
Abstract
Addition of a small peptide called ISG15 is known as ISGylation, which is an ubiquitin (ub)-like posttranslational modification. We currently show that focal ischemia induced by transient middle cerebral artery occlusion (MCAO) in adult mice significantly induces cortical protein ISGylation between 6 and 24 hours reperfusion. With two-dimensional western blotting, 45 proteins were observed to be significantly increased in ISGylation (by 1.8- to 9.7-fold) after focal ischemia compared with sham control. Immunochemistry showed that ISGylated proteins are localized in neurons within the ipsilateral striatum and in astroglia within the peri-infarct cortex of ischemic mice. When subjected to transient MCAO, ISG15−/− mice showed increased mortality, exacerbated infarction, and worsened neurologic recovery than did wild-type controls. In addition, mice lacking UBE1L (ub-activating enzyme E1-like protein, the first enzyme of the ISGylation cycle) also showed bigger infarcts when subjected to transient MCAO. Regional cerebral blood flow or other physiologic parameters were not significantly different in both knockouts compared with wild-type controls. These studies indicate that increased protein ISGylation might be an endogenous neuroprotective adaptation to minimize poststroke brain damage.
Introduction
Proteins undergo several posttranslational modifications (PTMs), which exponentially increase the functional diversity of the proteome (Baumann and Meri, 2004; Sunyer et al, 2008). Many PTMs are reversible and thus activate/deactive proteins like enzymes and second messenger signaling mediators. The ubiquitin (ub) class of PTMs that includes ubiquitination, SUMOylation, and ISGylation add a small protein or peptide to other proteins mediated by a set of specific ligases (Kerscher et al, 2006).
In particular, ISGylation is the conjugation of the protein ISG15 to other proteins. ISG15 is expressed by many mammalian and rodent tissues, but proteins will be ISGylated only based on the need. The C-terminal residue of ISG15 is a glycine, and the carboxyl group of this glycine is the site of attachment to substrates. Lysine side chains are the most common target sites within substrate proteins, resulting in an amide (or isopeptide) bond between ISG15 and substrate (Kerscher et al, 2006). When infected with bacteria or virus, protein ISGylation increases quickly by several fold, presumably stimulated by type I interferon released by virus and by lipopolysaccharide released by bacteria (Kim and Zhang, 2003). Despite its discovery 18 years ago (Loeb and Haas, 1992), the precise functional role of ISGylation in various cell types of the body is still not completely understood. As ISG15 is absent in lower organisms like nematodes and insects, ISGylation is considered to control higher-order cellular functions in eukaryotes (Dao and Zhang, 2005). As ISGylation is induced by bacterial/viral infections, it is considered a regulator of immune functions (Jeon et al, 2010).
ISGylation and de-ISGylation occur in a cyclical manner mediated by a set of ligases (Supplementary Figure 1). The E1 enzyme ub-activating enzyme E1-like protein (UBE1L) a.k.a. ub-like modifier-activating enzyme 7 adenylates ISG15 and forms a thioester bond with it. Activated ISG15 will be passed to the E2 ligase ub-conjugating enzyme H8 by transesterification. There are two E3 ligases for ISG15. The first is an estrogen-responsive finger protein (Efp) that brings the target protein to be ISGylated to the E2–ISG15 complex, and the second ligase is an HECT domain and RCC1-like domain-containing protein 5 that mediates transfer of ISG15 to the substrate protein (Lenschow et al, 2007). The de-ISGylating enzyme ub-processing protease-43 a.k.a. ub-specific peptidase-18 releases ISG15 from the ISGylated proteins (Malakhov et al, 2002).
Recent studies have shown that increased protein ubiquitination and SUMOylation seen after global ischemia, focal ischemia, and hypoxia are neuroprotective (Cimarosti et al, 2008; Lee et al, 2007; Vannucci et al, 1998; Yamashita et al, 1991; Yang et al, 2008a, 2008b). As the significance of ISGylation after cerebral ischemia is not yet studied, we presently evaluated the temporal and cellular patterns of protein ISGylation after transient middle cerebral artery occlusion (MCAO) in adult mice. We further studied the effect of transient MCAO in knockout mice that lack either ISG15 or UBE1L.
