
Abstract
Spreading depression (SD) is a slowly propagating neuronal depolarization that underlies certain neurologic conditions. The wave-like pattern of its propagation suggests that SD arises from an unusual form of neuronal communication. We used enzyme-based glutamate electrodes to show that during SD induced by transiently raising extracellular K+ concentrations ([K+]o) in rat brain slices, there was a rapid increase in the extracellular glutamate concentration that required vesicular exocytosis but unlike fast synaptic transmission, still occurred when voltage-gated sodium and calcium channels (VGSC and VGCC) were blocked. Instead, presynaptic
INTRODUCTION
Spreading depression (SD, also termed spreading depolarization) is a slowly propagating wave of neuronal depolarization that contributes to damage expansion after several neurologic conditions like malignant hemispheric stroke, subarachnoid hemorrhage, intracranial hemorrhage, and brain trauma.1,2 Although SD is mostly observed in damaged brains clinically, it can also be evoked in healthy tissues by various stimuli. A profound depolarization occurs at the SD wave front, which inactivates voltage-gated ion channels and causes SD of brain electrical activity.
3
Recent compelling anatomic evidence has demonstrated that NMDARs are expressed at presynaptic terminals in various regions including the neocortex and hippocampus.9,10 Electrophysiological recordings have shown that presynaptic NMDARs facilitate both spontaneous and evoked neurotransmitter release.9,11 Focal glutamate uncaging directly induced pre-synaptic NMDAR currents in the hippocampal pyramidal cell boutons. 10 Based on these findings, we hypothesized that activation of presynaptic NMDARs could directly trigger more glutamate release and further activate more presynaptic NMDARs thereby contributing to glutamate release during SD. Under some conditions, glutamate may diffuse to activate NMDARs on adjacent presynaptic terminals, evoking further release at these sites and thereby generating a propagating wave of regenerative glutamate release. Such a mechanism could mediate the propagation of SD under certain circumstances like high [K+]o.
In this study, we used enzyme-based glutamate electrodes in combination with electrophysiological recordings and intrinsic optical signals (IOS) to examine the contribution of different mechanisms to the glutamate release during an SD-like propa-gating wave triggered by high [K+]o in brain slices. We report that when SD was induced by high [K+]o solutions, SD propagation was mediated by vesicular glutamate release triggered by pre-synaptic NMDARs independent of VGSCs or VGCCs. Our observations support the notion that a presynaptic NMDAR-dependent mechanism can contribute to and under some circumstances underlie a vicious cycle of glutamate-induced glutamate release to promote the propagation of SD induced by high [K+]o treatments.
MATERIALS AND METHODS
Slice Preparation and Induction of Spreading Depression Experimental protocols were approved by the University of British Columbia Animal Care Committee and conformed to the Canadian Council on Animal Care and Use guidelines, and were evaluated and approved by the Institutional Animal Care and Use Committee of China Medical University according to Care of the Animals and Surgical Procedures of China Medical University Protocols. Rats were anesthetized with halothane, decapitated, and the brains removed into ice-cold slicing solution. Transverse hemi-sections, 400 μm thick, from the hippocampus or cortical hemi-sections from the neocortex/striatum were prepared from 19- to 23-day old Sprague-Dawley rats of either sex. Then the slices were transferred to a storage chamber and incubated with fresh artificial cerebrospinal fluid (ACSF). Spreading depression was induced by perfusing high [K+]o solution (containing 40 mmol/L K+) at 3 mL/minute.
Recording Spreading Depression Propagation
Intrinsic optical signals data were collected from the transmitted light of brain slices by a CCD camera (DAGE-MTI) connected to a DT3155 frame grabber (Data Translation) controlled by Imaging Workbench 5.0 software (INDEC, Santa Clara, CA, USA). The illumination source was a standard Zeiss tungsten bulb whose output was directed through a 750DF20 filter. In all the experiments, the IOS’ were recorded simultaneously with real time glutamate measurements to indicate the onset and propagation of SD (Figure 1). 8
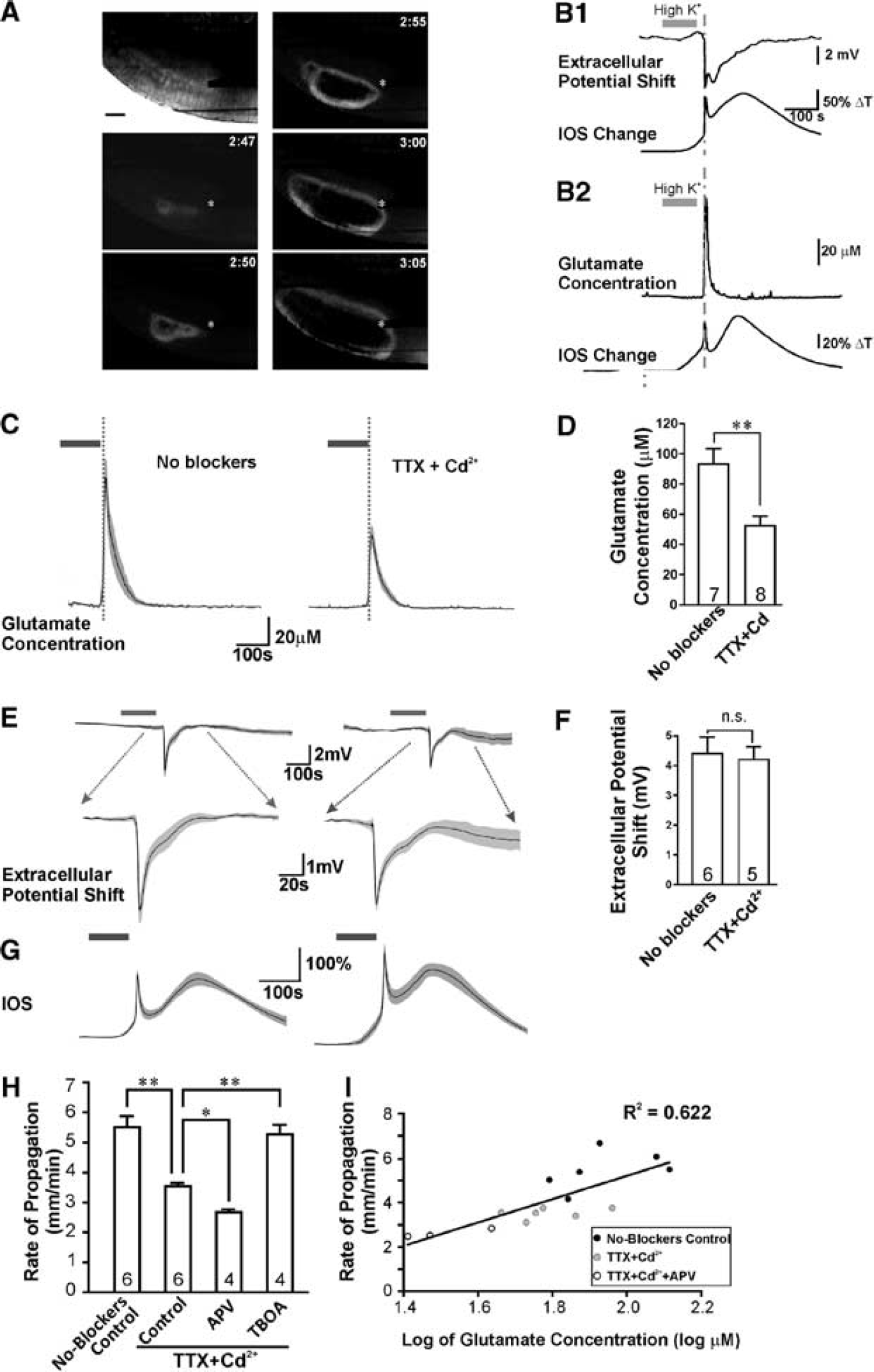
Glutamate transients during spreading depression (SD) were coincident with intrinsic optical signal (IOS) changes and extracellular potential shifts, and occurred independent of action potentials and voltage-gated calcium channels. (
