
Abstract
Sublethal injurious stimuli induce tolerance to subsequent lethal insults, a phenomenon termed preconditioning. Inducible nitric oxide synthase (iNOS) is essential for the preconditioning induced by transient bilateral common carotid artery occlusion (BCCAO) or by systemic administration of the endotoxin lipopolysaccharide (LPS). We used a model of brain injury produced by neocortical injection of N-methyl-d-aspartate (NMDA) to investigate the mechanisms by which iNOS-derived nitric oxide (NO) contributes to tolerance induced by LPS or BCCAO. We found that the tolerance is blocked by the iNOS inhibitor aminoguanidine, is not observed in iNOS-null mice, and is rescued by the NO donor DTPA NONOate. Lipopolysaccharide failed to induce preconditioning in mice lacking the nox2 subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, suggesting that superoxide derived from NADPH oxidase is needed for the induction of the tolerance. Because superoxide reacts with NO to form peroxynitrite, we investigated the role of peroxynitrite. We found that LPS induces the peroxynitrite marker 3-nitrotyrosine in cortical neurons and that the peroxynitrite decomposition catalyst FeTPPS abolishes LPS-induced preconditioning. These results suggest that the protective effect of iNOS-derived NO is mediated by peroxynitrite formed by the reaction of NO with NADPH oxidase-derived superoxide. Thus, peroxynitrite, in addition to its well-established deleterious role in ischemic brain injury and neurodegeneration, can also be beneficial by inducing tolerance to excitotoxicity.
Introduction
The terms ischemic preconditioning and ischemic tolerance define a phenomenon in which a sublethal ischemic insult confers protection from a subsequent lethal insult (Gidday, 2006). In brain, as in other organs, ischemic tolerance occurs in two distinct time frames after the preconditioning stimulus: early and delayed. Early ischemic tolerance occurs within minutes of the preconditioning stimulus, and confers short-lasting protection that does not require new protein synthesis. Delayed ischemic tolerance occurs after a latency of many hours to days, requires de novo protein synthesis, and exhibits a longer-lasting, albeit transient, protection (Gidday, 2006; Kirino, 2002; Nandagopal et al, 2001). Although early preconditioning has been described in brain (Atochin et al, 2003), delayed preconditioning has been investigated more extensively, because of the longer time window and therapeutic applicability (Kirino, 2002). We recently found that the immunologic or ‘inducible’ isoform of NOS (iNOS) is essential for the delayed tolerance to focal ischemia produced by transient bilateral occlusion of the common carotid arteries (BCCAO) or by the proinflammatory mediator lipopolysaccharide (LPS) (Cho et al, 2005). However, the downstream mediators through which iNOS-derived nitric oxide (NO) exerts its preconditioning effect have not been defined.
Nitric oxide exerts its biologic actions by reacting with transitional metal centers, oxygen or superoxide (Gross and Wolin, 1995; Ischiropoulos and Beckman, 2003). Thus, NO reacts with heme groups of enzymes, such as guanylyl cyclase, modulating their activity (Gross and Wolin, 1995), and with thiol groups producing post-translational modifications of proteins (nitrosylation) (Stamler et al, 1992). However, NO has the greatest affinity for the free radical superoxide, with which it reacts to form peroxynitrite, a highly reactive molecule that mediates many of the biologic effects of NO (Beckman et al, 1990; Ischiropoulos and Beckman, 2003; Klotz et al, 2002). Although superoxide is required for expression of the tolerance induced by hypoxia in neuronal cultures (Liu et al, 2005), the role of peroxynitrite and its relationships to iNOS-dependent preconditioning has not been investigated. Therefore, in this study, we used LPS and BCCAO as preconditioning stimuli in a model of excitotoxic brain injury to investigate the downstream mediators through which iNOS contributes to the preconditioning. The evidence indicates that the tolerance induced by LPS requires peroxynitrite formed from iNOS-derived NO and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-derived superoxide.
Materials and methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Experiments were performed in male mice lacking iNOS (MacMicking et al, 1995), cyclooxygenase-2 (COX-2) (Morham et al, 1995), or nox2, the catalytic subunit of NADPH oxidase (Pollock et al, 1995). Mice were obtained from in-house colonies (Iadecola et al, 2001, 1997; Park et al, 2004) and were studied at age 2–3 months. Inducible nitric oxide synthase (iNOS), COX-2, and nox2-null mice were congenic with the C57BL/6 strain and C57BL/6 mice were used as wild-type controls.
Ischemic Preconditioning and Lipopolysaccharide
Procedures for induction of tolerance either by LPS or bilateral common carotid artery occlusion (BCCAO) were identical to those previously described (Cho et al, 2005) and are summarized here. For BCCAO mice were anesthetized and the parietal bone was exposed. A flexible fiber-optic probe was glued to the skull (−2 mm AP, 5 mm ML to bregma) for continuous cerebral blood flow (CBF) monitoring using laser-Doppler flowmetry (Periflux System 5000, Perimed, Sweden). Through a midline neck-incision, 4-0 surgical sutures were loosely placed around both common carotid arteries. Preconditioning was induced by three episodes of 1-min occlusion of both common carotid arteries, each followed by 5 mins of reperfusion. After surgery, mice were returned to their cages. Sham-operated animals in which the carotids were exposed but not occluded served as controls (Cho et al, 2005). For LPS preconditioning, Salmonella typhimurium LPS (0.5 mg/kg, i.p.; Sigma-Aldrich, St Louis, MO, USA; Lot-No. 054K4010) was administered i.p. and mice were returned to their cages. Mice treated with vehicle (saline) served as controls. Mice were subjected to N-methyl-d-aspartate (NMDA) microinjection in the neocortex at different time points after BCCAO or LPS administration.
N-methyl-d-aspartate Microinjection in Neocortex and Lesion Volume Measurement
Procedures for microinjection of NMDA into the cortex were identical to those described previously (Iadecola et al, 2001). In brief, in isoflurane-anesthetized mice the dura overlying the parietal cortex was exposed and NMDA (20 nmol in 140nl of sterile 0.1 M phosphate-buffered saline, pH 7.4) or vehicle was loaded in a glass micropipette (tip 40 to 50 μm) connected to a microinjection device. The micropipette was inserted into the parietal cortex at a site 1.5 mm caudal to bregma, 4.0 mm from the midline, and 0.8 mm below the dural surface, and NMDA was injected. Twenty-four hours after NMDA injection, mice were killed and their brains were removed and frozen in isopentane (−30°C). Coronal forebrain sections (thickness 30 μm) were serially cut in a cryostat, collected at 180 μm intervals, and stained with cresyl violet for determination of lesion volume by an image analyzer (MCID; Imaging Research, St Catharines, Ontario, Canada).