Materials and methods
Knockout Mice
Both ISG15−/− and UBE1L−/− mice were developed on a C57BL/6 background. ISG15−/− mice were generated in Dr Klaus-Peter Knobeloch's laboratory in Charité Universitätsmedizin (Berlin, Germany) (Osiak et al, 2005) and subsequently bred in Dr Deborah Lenschow's laboratory at the Washington University (St Louis, MO, USA) (Lenschow et al, 2007). ISG15−/− mice used in this study were bred from Dr Lenschow's colony. UBE1L−/− mice were developed in Dr Dong-Er Zhang's laboratory (The Scripps Research Institute, La Jolla, CA, USA). UBE1L−/− mice used in this study were bred using breeders from Dr Zhang's laboratory. UBE1L−/− mice show normal levels of ISG15, but lack ISG15 conjugation capability (Kim et al, 2006). All surgical procedures were approved by the Research Animal Resources and Care Committee of the University of Wisconsin-Madison and animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, US Department of Health and Human Services Publication Number 86–23 (revised).
Focal Ischemia
Focal ischemia was induced in adult, male mice (weighing 23 to 26 g) by transient MCAO by an intraluminal suture method as described earlier (Kapadia et al, 2006; Tureyen et al, 2008). In brief, under isoflurane anesthesia (induction: 2%; maintenance: 1.2% in an oxygen, and nitrous oxide 50:50 mixture), the left femoral artery was cannulated for continuous monitoring of arterial blood pressure and to obtain measurements of pH, PaO2, PaCO2, hemoglobin, and blood glucose (i-STAT; Senor Devices, Waukesha, WI, USA). The rectal temperature was maintained at 37°C±0.5°C during surgery with a feedback-regulated heating pad. After a midline skin incision, the left external carotid artery was exposed, a surgical 6 to 0 monofilament nylon suture blunted at the end was introduced into its lumen, and gently advanced into the internal carotid artery until regional cerebral blood flow was reduced to ∼15% of the baseline (recorded by laser Doppler flowmeter; Vasamedics, LLC, St Paul, MN, USA) as described earlier (Vemuganti et al, 2004). After either 40 minutes or 1 hour occlusion, the suture was withdrawn (reperfusion confirmed by laser Doppler), the wound sutured, and the mouse allowed to recover from anesthesia and returned to the cage with
Infarct Volume Measurement
Ischemic infarct volume was measured as described earlier (Kapadia et al, 2006; Tureyen et al, 2008). In brief, mice subjected to transient MCAO were perfused transcardially with buffered paraformaldehyde at 3 days of reperfusion. Each brain was postfixed, cryoprotected, and sectioned (coronal; 40
Neurologic Function Tests
To evaluate postischemic sensory and motor functions, cohorts of mice were subjected to modified neurologic severity scoring test, adhesive-removal test, rotarod test, and beam balance test. All mice were tested by an evaluator blinded to the study groups at 2 days before MCAO and then on day 3 of reperfusion after MCAO. Postural reflex was measured by modified neurologic severity scoring test on a 5-point scale as described previously (Kapadia et al, 2006; Tureyen et al, 2008). A score of 0 suggests no neurologic deficit (normal), 1 suggests mild neurologic deficit (failure to extend the right forepaw fully), 2 suggests moderate neurologic deficit (circling to the right), 3 suggests severe neurologic deficit (falling to the right), and 4 suggests very severe neurologic deficit (the rat did not walk spontaneously and had a depressed level of consciousness). Sensory motor deficits were evaluated with the adhesive-removal test by placing small adhesive tapes (1 × 1 cm2) on each forepaw. The time taken by a mouse to remove tape from each paw was recorded in three trials (Bouet et al, 2009). Motor coordination and motor learning were measured by rotarod test (Jin et al, 2010). After habituation on a stationary rod, mice subjected to transient MCAO were tested with a cylinder that rotated at 8 r.p.m. for 3 minutes. The latency time to fall from the rod was measured automatically with a stop watch connected to a detector with a photo beam circuit. To examine the vestibulomotor reflex activity, ischemic mice were subjected to beam balance test (Windle et al, 2006). Mice were trained before MCAO to balance on a narrow beam (30 × 1.3 cm2) for 60 seconds. At 3 days of reperfusion after MCAO, they were scored on a scale of 0 to 6 (steady balance, 0; grasps side of the beam, 1; one limb falls down but hugs the beam, 2; two limbs fall down or spins on beam but will not fall down, 3; falls down between 40 and 60 seconds, 4; falls down between 20 and 40 seconds, 5; and falls down before 20 seconds, 6).