Glutamate Measurement
Real time glutamate concentration changes were detected by enzyme-based microelectrode arrays coated with glutamate oxidase and recorded by a FAST recording system (Quanteon, Nicholasville, KY, USA). The slope (nA/μmol/L) and R 2 of microelectrode arrays were calibrated with glutamate-containing solutions before experiments. To measure the extracellular glutamate changes, the tip of the microelectrode arrays was positioned with the glutamate oxidase coating site gently touching the surface of the brain slice. The change of glutamate concentration was calculated by the peak glutamate concentration during SD minus the baseline glutamate concentration. The baseline concentration was calculated by the averaged glutamate concentration during the 2 minutes before high [K+]o application. Because microelectrode arrays recording could not be combined with other electrophysiology recordings, all the glutamate measurement experiments were performed with simultaneous IOS imaging to indicate the occurrence of SD onset.
Electrophysiology
Field excitatory postsynaptic potentials (fEPSPs) were evoked by stimulation of the Schaffer collateral pathway of the hippocampus using bipolar tungsten stimulating electrodes and recorded from the CA1 stratum radiatum. Spreading depression-related extracellular direct current potentials were recorded from the cortex or the stratum radiatum of the hippocampus. The fEPSPs and direct current potentials were recorded with glass micropipettes filled with ACSF. Whole-cell currents of cortical L2/3 pyramidal neurons were recorded with glass pipettes filled with Cs-based internal solutions. Field excitatory postsynaptic potentials signals were amplified 1,000 times with an alternating current amplifier (A-M systems, Sequim, WA, USA) and acquired via a Digidata 1440A (Molecular Devices, LLC, Sunnyvale, CA, USA). Direct current potentials and whole-cell currents were monitored with MultiClamp 700B amplifier (Molecular Devices) and acquired via a Digidata 1440A. All data were analyzed with Clampfit 10.0 (Molecular Devices).
Data Analysis
In all bar graphs, values are reported as mean ± standard error of mean (s.e.m.). The gray regions of the averaged traces represent error bars as mean ± s.e.m. connected from each time point. R
2
is the coefficient of determination. Statistical analysis was performed using the Prism statistical analysis program (GraphPad, La Jolla, CA, USA). Normality was tested by D'Agostino-Pearson omnibus test. One-way analysis of variance with Newman-Keuls
For more details on methodologies, please refer to the Supplementary Information.
RESULTS
Glutamate is Released During Spreading Depression Without Fast Synaptic Transmission
We first determined whether glutamate release still occurred from brain tissue during SD when VGCCs, VGSCs, and evoked synaptic transmission were blocked. In normal ACSF, perfusing high [K+]o (40 mmol/L) solution for 90 to 120 seconds consistently triggered SD with a propagation velocity of 5.5±0.3 mm/minute in rat neocortical brain slices (Figure 1A). The SD wave was measured as an extracellular direct current potential shift of approximately 4 mV and a correlated propagating IOS transient, as previously reported (Figure 1B1).
8
Using an enzyme-based glutamate electrode positioned within the brain slice, we detected a significant increase in extracellular glutamate during SD, approximately 50 to 100 seconds in duration (peak during SD, 93.2 ± 10.2 μmol/L,
Glutamate is Released by Vesicular Exocytosis but not Transporters or Hemichannels During Spreading Depression
The large component of glutamate release that we still observed after blocking VGCCs and VGSCs with Cd2+ and TTX led us to examine whether the glutamate release observed during high [K+]o-induced SD was due to the exocytosis of glutamate-filled vesicles, or due to other possible non-vesicular mechanisms including glutamate transporters,
14
cystine-glutamate exchangers,
15
and pannexin or connexin hemichannels.
16
We tested the dependence on vesicular exocytosis by examining the effects of bafilomycin A1 and concanamycin A, both of which inhibit vacuolar H+ -ATPase and depress vesicular glutamate release by preventing glutamate transport into synaptic vesicles.
17
To confirm that these inhibitors can block vesicular release of glutamate, we observed that fEPSPs were blocked after slices were treated with either 10 μmol/L bafilomycin alone for >3 hours, or 4 μmol/L bafilomycin or 2 μmol/L concanamycin with brief addition (5 minutes) of 10 μmol/L veratridine to stimulate vesicle turnover.