Drug Treatment Protocols
The iNOS inhibitor aminoguanidine (AG; 100 mg/kg, i.p.; Sigma-Aldrich) or vehicle (saline) was administered 10 mins and 6 h after preconditioning with LPS or BCCAO (Figure 1). Aminoguanidine was chosen because it abolished the preconditioning effect of LPS or BCCAO in a model of focal cerebral ischemia (Cho et al, 2005). The timing of the administration was selected on the basis of the time course of iNOS mRNA expression induced by preconditioning stimuli in brain (see Results). The COX-2 inhibitor NS398 (20 mg/kg, i.p.; Cayman Chemical, Ann Arbor, MI, USA) or vehicle (H2O; pH 12) was administered 10 mins and 6 h after the preconditioning stimulus (Figure 1). The NO donor dipropylenetriamine (DPTA)-NONOate (1.36 μmol in 140 nl of Na2HPO4/phosphate-buffered saline, pH 7.2; Cayman Chemical), its inactive analog sulpho NONOate (Cayman Chemical), or the cGMP analog 8-bromo-cGMP (42 pmol in 140 nl; Sigma-Aldrich) or vehicle (phosphate-buffered saline) were microinjected into the neocortex 4 h after LPS (Figure 1). The peroxynitrite decomposition catalyst 5,10,15,20-tetrakis(4-sulfonatophenyl)-porphyrinato iron (III) (FeTPPS, Calbiochem, San Diego, CA, USA), its inactive version TPPS (0.084 nmol in 140 nl, Frontier Scientific, Logan, UT, USA) or vehicle (saline) was also microinjected into the neocortex 4 h after LPS (Figure 1). The 4 h time point for microinjection of DPTA NONOate, 8-bromo-cGMP, and FeTPPS was selected on the basis of the time course of NO production, inferred from the time course of 3-nitrotyrosine (3-NT) immunoreactivity after LPS (data not shown). Cortical microinjections of FeTPPS, NONOates, 8-bromo-cGMP were performed at the same stereotaxic coordinates as NMDA (see previous section).
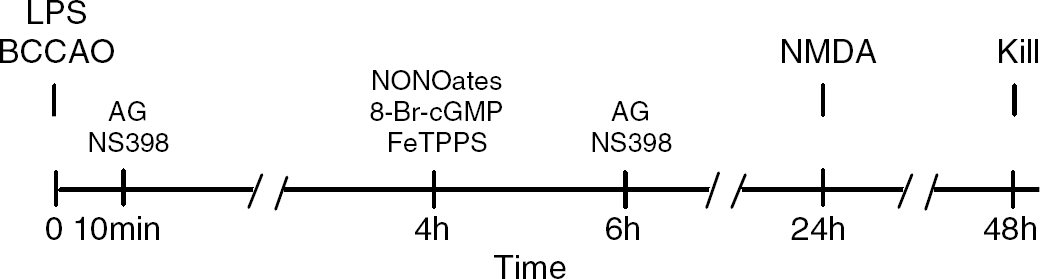
Preconditioning stimuli (LPS, BCCAO) or sham treatments (Vehicle, Sham surgery) were delivered at time 0. Twenty-four hours after preconditioning, NMDA was injected intracortically and mice were killed 24 h later (Kill). Aminoguanidine or NS398 was administered 10 mins and 6 h after the preconditioning stimulus. Dipropylenetriamine NONOate, sulpho NONOate, 8-Br-cGMP, or FeTPPS were microinjected intracortically 4 h after the preconditioning stimulus.
Real-Time Polymerase Chain Reaction
To assess the temporal profile of iNOS expression, mice were killed 2, 6, 12, 24, and 48 h after LPS administration and their brains were removed. Two-mm-thick coronal brain slices were collected and the parietal cortex was dissected out and frozen in liquid nitrogen. Total RNA was prepared from the samples using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). Quantitative determination of gene expression levels, using a two-step cycling protocol, was performed on Chromo 4 detector (Peltier Thermal Cycler MJ Research). Primers for iNOS (forward, 5′-CAGCTGGGCTGTACAAACCTT-3′; reverse, 5′-CATTGGAAGTCAAGCGTTTCG-3′) were purchased from Invitrogen Life Technologies. Two microliters of diluted cDNA (1:10) were amplified by Platinum SYBR green qPCR supermix UDG (Invitrogen Life Technologies). The reactions were incubated at 50°C for 2 mins and then at 95°C for 10 mins. A polymerase chain reaction cycling protocol consisting of 15 secs at 95°C and 1 mins at 60°C for 45 cycles was required for quantification. Relative expression levels were calculated by 2(−Delta Delta C(T)) method (Livak and Schmittgen, 2001). Quantities of all targets in test samples were normalized to the mouse HPRT housekeeping gene and values were normalized to vehicle-treated control samples.
3-Nitrotyrosine and NeuN Immunohistochemistry
Mice were perfused transcardiacally with heparin (1000 USP U/mL), and their brains removed and frozen. Brain sections (thickness: 14 μm) were cut through the parietal cortex and collected at 100 μm intervals. To assure uniformity of the immunolabel, sections from controls and experimental groups were processed together. Sections were fixed in ethanol and incubated with purified rabbit anti-mouse 3-NT (1:200; Upstate, Millipore Corporation, Billerica, MA, USA) followed by a fluorescein isothiocyanate-conjugated goat anti-rabbit secondary antibody (1:200; Molecular Probes, Invitrogen Corporation, Carlsbad, CA, USA). The specificity of the 3-NT stain was tested as previously described (Forster et al, 1999). For simultaneous visualization of 3-NT and the neuronal marker NeuN, sections were incubated with a mouse anti-NeuN monoclonal antibody (1:100, Chemicon, Millipore Corporation), followed by a Cy5-conjugated goat anti-rat secondary antibody (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). The specificity of the immunolabel was tested by processing sections without the primary antibody or after preadsorption with the antigen. Brain sections were examined with a Leica TCS SP5 confocal microscope (Leica, Mannheim, Germany).
Data Analysis
All data are expressed as means±s.e.m. Comparison between two groups was statistically evaluated by Student's t-test. Multiple comparisons were evaluated by oneway analysis of variance followed by Tukey's test. Differences were considered significant at P < 0.05.