Western Blotting
Western blotting was conducted as described earlier (Nakka et al, 2010). In brief, the ipsilateral cortical tissue was homogenized in ice-cold 200 mmol/L HEPES buffer (pH 7.4) containing 250 mmol/L sucrose, 1 mmol/L dithiothreitol, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.1 mmol/L phenylmethylsulfonyl fluoride, and 1% protease inhibitor cocktail (Sigma Chemical Co., St Louis, MO, USA). The homogenates were centrifuged at 10,000
Two-Dimensional Analysis of ISGylated Proteins
Two-dimensional (2D) electrophoresis and densitometric analysis were conducted as described earlier (Dhodda et al, 2004). In brief, ipsilateral cortical tissue (∼50 mg) from each mouse was homogenized in 1 mL of 10 mmol/L Tris-HCl buffer (pH 7.4) containing 0.3% SDS, 50 mmol/L magnesium chloride, 500
Immunohistochemistry
Mice subjected to 40 minutes MCAO were transcardially perfused with 4% buffered paraformaldehyde and the brains were sectioned (coronal; 35
Results
Focal Ischemia Induced Protein ISGylation
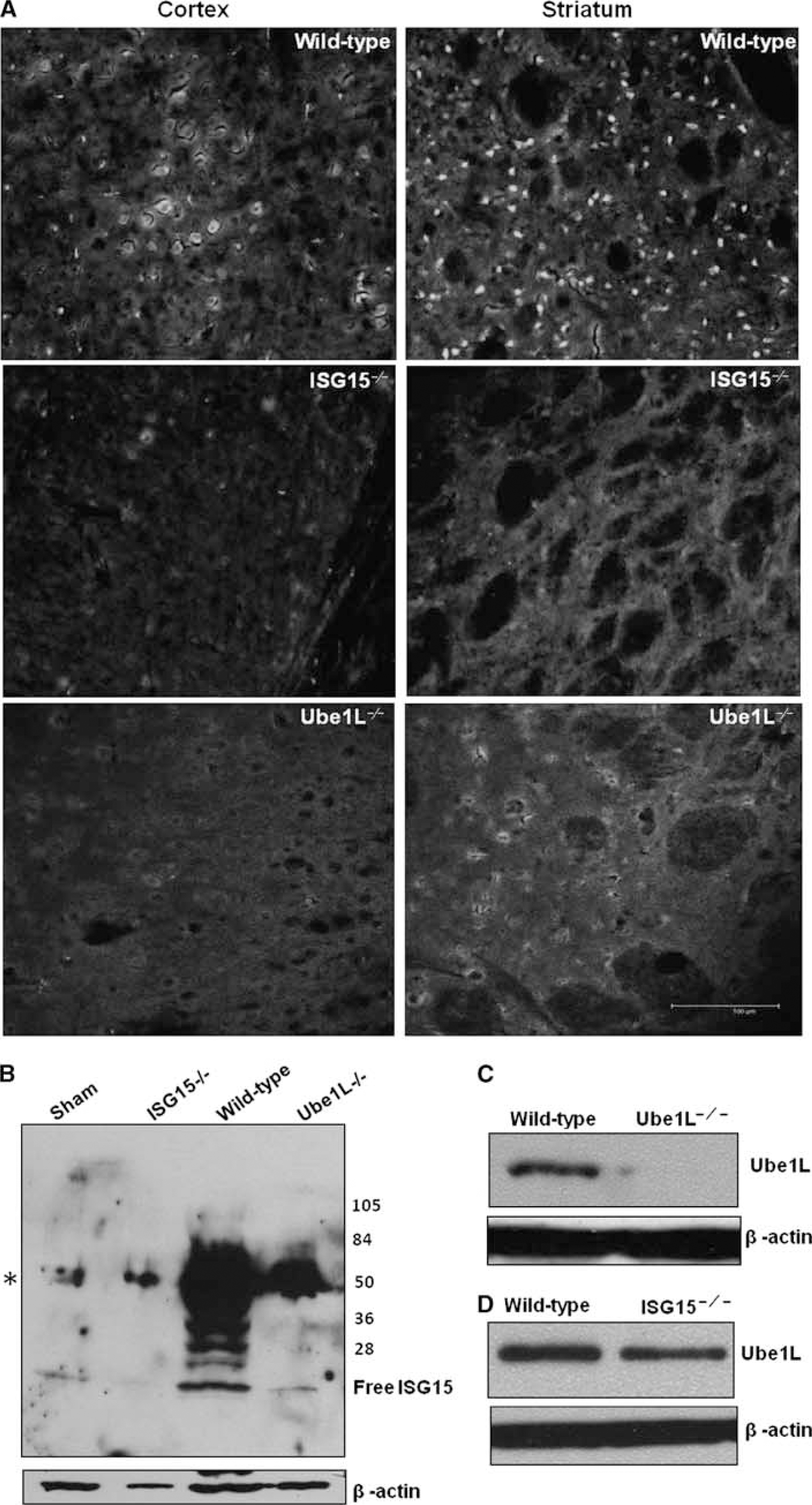