18
After blockade of fEPSPs with bafilomycin A1 (Figure 2A) or concanamycin A, we applied TTX and Cd2+ and evoked SD with perfusion of high [K+]o solution. Glutamate release during SD was greatly inhibited by 10 μmol/L bafilomycin (5.2 ±0.6 μmol/L,
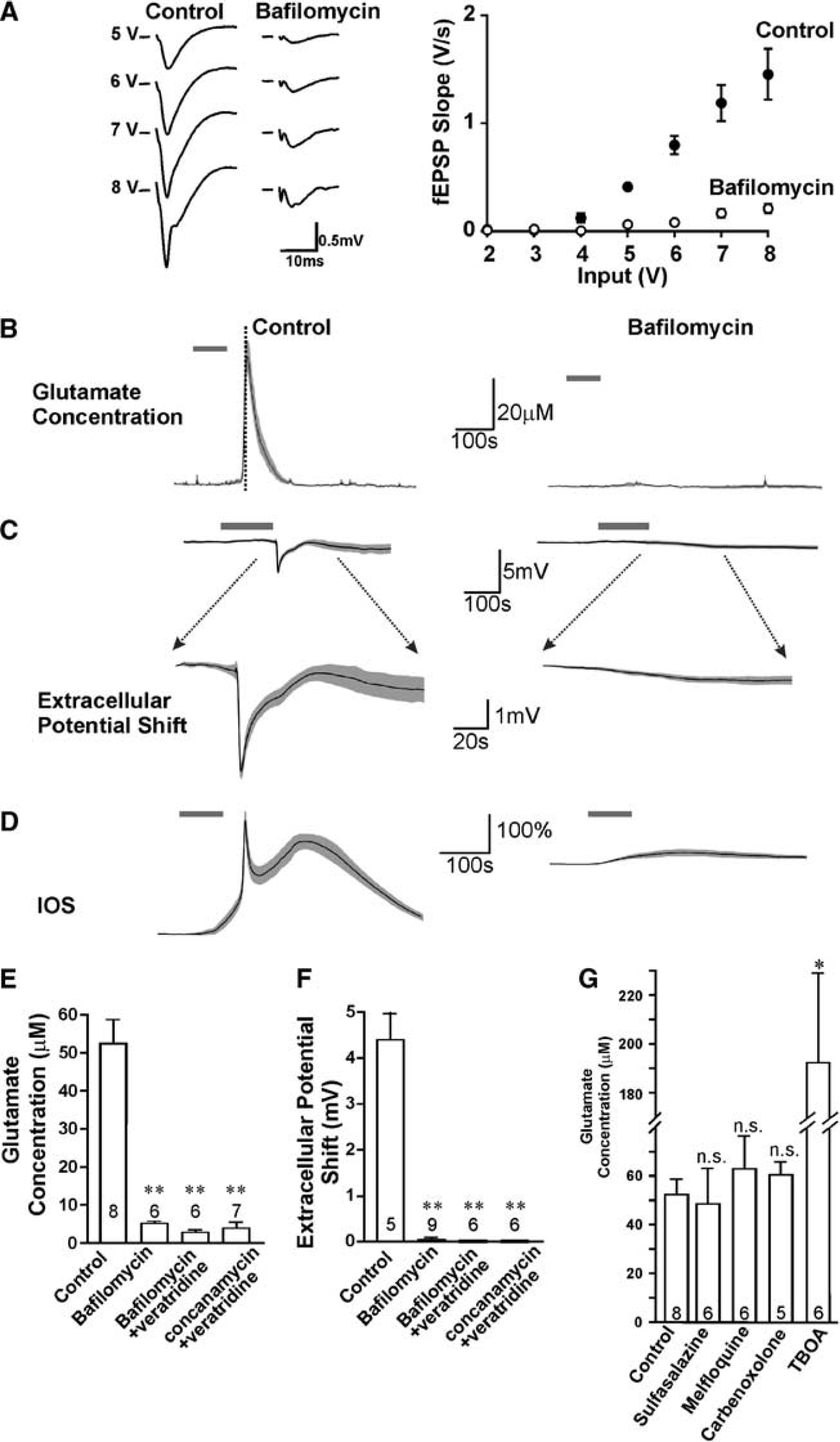
Both the glutamate release and waveforms of spreading depression (SD) required TTX- and Cd2+ -insensitive vesicular release. (
Glutamate Release During Spreading Depression is Dependent Upon NMDARs
Exocytosis of synaptic vesicles at presynaptic sites during synaptic transmission requires elevation of cytosolic Ca2+ that results from Ca2+ entry via VGCCs.
23
Considering that our results thus far have demonstrated substantial glutamate exocytosis during high [K+]o-induced SD when synaptic fEPSPs and VGCCs were blocked by Cd2+, we tested for alternative sources of Ca2+ entry. NMDARs are permeable to Ca2+ and their activation is critical for SD propagation. Recently, it has been reported in several studies that NMDARs are localized to presynaptic terminals and their activation can directly and exclusively promote excitatory neurotransmitter release in the neocortex and hippocampus.10,11 As such, we hypothesized that when VGCCs, VGSCs, and fEPSPs are blocked, activation of presynaptic NMDARs could still elicit glutamate release during high [K+]o-induced SD. Accordingly, blockade of NMDARs should not only inhibit SD-induced extracellular potential shifts, which have a substantial component mediated by postsynaptic NMDA glutamate receptors, but also suppress presynaptic vesicular glutamate release. Alternatively, if synaptic release is not dependent upon presynaptic NMDARs, then NMDAR antagonists should only affect postsynaptic depolarization but not the presynaptic glutamate release. We tested this hypothesis by applying NMDAR antagonists D-(-)-2-amino-5-phosphonopentanoic acid (D-APV) (50 μmol/L), 7-chlorokynurenic acid (7-CKA, 100 μmol/L) or MK-801 (10 μmol/L). D-APV and 7-CKA had minimal effect on evoked fEPSPs in the hippocampal slices (Figure 3A), indicating preservation of normal synaptic transmission; however, both glutamate release (1.6±0.4 μmol/L,
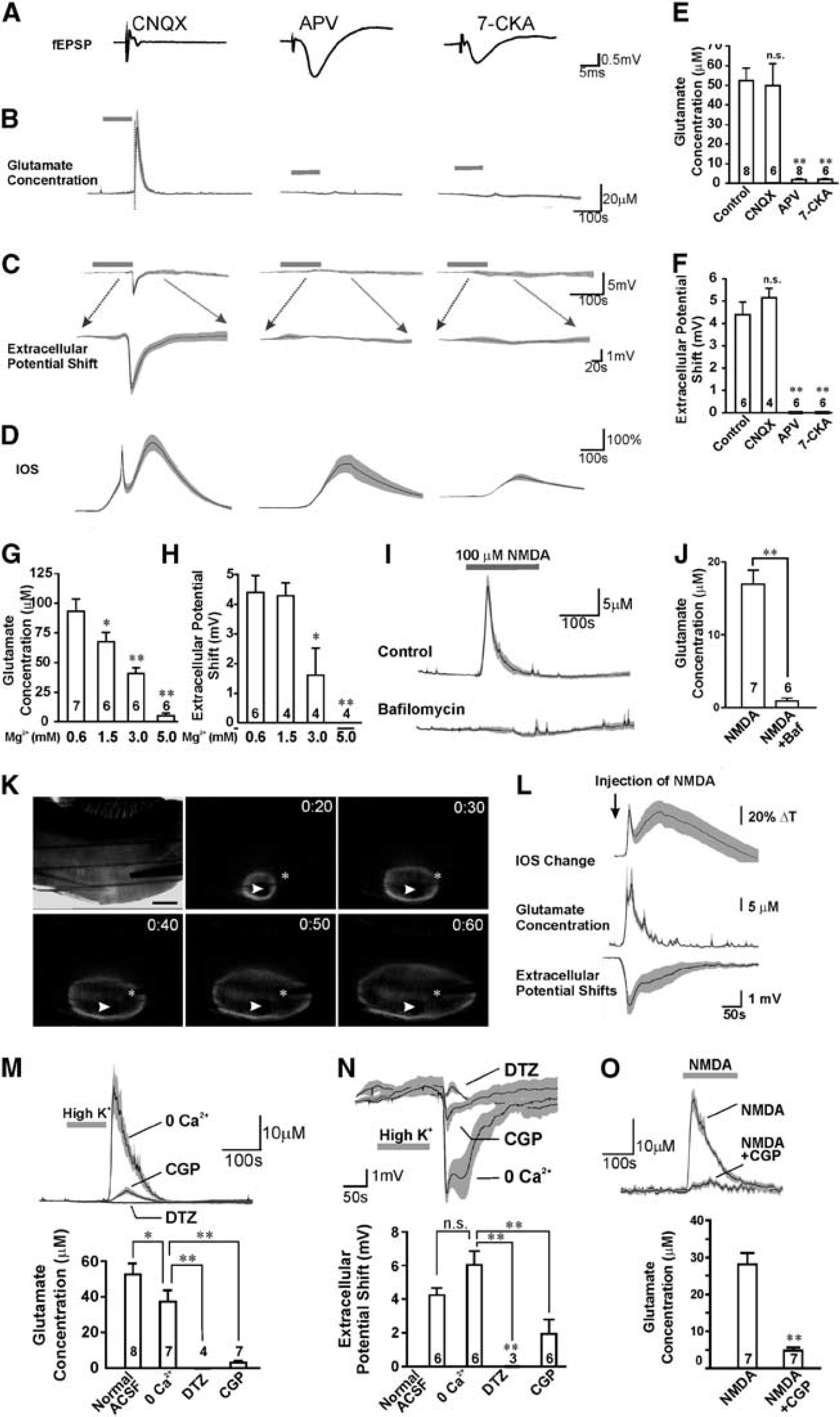
For legend see page 1588. Activation of
To further confirm the NMDAR-dependence of glutamate release, we evoked high [K+]o-induced SD in solutions containing different concentrations of Mg2+, the voltage-dependent blocker of NMDARs.
24
Mg2+ will also differentiate between neuronal NMDARs that are sensitive to extracellular Mg2+ and astrocytic NMDARs that are Mg2+-insensitive at concentrations up to 10 mmol/L.
25
Both SD-induced potential shifts and glutamate release were inhibited by Mg2+ in a dose-dependent manner with a complete block observed at 5 mmol/L (