Results
Lipopolysaccharide or Bilateral Common Carotid Artery Occlusion Induce Tolerance to N-methyl-d-aspartate Lesions
First, we sought to determine whether LPS or BCCAO induces tolerance to NMDA lesions. Mice received LPS or BCCAO and were subjected to neocortical injection of NMDA 4, 12, 24, or 48 h later. Lesion volume was determined 24 h after NMDA injection. As illustrated in Figure 2, the lesion induced by NMDA was significantly reduced 24 h after LPS (−23%; P < 0.05; Figure 2A) or BCCAO (−28%; P < 0.05). However, a transient increase in lesion volume was observed 12 h after LPS (Figure 2A), an observation consistent with reports of increased susceptibility to injury in the period preceding the development of the tolerance (Eklind et al, 2005; Kirino, 2002). Thus, LPS or BCCAO are able to induce transient tolerance to NMDA lesions.
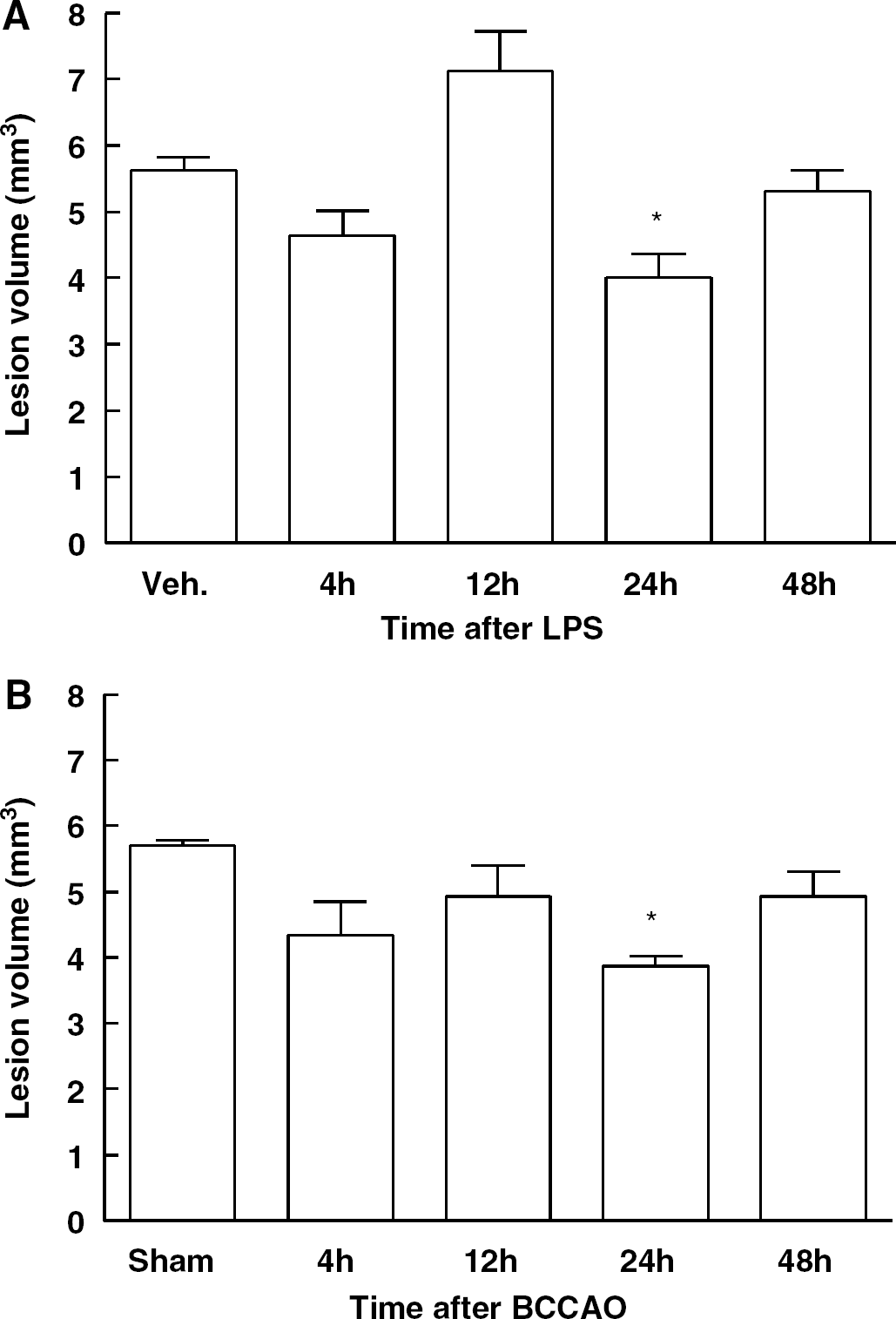
Temporal profile of NMDA-induced lesion volume at different time points after LPS injection (
Inducible Nitric Oxide Synthase is Expressed in Brain after Lipopolysaccharide and is Essential for the Tolerance
Next, we investigated whether iNOS contributes to the tolerance to NMDA lesions. We have previously established that BCCAO induces iNOS expression in brain (Cho et al, 2005). To determine whether also LPS induces iNOS, we used real-time polymerase chain reation. Inducible nitric oxide synthase mRNA increased 2 h after LPS, was reduced at 12 h, and returned to baseline at 24 to 48 h (Figure 3A). We then examined whether the preconditioning is dependent on iNOS. Administration of the iNOS-inhibitor AG 10 mins and 6 h after LPS or BCCAO blocked the tolerance (Figures 3B and 3C). Aminoguanidine did not affect the NMDA lesion when administered without LPS or BCCAO (Figures 3B and 3C) and did not block LPS preconditioning if it was administered 30 mins before NMDA (i.e., 23.5 h after LPS; Figure 3D). LPS and BCCAO failed to induce preconditioning in iNOS-null mice (Figures 3E and 3F). Thus, iNOS is expressed after LPS preconditioning and contributes to the tolerance to NMDA lesions induced by LPS or BCCAO.
