 2001). In experimental spinal cord injury, pharmacological or genetic inhibition of Fas ligand or Fas receptor improved measures of apoptotic cell death, axonal injury, tissue damage, and functional outcome (Casha et al, 2005; Demjen et al, 2004). These studies suggest a greater degree of involvement of Fas signaling in stroke and spinal cord injury models compared with CCI.
Previous studies have showed magnified effects of apoptotic death mechanisms, inflammation, and neuroprotective therapies after experimental stroke and TBI in immature versus adult animals (Bittigau et al, 2004; Cheng et al, 1997; Graham et al, 2004; Pohl et al, 1999). In this study, TNFα/Fas inhibition reduced tissue damage to a similar degree in immature and adult mice, suggesting that the protective effects of TNFα/Fas—/— are long lasting and age-independent within the age range (3 to 16 weeks) studied. In contrast, only transiently reduced tissue damage was observed in TBI studies inhibiting intrinsic cell death mechanisms lacking an inflammatory component (Bermpohl et al, 2006; Clark et al, 2007). Mechanisms leading to brain tissue atrophy after TBI, although not well understood, involve persistent upregulation of brain TNFα in the chronic phase (Holmin and Mathiesen, 1999). Our data suggest that redundant TNFα and Fas signaling may contribute to the chronic, progressive tissue loss, which occurs in adults and children after severe TBI.
In agreement with decreased tissue damage, we also found decreased numbers of cells with acute plasmalemma permeability to PI at 6 h after CCI. Loss of plasmalemma integrity is a hallmark of ischemic and traumatic neuronal cell injury and death (Farkas et al, 2006; Unal Cevik and Dalkara, 2003). Tumor necrosis factor-alpha and Fas may initiate caspase-mediated apoptosis as well as necrosis, each of which might be associated with time-dependent loss of plasmalemma integrity in injured cells. Recently, TNFα and Fas have also been shown to initiate a novel form of programmed necrosis, termed ‘necroptosis,‘ which also features plasmalemma permeability to PI (Degterev et al, 2005). Thus, one possible interpretation of our data is that decreased PI-positive cells reflect a reduction in acute necroptotic cell death, and perhaps other modes of cell death as well. However, membrane poration can also be seen in cells with normal ultrastructure and may be reversible, as reported in a diffuse TBI model that lacks overt tissue damage (Farkas et al, 2006). We reported previously decreased PI-positive cortical cells at 6 h after CCI in mice deficient in Bid (Bermpohl et al, 2006). Other secondary injury mechanisms leading to loss of cellular plasmalemma integrity may include excitotoxicity, oxidative stress, and energy failure, among others (Lee et al, 1999; Kristensen et al, 2001). Data from this study suggest that TNF and Fas also play a role after CCI.
We found significant improvement in early post-traumatic motor performance in mice deficient in TNFα/Fas compared with TNFα—/—, Fas—/—,orWT mice. The observed improvement in TNFα/Fas—/—mice was reversible by reconstitution with recombinant TNFα, proving that TNFα and Fas contribute redundantly to post-traumatic motor dysfunction after CCI. The differences in motor dysfunction between WT animals treated with PBS in experiments shown in Figure 3A versus Figure 3E are likely due to intralaboratory variability in injury severity occasionally produced by CCI models (Clark et al, 2000). Motor performance was similar in a subgroup of TNFα/Fas—/— and WT mice tested at 38 days after injury (data not shown), and we did not observe obvious deterioration in motor function at 4 or more weeks after injury in any of the single knockout mice studied (data not shown), as has been previously reported in TNFα—/— mice (Scherbel et al, 1999). One reason for the discrepancy between our findings and those reported previously for TNFα—/— may be that our wire grip test is less sensitive to subtle changes in vestibulomotor function detected using composite neuroscores (Scherbel et al, 1999).