Taken together, these data indicate that the level of glutamate release during high [K+]o-induced SD is correlated with the extent of NMDAR opening. Moreover, NMDAR-dependent glutamate release from neurons but not astrocytes is critical for SD propagation.
Activation of NMDARs Triggers Vesicular Glutamate Release After Action Potentials and Voltage-Gated Calcium Channels are Blocked
If exocytosis of glutamate-containing vesicles during high [K+]o-induced SD is directly caused by presynaptic NMDAR opening, we predicted that NMDAR agonists should trigger glutamate release even when synaptic transmission involving action potentials and VGCC-mediated calcium entry is blocked. Consistent with this idea, bath application of 100 μmol/L NMDA in the presence of TTX and Cd2+ induced significant glutamate release from brain slices (16.9±1.9 μmol/L,
NMDA-Dependent Glutamate Release Involves Both Ca2+ Influx and Activation of the Mitochondrial Na+/Ca2+ Exchanger (NCXmito)
NMDAR activation may induce glutamate release either by directly producing a calcium influx across cell membranes, or by activating secondary calcium-permeable channels or transporters close to the vesicle release sites. To test the contribution of calcium influx, we examined whether we could induce SD by applying high [K+]o in solutions containing 0 mmol/L extracellular Ca2+ with 2 mmol/L EGTA (0 [Ca2+]o solution) in the presence of TTX and Cd2+. Surprisingly, we still observed propagating SD that occurred concurrently with a transient, albeit decreased, glutamate release (37.3±6.7 μmol/L,
Mitochondria are typically found in presynaptic terminals adjacent to synaptic vesicles at active zones
27
and previous studies have shown that substantial neurotransmitter release occurs when increased cytosolic Na+ triggers NCXmito dependent Ca2+ release from mitochondria in presynaptic terminals.
28
To investigate the involvement of NCXmito, we tested the effect of CGP-37157 (CGP, 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one), which is a specific NCXmito blocker,
29
on SD-induced glutamate release under the conditions of 0 [Ca2+]o solutions containing TTX and Cd2+. CGP-37157 (20 μmol/L), inhibited both SD-induced glutamate transients (3.1±1.1 μmol/L,
Presynaptic NMDARs are Activated During Spreading Depression
From the results above, we inferred that a significant component of glutamate release during high [K+]o-induced SD is caused by activation of presynaptic NMDARs. Several studies have employed electrophysiological recordings of minitature excitatory postsynaptic currents (mEPSCs) to demonstrate the facilitatory effect of presynaptic NMDARs on spontaneous neurotransmitter release.