The number and content of ISG15-conjugated proteins (reactive with ISG15 antibodies) increased progressively from 6 to 24 hours reperfusion in the ipsilateral cortex of mice subjected to transient MCAO compared with sham control (Figure 1A). Most of the ISGylated proteins in the postischemic brain were observed to be in the MW range of 25 to 250 kDa. There was a significant increase in the total ISGylated protein content at 6 hours (by 242%±33%,
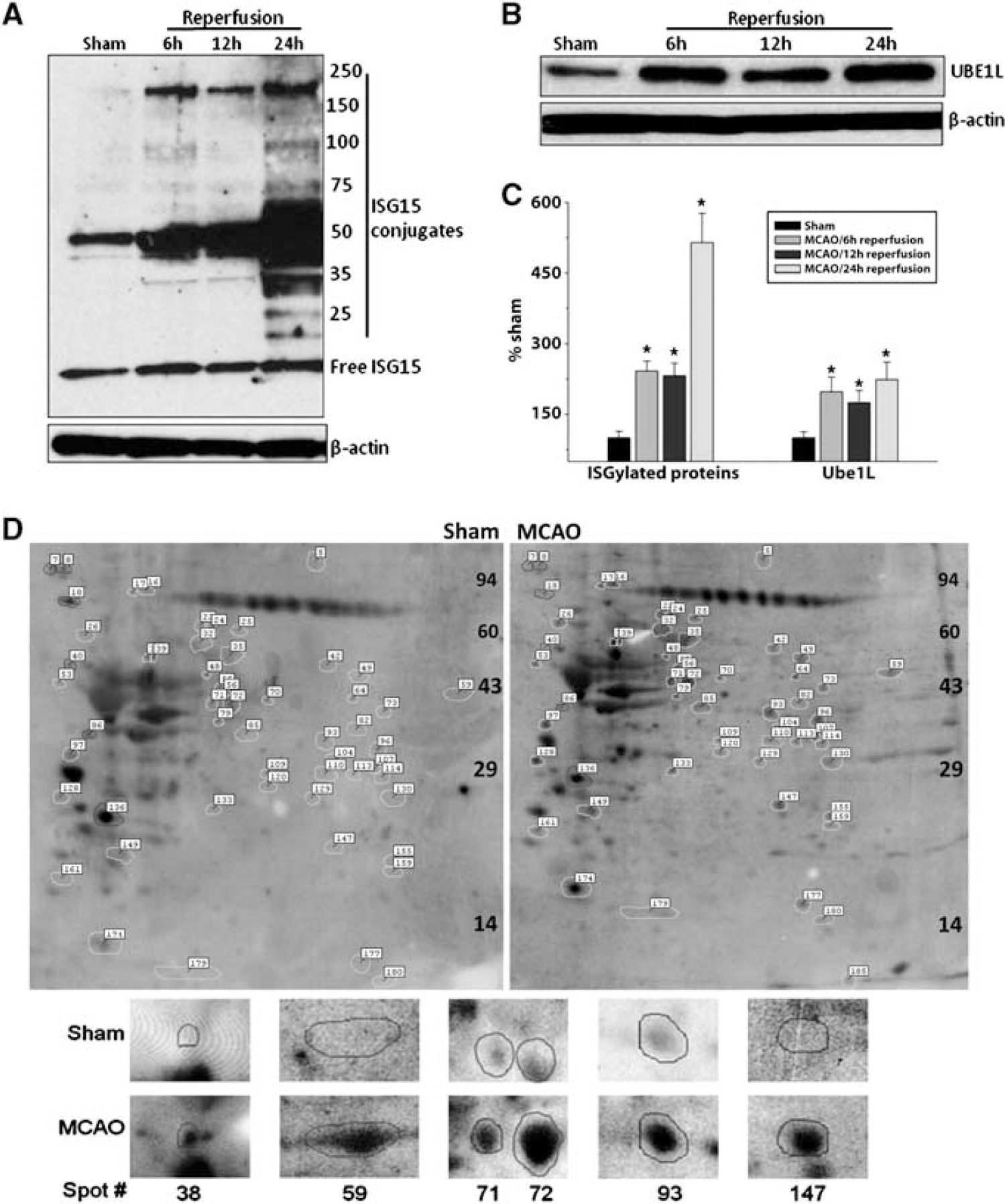
Transient MCAO increased the ISGylation of proteins in the molecular weight range of 25 to 200 kDa (
Increased protein ISGylation in the ipsilateral cortex of mice subjected to transient MCAO or sham surgery was also confirmed with 2D western blotting. With laser scanning densitometry combined with analysis by Progenesis PG240 software/TT900, 186 protein spots reacted with ISG15 antibodies (those that can be reliably identified in all 2D western blots of either MCAO or sham group) were analyzed. Of the 186 spots, 45 showed significantly increased and 6 showed significantly decreased density in the MCAO group than in the sham group (Supplementary Table 1). The sum of densities of all the ISG15+ spots was 5.34±1.21 for the sham and 13.38±1.72 for the MCAO groups (