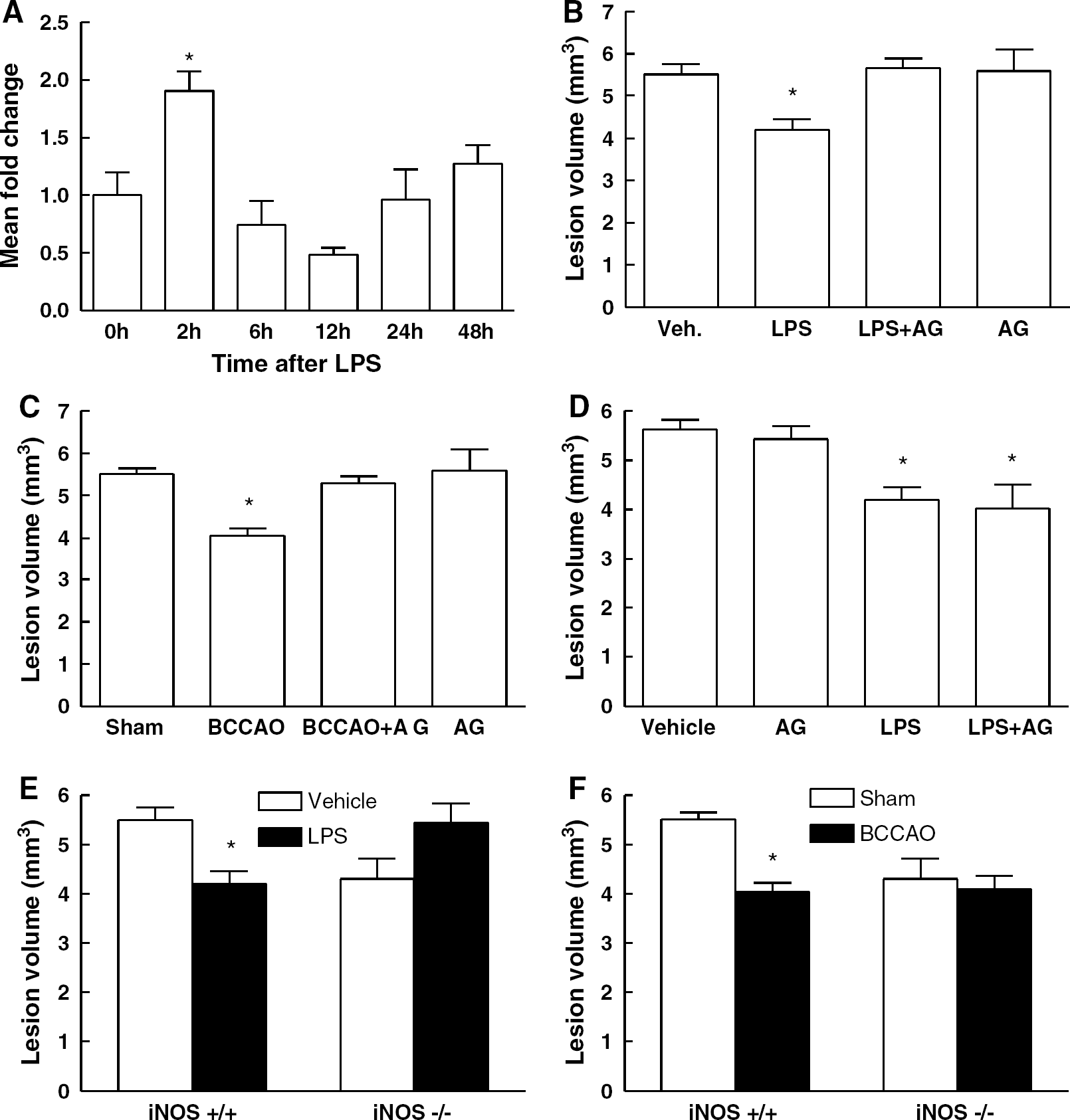
Inducible nitric oxide synthase contributes to preconditioning against NMDA lesions. Lipopolysaccharide induces iNOS mRNA expression (
Effect of Lipopolysaccharide on Rectal Temperature in iNOS +/+ and −/− Mice
Body temperature exerts a profound influence on the outcome of excitotoxic brain lesions (Suehiro et al, 1999). Therefore, we investigated the effect of LPS on rectal temperature in wild type and iNOS null. In vehicle-injected mice, rectal temperature remained essentially stable during the 24 h after LPS administration (Figure 4A). In wild-type mice treated with LPS, temperature decreased at 4, 6, and 10 h compared with pre-LPS levels (P < 0.05; analysis of variance and Tukey's test; Figure 4A). In iNOS-null mice treated with LPS, the reduction in temperature was more marked and sustained (P < 0.05 between 2 and 16 h). These findings are in agreement with previous studies on the effect of LPS on body temperature in wild type and iNOS-null mice (Saia and Carnio, 2006).
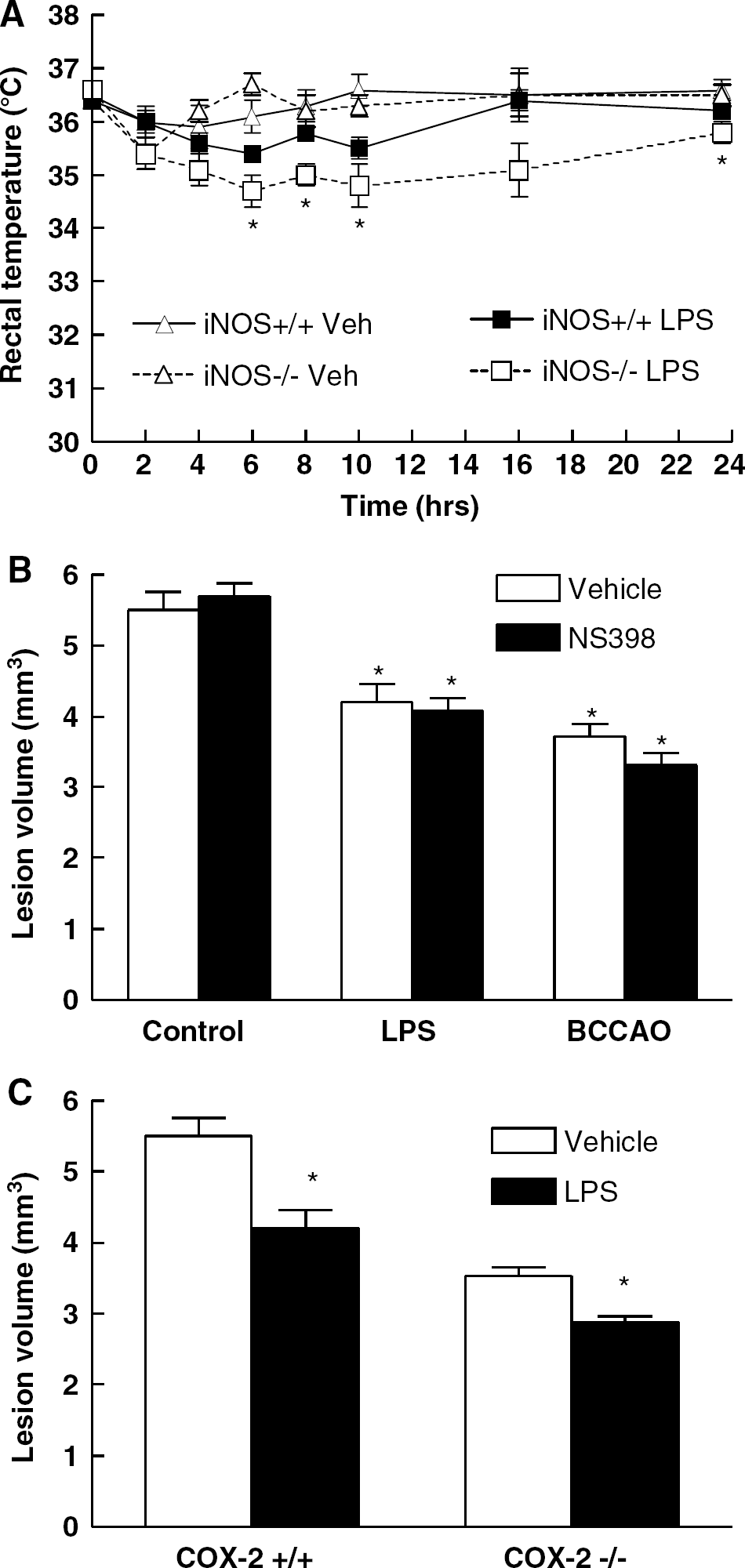
Time course of the changes in rectal temperature induced by LPS or vehicle (Veh) in wild type (iNOS +/+) and iNOS-null mice (iNOS−/−) (n = 5/group). Lipopolysaccharide administration reduced temperature in iNOS + / + mice at 4, 6, and 10 h (P < 0.05 from time 0; analysis of variance) and in iNOS−/− mice at all time points between 2 and 16 h (P < 0.05 from time 0). The reduction in temperature was more marked in iNOS−/− mice at 6, 8, 10, and 24 h (*P < 0.05 from iNOS−/− Veh) (
COX-2 is not Involved in the Tolerance induced by Lipopolysaccharide or Bilateral Common Carotid Artery Occlusion