11
In these studies, using cortical neurons where postsynaptic NMDARs were blocked, the NMDAR antagonist D-APV decreased the frequency but not the amplitude of mEPSCs. We investigated whether presynaptic NMDARs were indeed activated during SD by applying MK-801 during SD induction. MK-801 is an irreversible, open channel blocker of NMDARs, and will only block channels that are activated during SD. We predicted that if presynaptic NMDARs were stimulated during SD then MK-801 would occlude the effects of a subsequent D-APV application on decreasing the frequency of mEPSCs. In our experiments, postsynaptic NMDARs were first blocked by inclusion of MK-801 (1 mmol/L) in the intracellular recording solution (Figure 4A2), and the effects of presynaptic NMDAR block on mEPSCs were tested by subsequently applying D-APV (Figure 4A3). In control slices with no MK-801 added during SD, mEPSCs were recorded in cortical layer 2/3 pyramidal neurons 30 minutes after SD was induced (Figure 4A1) to ensure that SD itself did not obscure presynaptic actions of NMDAR activation. Consistent with previous studies, D-APV (100 μmol/L) significantly reduced the frequency of mEPSCs (62.1±5.5% of baseline,
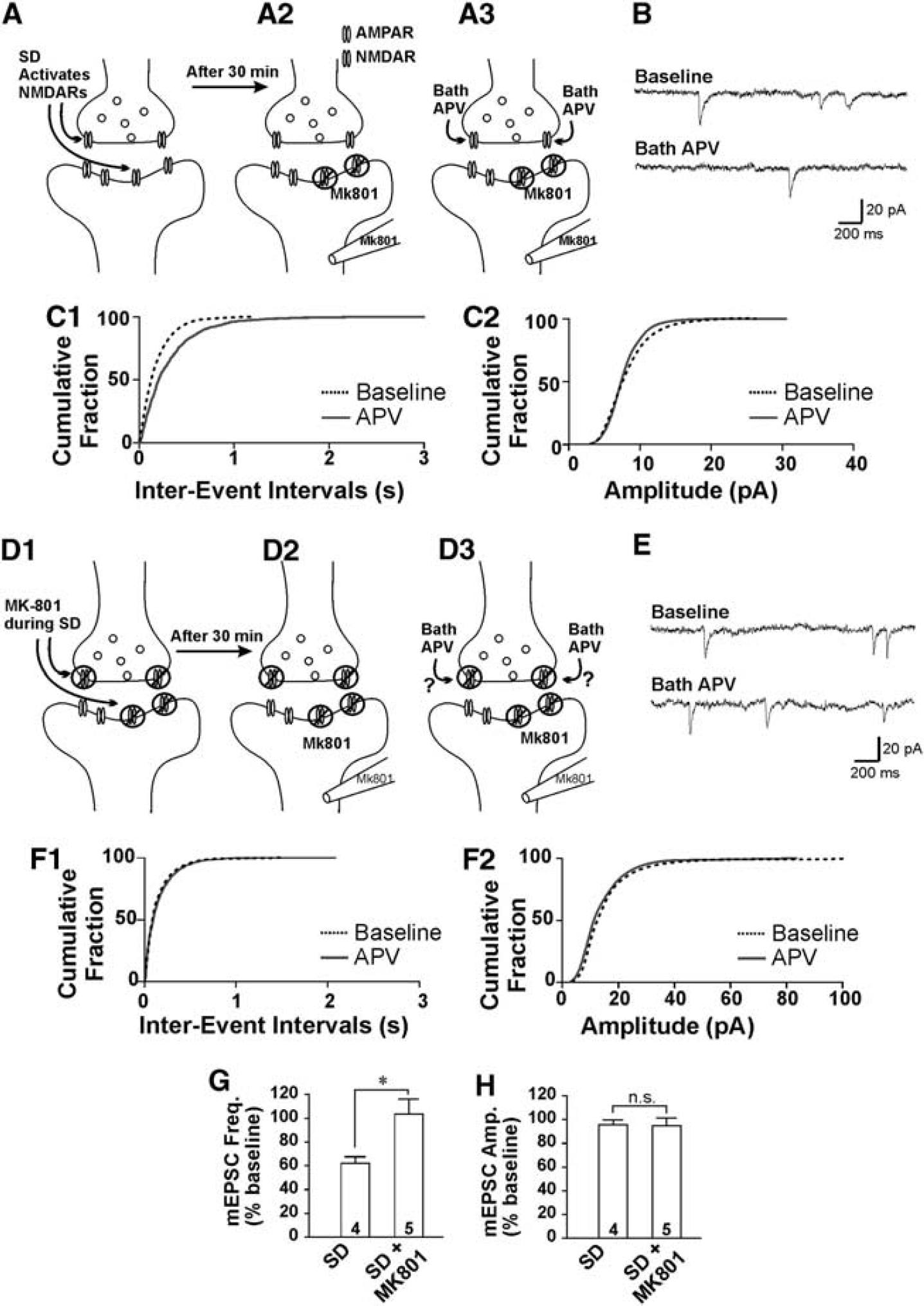
Activation of presynaptic
Glutamate Release During Spreading Depression Requires Presynaptic NMDARs
The requirement of presynaptic versus postsynaptic NMDAR activation for eliciting glutamate release during SD was further investigated using a strategy to block either post-or presynaptic NMDARs pharmacologically before evoking SD. We designed two strategies to selectively depolarize either presynaptic terminals or postsynaptic dendrites with co-application of MK-801 to selectively and irreversibly block NMDARs at either site. The first strategy to selectively stimulate and block postsynaptic NMDARs was as follows: presynaptic release was inhibited by baclofen (to activate GABAB receptors) in addition to blocking action potentials with TTX, and VGCCs with Cd2+. In addition, NMDAR channel activity at resting membrane potentials was decreased with higher extracellular Mg2+ (1 mol/L). Therefore, we used 1 μmol/L TTX, 30 μmol/L Cd2+, and 10 μmol/L baclofen to inhibit presynaptic depolarization, together with 1 μmol/L AMPA to depolarize postsynaptic dendrites and 10 μmol/L MK-801 to irreversibly block postsynaptic NMDARs when they are activated by spontaneous glutamate release (Figure 5A, Supplementary Figures 3A, B). We also confirmed that the combined treatment did not affect presynaptic NMDARs as shown by blocking NMDARs with D-APV on mEPSCs recordings with intracellular MK-801 (Supplementary Figure 4). As the combination of TTX, Cd2+, baclofen, AMPA, and MK-801 only blocked postsynaptic NMDARs and did not affect presynaptic NMDARs, we used this treatment to examine whether blockade of postsynaptic NMDARs would impair SD. Blocking postsynaptic NMDARs significantly reduced the extracellular potential shifts (2.40±0.30 mV,