A striking finding of this study is that dual inhibition of TNFα/Fas significantly improved cognitive performance after CCI. Compared with injured immature and adult WT mice, which failed to significantly improve their performance, acquisition times in injured TNFα/Fas—/— mice were almost as low as those of uninjured mice by the end of the (hidden platform) testing period. Tumor necrosis factor-alpha and Fas functioned redundantly in this regard, and the beneficial effects of TNFα/Fas inhibition in immature mice persisted into adulthood, suggesting a permanent protective effect. This finding is particularly significant in light of recent evidence that the severely injured immature brain may have much less plasticity and potential for recovery of cognitive function than previously considered (Anderson et al, 2005). Although improved MWM performance in TNFα/Fas—/— mice was associated with decreased post-traumatic lesion size, previous studies do not support a direct correlation between histopathology and functional outcome after CCI (Bermpohl et al, 2006). In our study, administration of anti-Fas ligand antibodies to TNFα—/— mice improved MWM performance after injury without reducing lesion size (versus similarly treated WT mice). Thus, mechanisms in addition to reduced tissue damage likely account for the beneficial effects of TNFα/Fas inhibition on after injury MWM performance.
Tumor necrosis factor-alpha and other cytokines have been postulated to impair learning and memory in a number of neuropathological conditions including CCI (Pickering et al, 2005; Scherbel et al, 1999), and to play a physiological role in normal brain function by modulating key elements of synaptic transmission involved in memory formation and learning (Albensi and Mattson, 2000). Mice with non-functional Fas on the MRL background have spatial learning deficits associated with autoimmunity and brain mononuclear infiltrates (Sakic et al, 1997); however, direct evidence that Fas participates in physiological or pathological learning and memory has not, to our knowledge, been reported.
Somewhat surprisingly, we found no differences in brain neutrophils, astrocytosis, or microgliosis between WT and TNF/Fas knockout mice. Thus, we conclude that reduced histopathology in knockout versus WT mice does not relate in a simple way to effects on generalized neuroinflammation after CCI. However, because MMP-9 activation may be considered a marker of neuroinflammation, effects on MMP-9 and perhaps other specific inflammatory mediators influenced by TNF/Fas may have contributed to differences in outcome between the two groups. Previous work has shown that MMP-9 mediates brain edema, motor impairment, and tissue damage after CCI in mice (Wang et al, 2000), and contributes to spatial learning deficits after meningitis in rats (Leib et al, 2001). Thus, MMP-9 may be one mechanistic link between TNFα/Fas signaling and histopathological and functional outcome after CCI. In our study, post-traumatic brain edema (assessed at 24 h) would not have been expected to differ between groups on the basis of decreased MMP-9 activity, because MMP-9 activity was similar in WT and TNFα/Fas—/— mice at the time brain edema was assessed (24 h).
From a clinical standpoint, it could be argued that our CCI model may not be relevant to patients who develop significant intracranial hypertension, as we did not replace the cranial bone after injury. Rather, our CCI model may be more representative of those patients with focal brain injury who undergo early decompressive craniectomy, or those patients who develop functional deficits in the absence of intracranial hypertension. In this regard, our model may be particularly clinically relevant if early decompressive craniectomy becomes standard of care in the management of patients with severe focal TBI. Significant histopathology and functional deficits are characteristics of CCI models with or without replacement of the craniotomy (Bermpohl et al, 2006; Whalen et al, 2000). Nonetheless, whether TNFα/Fas inhibition would be similarly protective in a CCI (or other TBI) model featuring significant intracranial hypertension is an important question for future studies.
Our findings that TNFα/Fas inhibition is protective after CCI may have important implications for treatment of patients, particularly young children, who suffer lifelong cognitive and other neurological sequelae after TBI. However, given the rapid upregulation of TNFα and Fas receptor in experimental TBI models (Beer et al, 2000; Qiu et al, 2002), inhibitors targeting TNFα and Fas might have a short therapeutic window as well as undesirable effects on immune and other homeostatic functions. Thus, mechanisms downstream of TNFα/Fas should be identified and investigated as therapeutic targets for developing specific treatment for patients with traumatic and perhaps other forms of acute brain injury.