The interaction between iNOS and COX-2 has been implicated in preconditioning in the myocardium (Shinmura et al, 2002). Therefore, we investigated whether COX-2 is involved in the tolerance induced by LPS or BCCAO. Treatment with the selective COX-2 inhibitor NS398 10 mins and 6 h after LPS or BCCAO did not block preconditioning (Figure 4B). Similarly, LPS-induced preconditioning was still observed in mice lacking COX-2 (Figure 4C). Therefore, COX-2 is not involved in the tolerance to NMDA lesions produced by LPS or BCCAO.
The NO Donor Dipropylenetriamine NONOate Rescues Lipopolysaccharide Preconditioning after iNOS Inhibition or in iNOS null Mice
To provide more direct evidence that NO is involved in the mechanisms of preconditioning, we tested whether exogenous NO could re-establish the tolerance in mice treated with AG or in iNOS-null mice. To simplify the experimental protocol, in this and subsequent experiments we used only LPS as the preconditioning stimulus. Neocortical microinjection of the NO donor DPTA NONOate 4 h after LPS re-established LPS preconditioning in mice treated with AG (Figure 5A). In contrast, administration of the inactive NO donor sulpho NONOate was ineffective (Figure 5A). Dipropylenetriamine NONOate, but not sulpho NONOate, was also able to rescue LPS tolerance in iNOS−/− mice (Figure 5B). Administration of DPTA NONOate or sulpho NONOate without LPS had no effect on the lesion produced by NMDA (Figure 5C). To determine whether NO exerts its effects by activating soluble guanylyl cyclase and increasing cGMP we examined whether the cell-permeable cGMP analog 8-bromo-cGMP could counteract the effect of iNOS inhibition on LPS preconditioning. However, neocortical microinjection of 8-bromo cGMP 4 h after LPS was unable to re-establish preconditioning in mice treated with AG (Figure 5D). These observations, collectively, indicate that NO is required for LPS preconditioning, but that its effect is not mediated through guanylyl cyclase and cGMP.