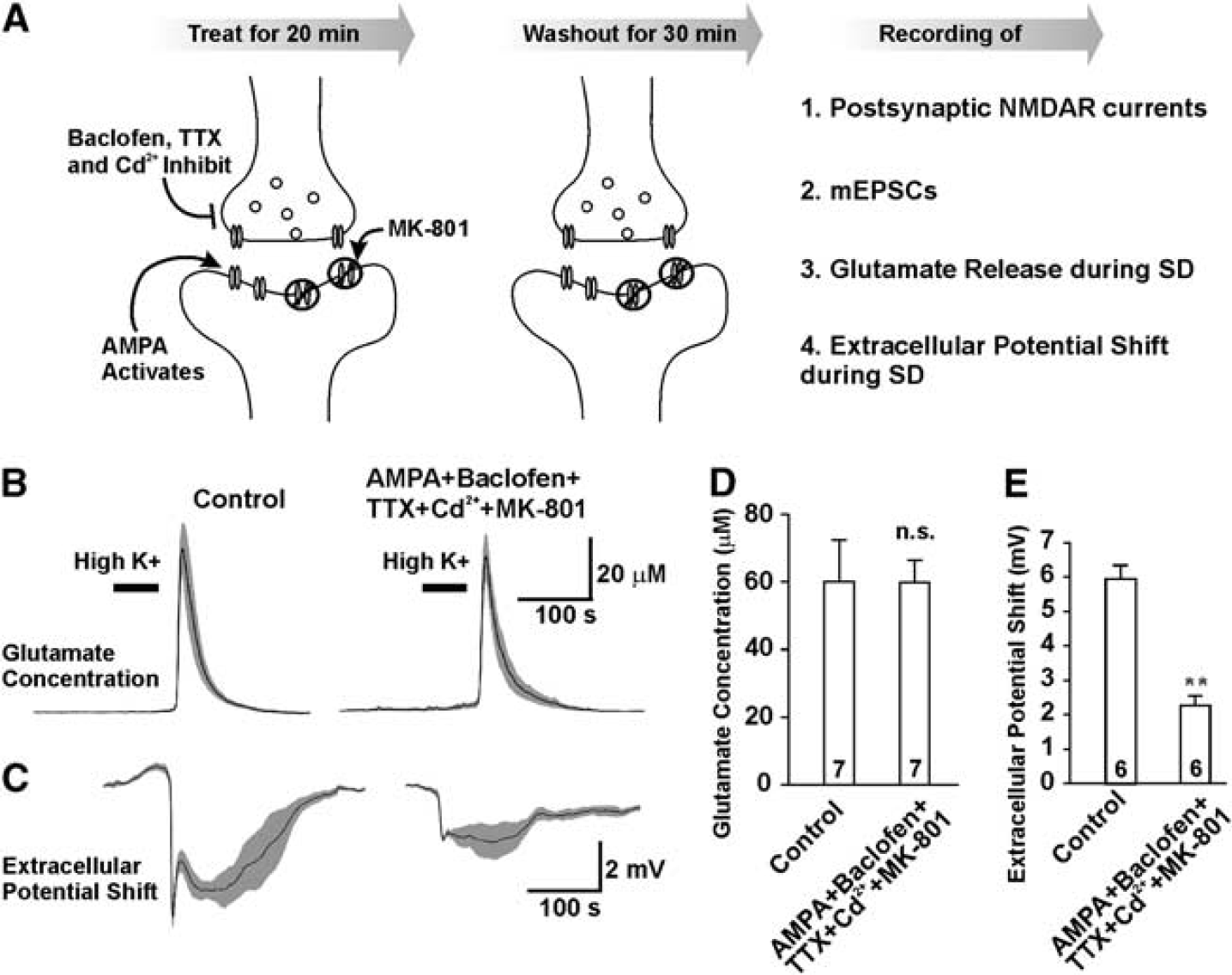
Postsynaptic
The second strategy was aimed to block presynaptic NMDARs by stimulating presynaptic terminals in the presence of MK-801. We used CNQX to inhibit postsynaptic activities, together with nicotine to depolarize presynaptic compartments and MK-801 to irreversibly block presynaptic NMDARs activated by spontaneous glutamate release stimulated by the presynaptic actions of nicotine (Figure 6A). These manipulations would reduce postsynaptic depolarization, thereby decreasing the ability of MK-801 to block postsynaptic NMDARs and increase its ability to block presynaptic NMDARs. The slices were treated with the combination of 20 μmol/L CNQX, 1 μmol/L nicotine, and 10 μmol/L MK-801 for 20 minutes followed by a 30-minute wash. Our results showed that the postsynaptic NMDAR currents were not significantly affected (Supplementary Figures 3C, D). Instead, the presynaptic NMDAR components were inhibited by the combined treatment (Supplementary Figure 4). After specifically blocking pre- but not postsynaptic NMDARs, glutamate release was significantly reduced (19.29±2.85 μmol/L,
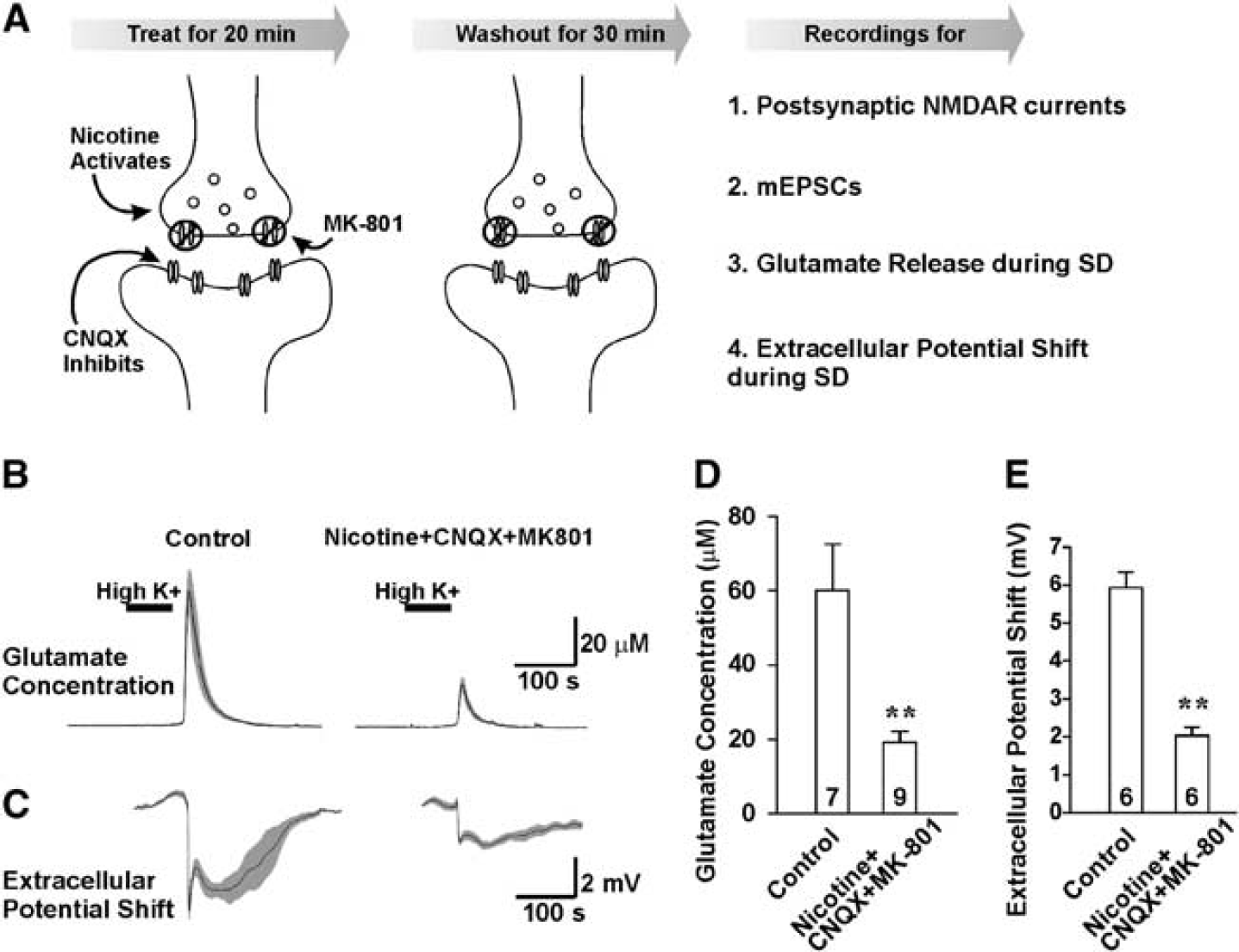
Blocking presynaptic
NMDA Triggers Propagating Spreading Depression and Glutamate Release in the Striatum That Lacks Glutamatergic Neurons
Our results in the cortex suggest that presynaptic NMDARs are required for the regenerative glutamate release in high [K+]o-induced SD that occurs when action potentials are blocked by TTX and VGCCs are blocked by Cd2+. It is still possible that the substantial postsynaptic dendritic depolarization of glutamatergic cortical neurons could passively depolarize presynaptic terminals of recurrent collaterals and thereby contribute to glutamate release. We performed experiments in another brain region to test whether NMDAR actions on presynaptic terminals were sufficient to trigger SD-induced glutamate release where there was no possible contribution from neuronal depolarization and subsequent passive depolarization of axons and presynaptic terminals to activate calcium influx at distal sites. We investigated glutamate release during SD in the isolated striatal brain slices that lack glutamatergic cell bodies but only contain glutamatergic presynaptic terminals from neocortical projections to the principal GABAergic striatal neurons.