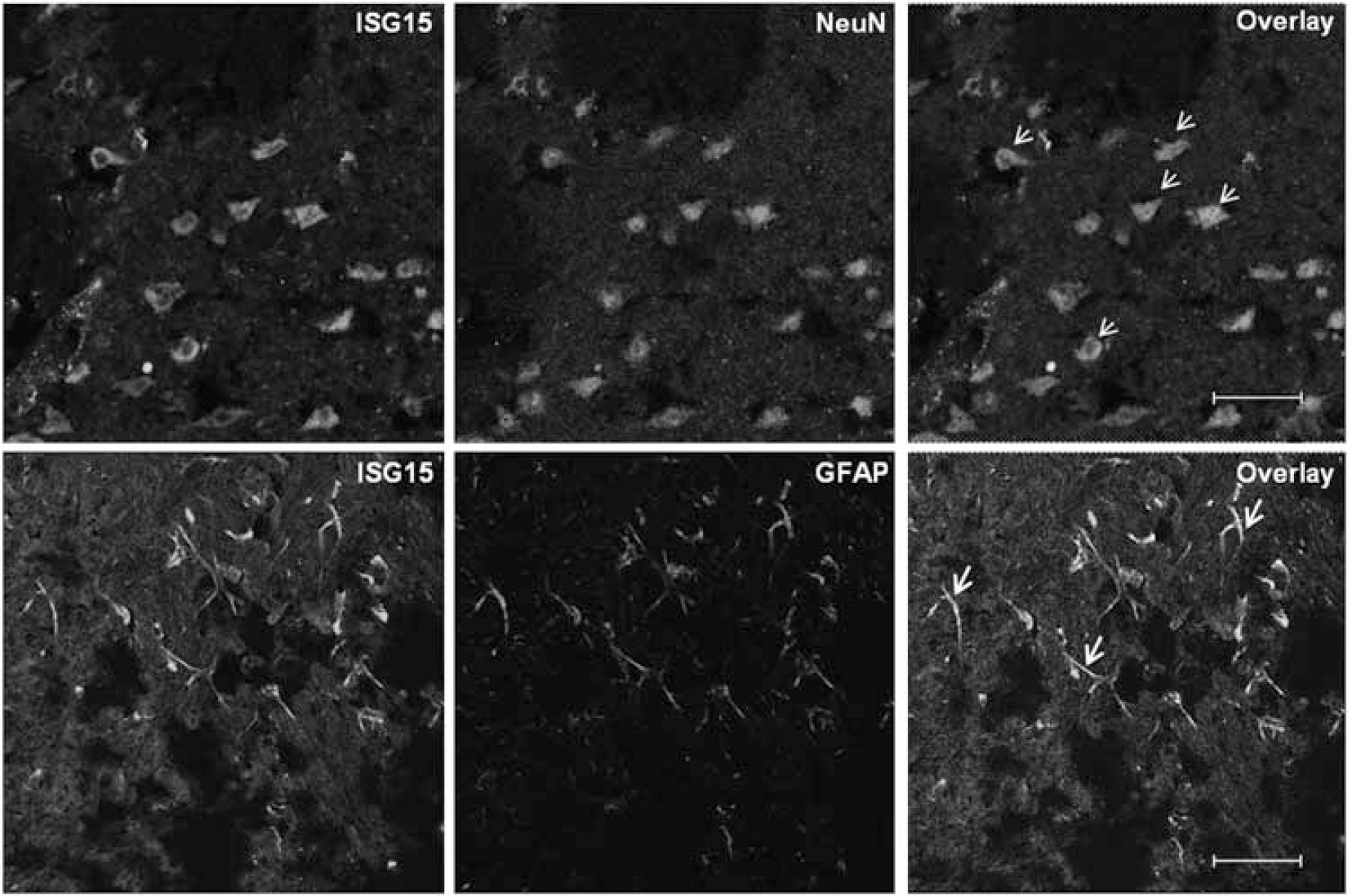
Colocalization of ISG15 protein expression with neuronal (NeuN) and astroglial (GFAP) markers in ischemic brain after 40 minutes MCAO, followed by 24 hours reperfusion. Double fluorescence labeling with rabbit polyclonal anti-ISG15 (top and bottom row, green), and mouse monoclonal anti-NeuN (top row red), or mouse monoclonal anti-GFAP (bottom row, red) was performed. Neuronal images were taken from the ipsilateral striatum and astrocytic images were taken from the peri-infarct cortex. Colocalization of ISG15 with NeuN (top row) and GFAP (bottom row) was shown by arrows. Scale bar for top and bottom rows is 31.1 and 47
Increased Postischemic Brain Damage in ISG15−/− and UBE1L−/− Mice
As 1 hour transient MCAO induced ∼45% mortality in ISG15−/−, the occlusion period was decreased to 40 minutes to measure infarction and neurologic function. Both ISG15−/− and UBE1L−/− mice developed bigger