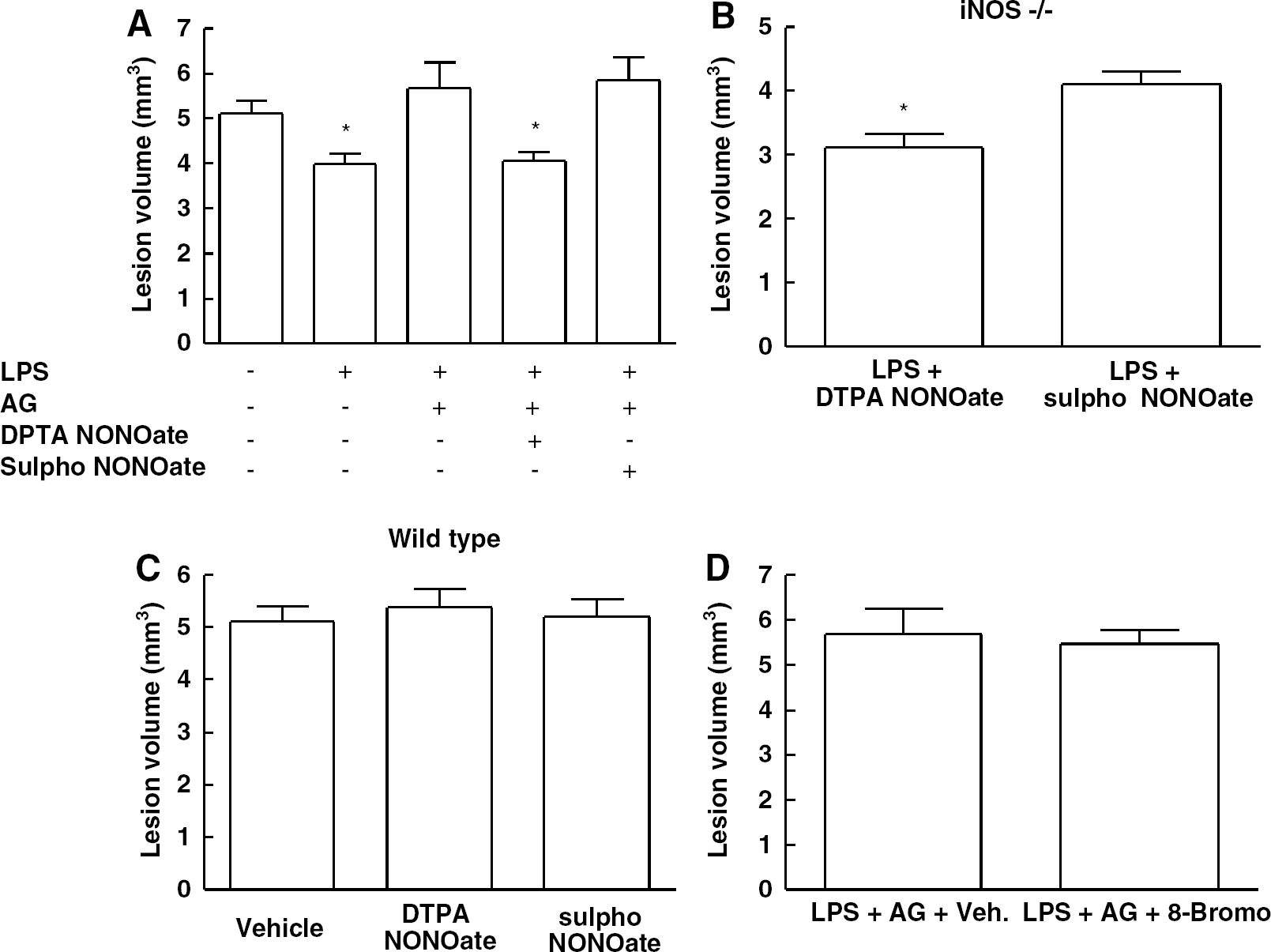
The NO donor DTPA NONOate, but not its inactive analog sulpho NONOate, reverses the inhibition of LPS preconditioning induced by AG (
Lipopolysaccharide Induces 3-nitrotyrosine Immunoreactivity in Cortical Neurons
The observation that NO does not mediate preconditioning through cGMP raises the possibility that the effects are mediated by peroxynitrite, the product of the reaction of NO with the ROS superoxide (Beckman et al, 1990). Therefore, we used 3-NT as a marker of peroxynitrite to determine whether LPS induces nitration in the cerebral cortex. In vehicle-treated mice, light 3-NT staining was present in the neocortex (Figure 6). 3-Nitrotyrosine immunoreactivity was also observed in cerebrovascular cells (data not shown). Double-label with the neuronal marker NeuN revealed that most 3-NT-positive cells were neurons (Figure 6). Twenty-four hours after LPS injection, 3-NT immunoreactivity was markedly upregulated mainly, but not exclusively, in NeuN positive cells (Figure 6). LPS-induced nitration was attenuated in iNOS-null mice and was virtually abolished in mice lacking the nox2 subunit of the superoxide producing enzyme NADPH oxidase (Figure 6). These data indicate that the nitration induced by LPS requires iNOS-derived NO and nox2-derived superoxide.
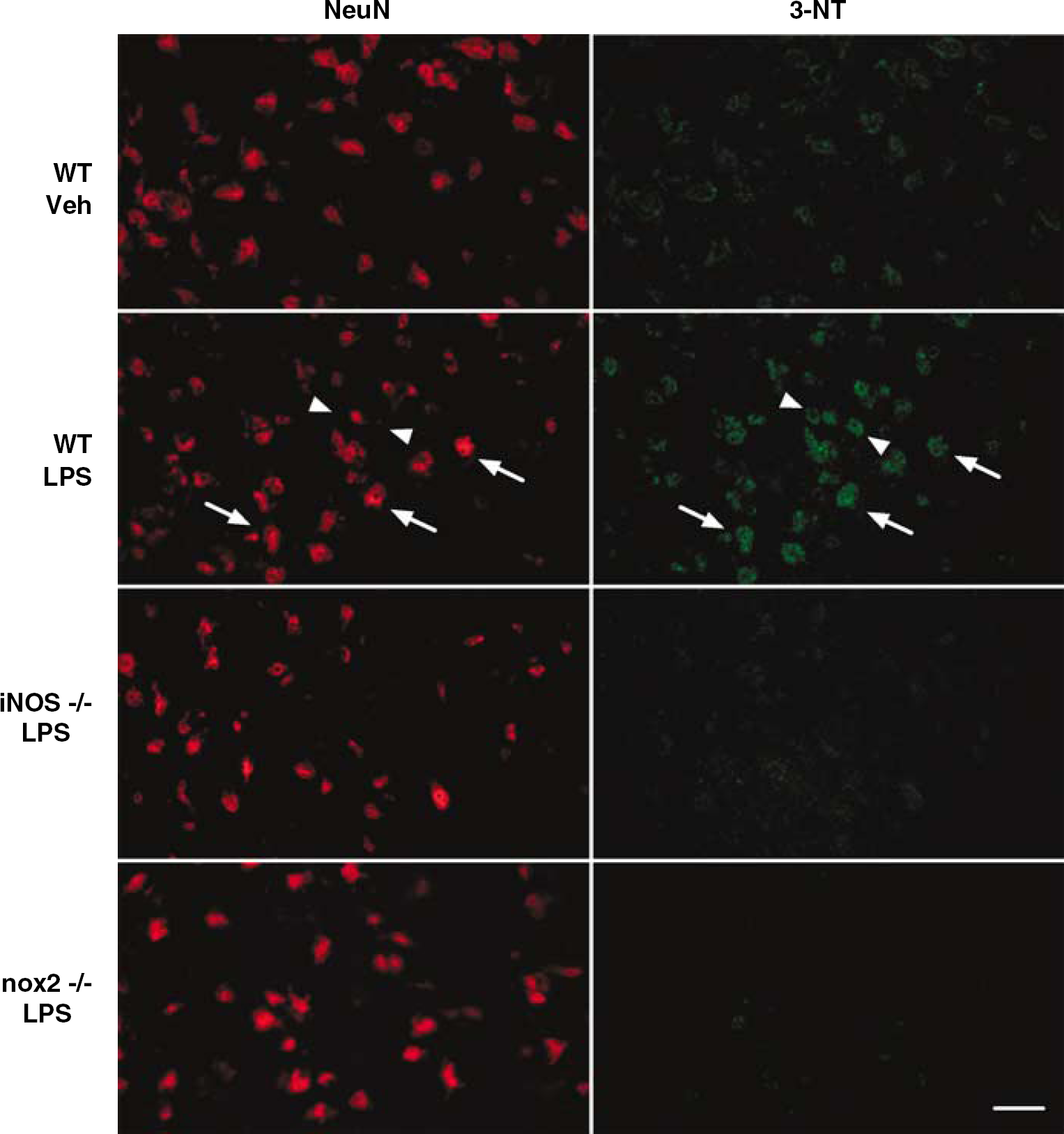
Confocal images of double label for 3-NT immunoreactivity and the neuronal marker NeuN in parietal cortex of mice 24 h after LPS. Lipopolysaccharide treatment increases 3-NT immunoreactivity mainly (arrows), but not exclusively (arrowheads), in neurons. 3-nitrotyrosine immunoreactivity is attenuated in iNOS and nox2-null mice. These are representative images from 3 mice/group. Calibration bar: 25 μm.