30
NMDAR-dependent glutamate release during SD propagation in this tissue would only be from glutamate diffusion between adjacent terminals and could not be due to passive propagation of postsynaptic depolarizations to presynaptic terminals. We repeated our SD experiments with focal NMDA application in isolated striatal slices where surrounding regions such as neocortex and the septal area were carefully removed (Figure 7A). We found that in the presence of 1 μmol/L TTX and 30 μmol/L Cd2+, focal ejection of 500 μmol/L NMDA from a glass pipette evoked a SD-like waveform that propagated at a rate of 1.9±0.2 mm/minute from the initiation site (indicated by arrow head,
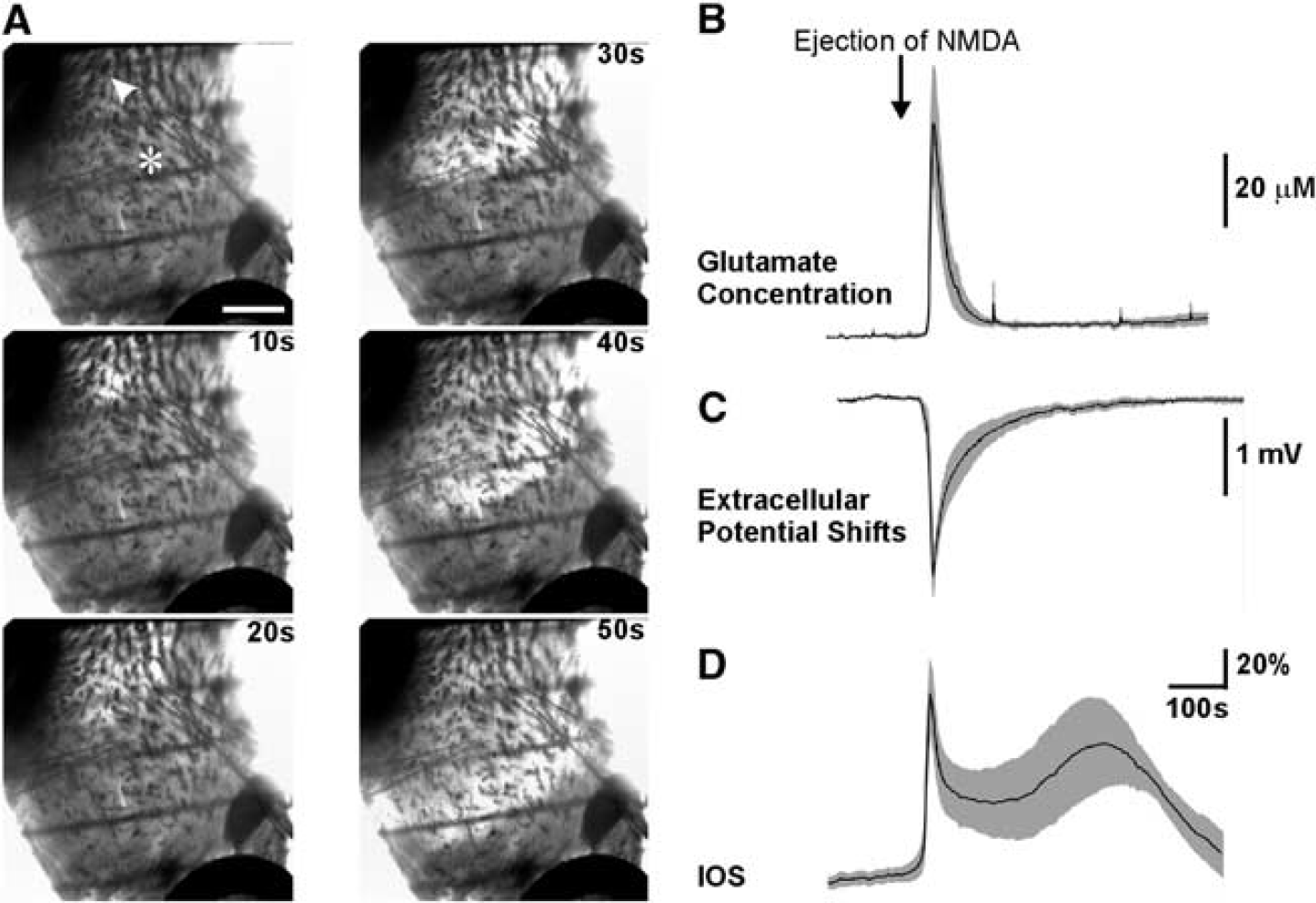
DISCUSSION
In this study, we show that an unusual form of regenerative glutamate release that occurs during high [K+]o-induced SD is sufficient for SD propagation. We conclude that presynaptic NMDARs are required for this regenerative glutamate release because: (1) glutamate release during SD was blocked by NMDAR antagonists but not by inhibiting pannexins, connexins, AMPA/kainate receptors, glutamate transporters, cystine-glutamate exchangers, VGSCs or VGCCs; (2) NMDAR-dependent glutamate release during SD requires vesicular exocytosis, as it is sensitive to bafilomycin and concanamycin; (3) when VGSCs and VGCCs were blocked, directly applying NMDA itself still induced bafilomycin-sensitive glutamate release that could trigger SD wave propagation in neocortical and striatal slices. In addition, our strategy to selectively inhibit presynaptic NMDARs dramatically reduced glutamate release during high [K+]o SD whereas selectively blocking postsynaptic glutamate receptors had no effect on glutamate release. Our data indicate that regenerative glutamate release during high [K+]o-induced SD can occur independently of canonical fast transmission across synapses that requires action potential invasion in presynaptic terminals and calcium influx through VGCCs. In contrast, our evidence indicates that a substantial fraction of glutamate release during SD triggered by high [K+]o requires opening of presynaptic NMDARs that induces vesicular exocytosis of glutamate. We propose that during high [K+]o-induced SD glutamate can diffuse to adjacent synapses to activate more NMDARs and trigger a self-propagating regenerative process that underlies the propagation of this form of SD.