infarcts than did wild-type mice (Figure 3, top panel). The regional cerebral blood flow measured during MCAO and reperfusion was observed to be similar between the three genotypes (Figure 3, middle panel). There were no significant differences in the physiologic parameters (brain and body temperature, hemoglobin levels, blood glucose, pH, PaCO2, and PaO2) measured before and during occlusion and during reperfusion between the three groups (data not shown). In wild-type mice, transient MCAO/reperfusion (3 days) resulted in an infarct with a total volume of 14.1±2.3 mm3 (Figure 3, bottom panel). The infarct volume was observed to be significantly higher in ISG15−/− mice (by 136%;
Exacerbated Neurologic Deficits in ISG15−/− Mice after Transient Middle Cerebral Artery Occlusion
The average postischemic neuroscore at 3 days of reperfusion after transient MCAO was observed to be significantly higher (
Worsened postischemic neurologic dysfunction in ISG15−/− mice
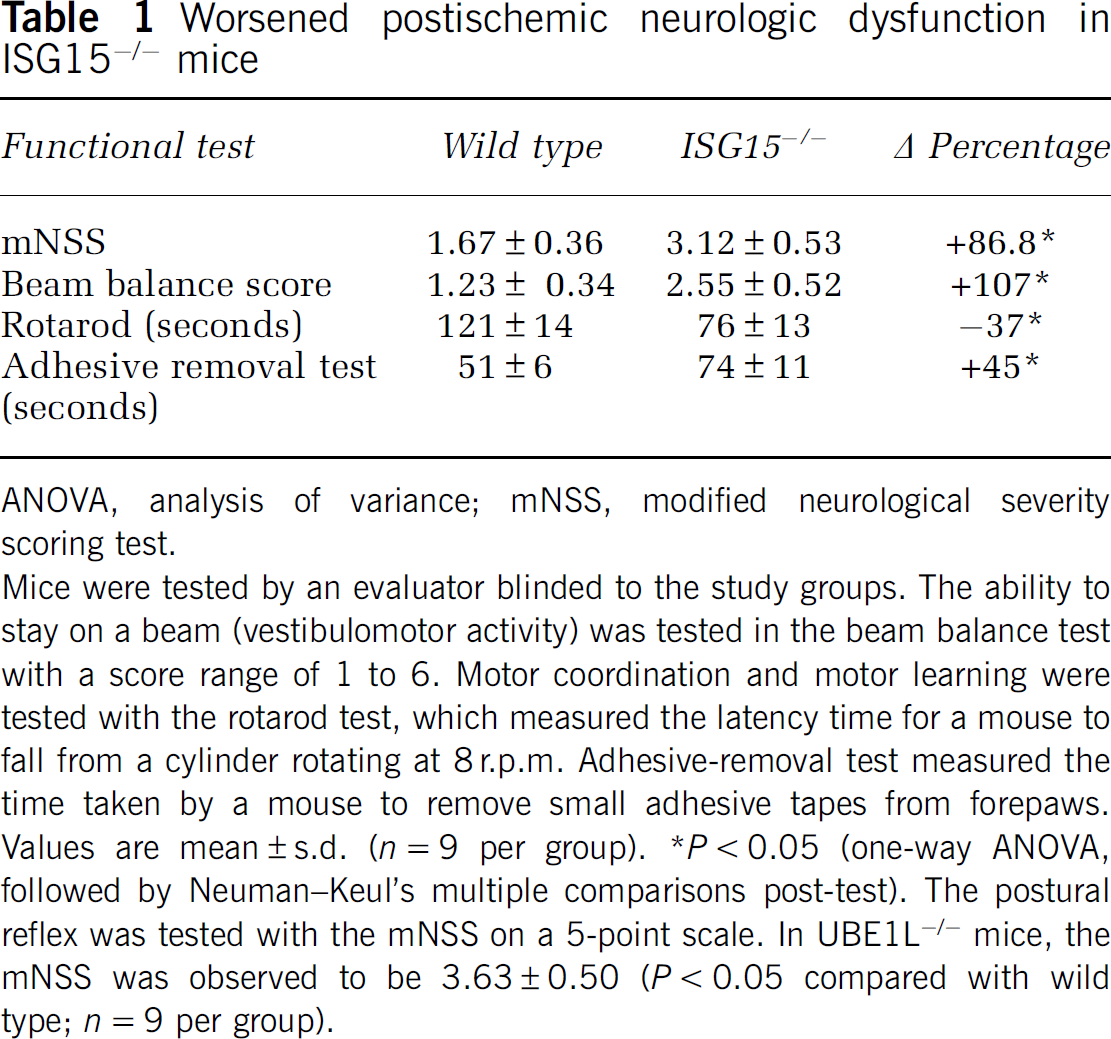
ANOVA, analysis of variance; mNSS, modified neurological severity scoring test.