Preconditioning Depends on Radicals Derived from nox2
The findings presented in the previous section indicate that LPS-induced nitration depends on nox2-derived superoxide. A NADPH oxidase type containing nox2 in the catalytic domain has emerged as an important source of superoxide in brain (Infanger et al, 2006), and participates in preconditioning in other organs (Bell et al, 2005). Therefore, we used nox2-null mice to determine whether nox2 is involved in the tolerance to NMDA lesions produced by LPS. We found that LPS does not induce tolerance to NMDA in nox2-null mice (Figure 7A). These findings provide further evidence implicating NADPH oxidase-derived superoxide in the mechanisms of LPS preconditioning.
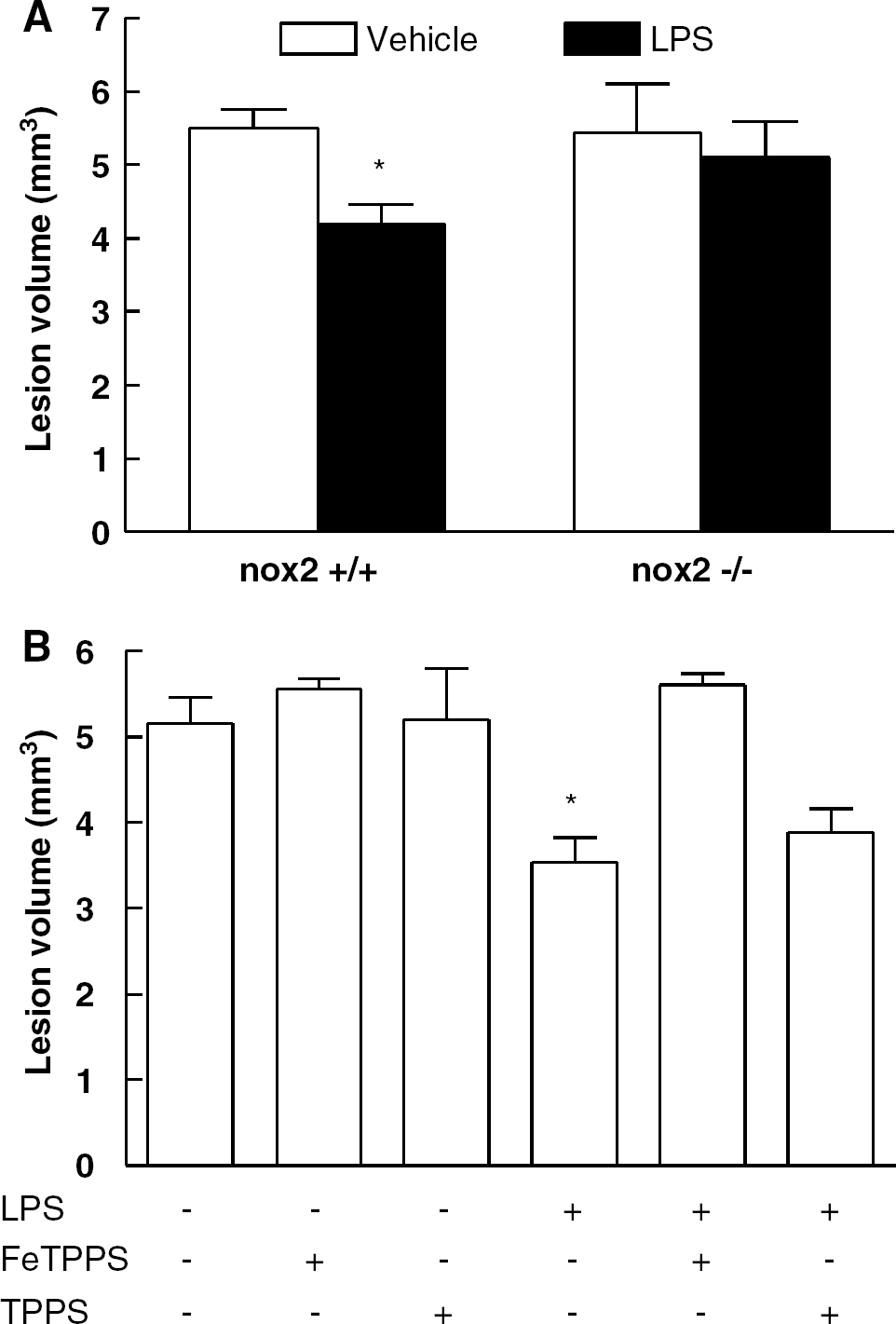
Lipopolysaccharide does not induce preconditioning in nox2-null mice (*P < 0.05 from vehicle; Student's t test; n = 7 to 8/group) (
The Peroxynitrite Decomposition Catalyst FeTPPS Blocks Lipopolysaccharide-induced Tolerance to NMDA Lesions
The observation that LPS preconditioning depends on NADPH oxidase-derived ROS supports the hypothesis that the effects of NO are mediated by peroxynitrite. To provide further evidence for this hypothesis, we used the peroxynitrite decomposition catalyst FeTPPS. FeTPPS, microinjected into the neocortex 4 h after LPS injection, blocked LPS preconditioning (Figure 7B). In contrast, TPPS, the inactive analog of FeTPPS, had no effect on LPS-induced tolerance (Figure 7B). FeTPPS or TPPS did not influence NMDA-induced lesions in the absence of LPS (Figure 7B).