Previous studies have shown that both SD initiation and propagation are inhibited by NMDAR antagonists. Although NMDARs are more abundantly distributed at postsynaptic sites, the ability of NMDA to evoke glutamate release in the presence of VGCC and VGSC antagonists, which blocked evoked fEPSPs, argues against postsynaptic depolarization triggering action potential propagation from soma to axon terminals. Our observations that SD propagates in the isolated striatum, where there are no glutamatergic cell bodies only presynaptic terminals, indicate that glutamate release is not due to passive propagation of postsynaptic depolarizations in glutamatergic neurons causing release from presynaptic terminals. Although dendritic release of glutamate can occur from olfactory mitral cells, 31 it is unlikely that our observations are due to such a mechanism, because dendritic glutamate release requires specialized dendrodendritic interactions and dendritic glutamate-containing vesicles but neither of these morphologies has been observed in the cortex or hippocampus. 32 The simplest explanation, in light of the high levels of functional expression of NMDARs in axons and presynaptic terminals in the cortex and hippocampus, is that NMDA acting on receptors on presynaptic axons and terminals is sufficient for the release of glutamate-containing vesicles. The enhanced release of vesicles from terminals could also explain the observed release of other SD-related neurochemicals like zinc that are found in vesicles. 5 Our results also suggest that both Ca2+ and Na+ influx via NMDARs is important for this process as Ca2+ release from mitochondria can have a role through the action of NCXmito. Mitochondria are typically found at metabolically active presynaptic terminals27,33 and are increasingly recognized as important for buffering and releasing calcium in neurons and at synapses.34,35 At present, because of the dramatic swelling and subsequent movement of all presynaptic and dendritic cellular components that occurs rapidly at the onset of SD as previously reported, 8 we are unable to track mitochondrial Ca2+ signals in presynaptic terminals during SD. Nevertheless, the role for Ca2+ release via NCXmito is indicated by our data that both SD- and NMDA-induced glutamate releases were inhibited by the selective NCXmito blocker CGP-37157 and by diltiazem when extracellular Ca2+ was removed. The presynaptic role of NMDARs is further demonstrated by our findings that presynaptic NMDARs are indeed activated during high [K+]o SD, and that blocking presynaptic NMDARs reduces glutamate release. Activation of presynaptic NMDARs was tested by the irreversible, use-dependent NMDAR blocker MK-801. When MK-801 was applied during SD to block NMDAR activated during SD, we no longer observed a decrease in mEPSC frequency by the NMDAR antagonist D-APV indicating that the facilitative effects of presynaptic NMDARs on neurotransmitter release were occluded. Using MK-801 to block NMDARs at specific synaptic locations also suggests that only presynaptic receptors are required for glutamate release, and that postsynaptic NMDARs partially contribute to the slow potential shifts. These results strongly suggest that presynaptic NMDAR-mediated glutamate release is a critical step in the positive feedback cycle required for SD propagation triggered by high [K+]o.
Our observations that SD propagation rates are linearly correlated with the logarithm of glutamate concentrations indicate that SD depends on diffusion of glutamate between release sites rather than action potential propagation between synaptic connections. Mechanisms that increase glutamate release at synapses will accelerate propagation speed by both increasing concentration gradients and by activating more presynaptic NMDARs thereby facilitating the regenerative glutamate release. In line with this speculation, our prior work has demonstrated that glutamate release through volume-activated channels in astrocytes contributes to the propagation of SD, suggesting that such a mechanism might be important in facilitating glutamate diffusion in at least some forms of SD. 36 The present study shows that other known mechanisms of astrocytic and neuronal glutamate release are not likely involved, including hemichannels, P2X7 receptor channels, reversed operation of glutamate transporters, or cystine-glutamate antiporters. 37 Our data that Mg2+ dose-dependently inhibited glutamate release also suggest that NMDAR-dependent glutamate release is from neurons rather than astrocytes.
Prolonged increases of extracellular K+ levels have been observed after several pathologic conditions, like subarachnoid hemorrhage, intracerebral hemorrhage, and traumatic brain injury,38-40 and may contribute to SD incidence under these conditions. Raised [K+]o will inhibit or reverse glutamate transporters and reduce the efficiency of glutamate uptake, which might facilitate spillover of glutamate from the synaptic release sites and increase the diffusion. 41 The shrinkage of interstitial space caused by raising [K+]o will also increase the concentrations of extracellular glutamate. Elevated [K+]o can also depolarize presynaptic terminals and remove the Mg2+ blockade of presynaptic NMDA receptors. These multiple factors could all contribute to maintain the positive feedback cycle of presynaptic NMDAR activation during SD. This is in line with a previous study where the threshold of SD triggered by high [K+]o was increased by NMDAR antagonists. 42
Brief focal application of a small volume of high [K+]o to cortical surfaces
The incidence of SD in the human cortex is highly correlated with exacerbation of brain damage after malignant hemispheric stroke, delayed ischemia induced by subarachnoid hemorrhage, intracranial hemorrhage, and traumatic brain injury.1,2 Blocking SD by NMDAR antagonists was observed to be neuroprotective after brain injury in clinical studies. 45 Our study reveals that a novel form of presynaptic NMDAR-dependent regenerative glutamate release that is independent of VGSC and VGCC activation contributes to and in some circumstances is sufficient for SD propagation. Such a mechanism may explain how SD propagates in the ‘isoelectric’ brain when typical synaptic transmission through action potential propagation is already suppressed. 46 It is possible that selectively targeting antagonists against presynaptic NMDARs could reduce SD-related neurologic conditions.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.