Mice were tested by an evaluator blinded to the study groups. The ability to stay on a beam (vestibulomotor activity) was tested in the beam balance test with a score range of 1 to 6. Motor coordination and motor learning were tested with the rotarod test, which measured the latency time for a mouse to fall from a cylinder rotating at 8 r.p.m. Adhesive-removal test measured the time taken by a mouse to remove small adhesive tapes from forepaws. Values are mean±s.d. (
Discussion
In brief, results of this study showed that transient focal ischemia induces significant protein ISGylation in the brain; and ISGylated proteins are localized in both neurons and astrocytes. Furthermore, transgenic mice that lack the capability to ISGylate proteins show exacerbated ischemic brain damage indicating that postischemic ISGylation might be an endogenous neuroprotective response to an ischemic insult.
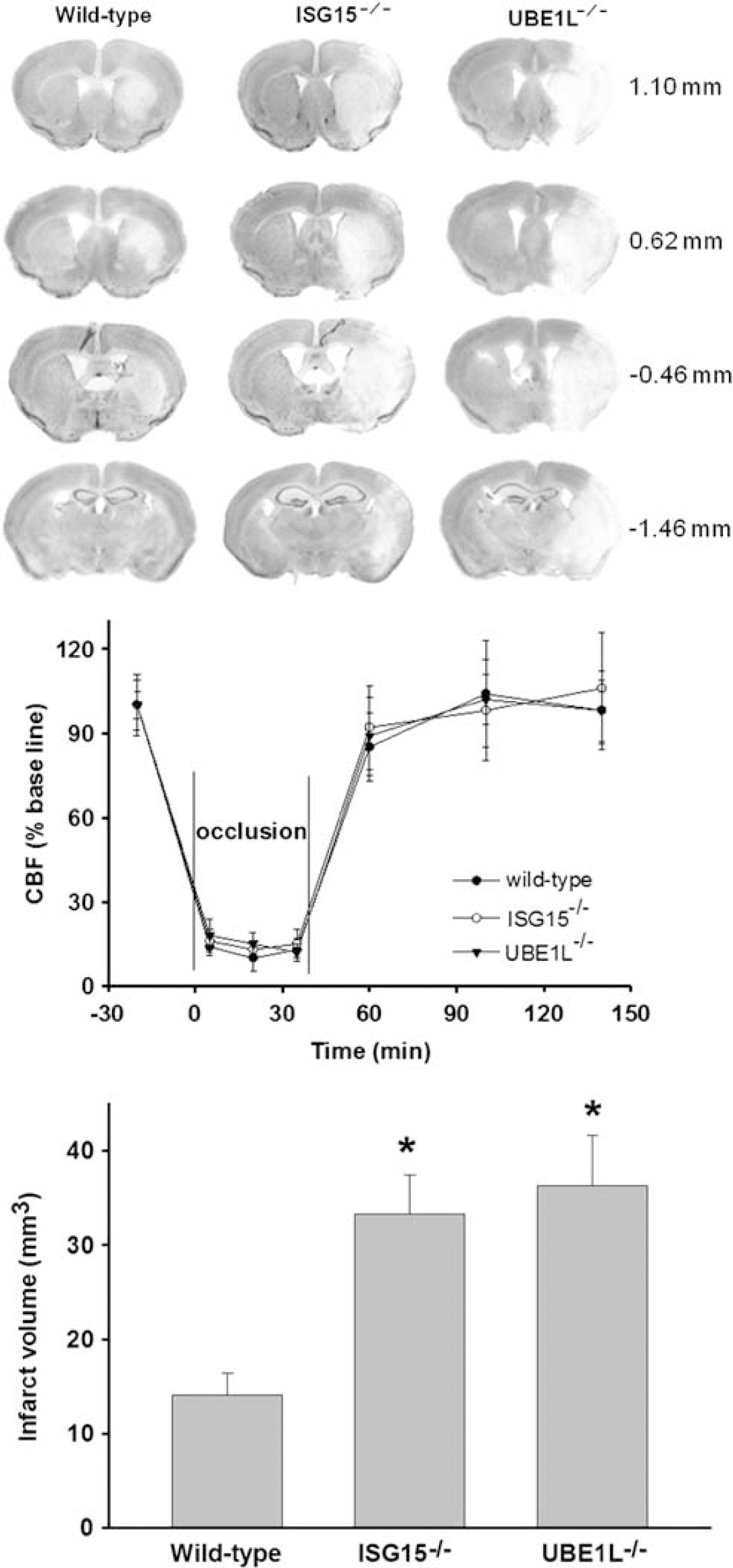
Cresyl violet-stained serial brain sections (35
(
We along with others showed that focal ischemia leads to extensive changes in the cerebral mRNA and microRNA expression in rodents (Dharap et al, 2009; Kapadia et al, 2006; Lu et al, 2003; Soriano et al, 2000; Vemuganti et al, 2002). This altered expression of various families of genes has an important role in promoting postischemic pathologic mechanisms like inflammation, ionic imbalance, edema, and receptor dysfunction that precipitate neuronal death after focal ischemia. Many studies showed that pharmacological manipulations can induce neuroprotection after focal ischemia by affecting the gene expression (Chen et al, 2006; Tureyen et al, 2007; Wang et al, 2004; Xu et al, 2005). However, most if not all proteins undergo PTMs that either activate or inactive them and increase the functional diversity of the proteome (Baumann and Meri, 2004; Sunyer et al, 2008). There are several types of PTMs including (1) addition of a small functional group (e.g., phosphorylation, oxidation, hydroxylation, etc.), (2) chemical modification of an amino acid (e.g., deamidation of glut to gln), (3) change in the structure (e.g., disulfide bonding of two cysteines), and (4) addition of a small protein or peptide (e.g., ubiquitination, SUMOylation, and ISGylation) (Kerscher et al, 2006). Although some of these like phosphorylation are well studied, the functional significance of ub class of PTMs in normal and pathologic brains is not evaluated in detail. Furthermore, very few studies evaluated whether cerebral ischemia induces PTMs and if yes, they have a physiologic significance in deciding the functional outcome after stroke. Previous studies showed that protein ubiquitination and SUMOylation increased after global ischemia, focal ischemia, and hypoxia might induce ischemic tolerance (Cimarosti et al, 2008; Lee et al, 2007; Vannucci et al, 1998; Yamashita et al, 1991; Yang et al, 2008a). Conversely, this is the first report that showed increased protein ISGylation after cerebral ischemia. Our studies showed exacerbated ischemic brain damage in knockout mice that lack either ISG15 or UBE1L (the E1 ligase of ISGylation cycle). When subjected to transient MCAO, the two knockout strains that lack the capability to ISGylate proteins showed bigger infarcts and worsened neurologic recovery as measured by sensorimotor deficits, postural reflex, vestibulomotor function, and motor coordination/motor learning compared with wild-type mice. This indicates that similar to other ub class of PTMs, ISGylation is also a neuroprotective adaptation to minimize postischemic brain damage.
In the postischemic brain, ISGylated proteins were observed to be localized in both astrocytes and neurons. Previous studies showed that SUMOylation (an ub-like PTM similar to ISGylation) also increases in the brain by 30 minutes to 6 hours of reperfusion after transient MCAO in adult rats. We observed that protein ISGylation starts to increase as early as 6 hours of reperfusion and progressively more proteins were ISGylated at 1 and 24 hours reperfusion. Although both SUMOylation and ubiquitination preferentially modify mostly the higher MW proteins, ISGylation seem to be specific towards mid-MW range proteins (Yang et al, 2008a; Vannucci et al, 1998). Thus, the targets of SUMOylation and ISGylation seem to be different and hence the mechanisms of neuroprotection induced by these two PTMs might also be different.
To conclude, results of this study showed that focal ischemia significantly increases protein ISGylation in the brain and prevention of postischemic ISGylation exacerbates brain damage. At present, the significance of proteins being ISGylated in the ischemic brain is not known. Future studies are required to show whether modulation of specific ISGylated proteins can be a therapeutic option to prevent postischemic pathophysiological events and/or to promote plasticity and regeneration.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.