Discussion
We sought to provide further insight into the role of iNOS in the delayed tolerance induced by BCCAO or LPS. Because NMDA receptor-mediated excitotoxicity is of crucial importance in ischemic brain damage (Lo et al, 2003), we used neocortical injection of NMDA as an injury model. First, we examined whether LPS and BCCAO were able to confer tolerance to brain lesions induced by NMDA. We found that LPS and BCCAO reduce the lesion produced by NMDA, an effect observed 24 h after the preconditioning stimulus. We also found that the preconditioning is blocked by pharmacological inhibition of iNOS and is not observed in iNOS-null mice. The tolerance was abrogated when the iNOS inhibitor was administered 10 mins and 6 h, but not 23.5 h, after LPS. This observation indicates that the critical period for the induction of the tolerance by iNOS-derived NO is early after LPS administration. In agreement with this prediction, an NO donor administered 4 h after LPS was able to re-establish LPS-induced tolerance after iNOS inhibition and in iNOS-null mice. Thus, LPS and BCCAO induce tolerance to NMDA lesions and iNOS-derived NO is essential for the expression of such tolerance.
We then focused on the downstream mediators by which iNOS-derived NO contributes to the tolerance. Nitric oxide exerts many of its biologic effects by activating soluble guanylyl cyclase and increasing cGMP (Moncada and Bolanos, 2006). However, we found that LPS-induced preconditioning does not depend on cGMP because a cGMP analog was unable to re-establish the tolerance after iNOS inhibition. Another mediator of NO's biologic actions is peroxynitrite formed by the reaction of NO with superoxide (Beckman et al, 1990). We found that LPS induces 3-NT immunoreactivity, a marker of peroxynitrite, in cortical neurons. Increased nitration and preconditioning were not observed in mice lacking the nox2 subunit of the superoxide-generating enzyme NADPH oxidase. Furthermore, the peroxynitrite decomposition catalyst FeTPPS blocked the tolerance. These findings, collectively, provide strong evidence that peroxynitrite, formed by iNOS-derived NO and NADPH oxidase-derived superoxide, is essential for the tolerance to NMDA lesions.
The findings of the present study cannot be attributed to differences in the experimental conditions among the different groups or to artifacts related to the methods used. Depth and duration of anesthesia, important variables in studies of preconditioning (Kapinya et al, 2002; Zhao and Nowak, 2006), were monitored and kept constant in all experiments. Body temperature, a factor that has powerful effects on NMDA-mediated brain injury (Buchan and Pulsinelli, 1990), was controlled during and up to 4 h after NMDA injection, the critical period for the development of the lesion (Manabe et al, 2004). Furthermore, the changes in temperature induced by LPS are unlikely to play a role because body temperature returned to normal by the time NMDA was injected. LPS induced a more sustained reduction in temperature in iNOS-null mice, but LPS induced hypothermia cannot explain the absence of protection in iNOS nulls. This is because a reduction in temperature would be expected to enhance the protection, while in iNOS nulls the protection induced by preconditioning was abolished. We also found that LPS or BCCAO did not attenuate the lesion in iNOS-null mice. One potential explanation for the lack of preconditioning in iNOS-null mice is that the NMDA lesion was already reduced as much as possible and could not be further attenuated. However, this is unlikely because DTPA NONOate was able to reduce the lesion further in iNOS nulls. It is of interest that in iNOS nulls treated with LPS and sulpho NONOate lesions appear to be smaller than those in LPS treated iNOS nulls. Although we cannot exclude a small protective effect in the nulls, the finding that sulpho NONOate did not reduce the lesion in wild-type mice, with or without LPS, does not support this possibility. Another potential confounding factor in experiments in which 3-NT immunocytochemistry is used as a marker of peroxynitrite is that protein nitration can be induced also by agents other than peroxynitrite (Halliwell and Whiteman, 2004). However, the 3-NT immunoreactivity induced by LPS was not observed in iNOS or nox2-null mice, supporting the hypothesis that peroxynitrite formed from iNOS-derived NO and nox2-derived superoxide is the nitrating agent. It must be noted that although our data implicate NADPH oxidase-derived ROS in the mechanisms of the tolerance, we cannot rule out that there is also a component of mitochondrial ROS triggered by NADPH oxidase-derived ROS, as suggested by studies in the myocardium (Brandes, 2005).
Nitric oxide has been shown to participate in the mechanisms of preconditioning in brain (Huang 2004; Nandagopal et al, 2001). Gidday et al (1999) demonstrated that the nonselective inhibitor of NOS nitro-l-arginine blocked anoxic preconditioning in a neonatal model of ischemic brain injury. Using an in vitro model, Gonzalez-Zulueta et al (2000) demonstrated that ischemic tolerance induced by oxygen-glucose deprivation depends on nNOS-derived NO. Furthermore, Atochin et al (2003) showed that early ischemic preconditioning to focal cerebral ischemia is dependent on endothelial NOS and nNOS. More recently iNOS-derived NO has also been implicated in the mechanisms of preconditioning. We have demonstrated that iNOS plays a critical role in the mechanisms by which BCCAO or LPS induce tolerance to focal cerebral ischemia (Cho et al, 2005). Furthermore, Kapinya et al (2002) reported that iNOS is involved in the preconditioning induced by volatile anesthetics. Inducible nitric oxide synthase has also been implicated in the mechanisms of ischemic tolerance in neonatal ischemic stroke (Zhao and Zuo, 2004). The findings of the present study further expand our understanding of the role of iNOS in preconditioning by demonstrating that iNOS is also essential for the tolerance to excitotoxic brain lesions.
To our knowledge, this is the first report demonstrating a key role of peroxynitrite in the phenomenon of delayed tolerance in brain. Thus, peroxynitrite, in addition to its deleterious role in a variety of brain pathologies (Carlson and Rose, 2006; Iadecola, 2004; Jenner, 2003; Keynes and Garthwaite, 2004), can also have beneficial effects by promoting tolerance to excitotoxic brain injury. Studies of cardiac preconditioning support a beneficial role of peroxynitrite in the heart as well (Altug et al, 2000). However, the mechanisms by which peroxynitrite can be both toxic and protective remain unclear. One possibility is that the effects are dependent on the amount of peroxynitrite produced. Thus, large amounts could be toxic whereas smaller amounts could be protective. In support of this hypothesis, nanomolar to low micromolar concentrations of peroxynitrite are protective in models of cardiac ischemia–reperfusion, while high micromolar concentrations are deleterious (Lefer et al, 1997; Schulz and Wambolt, 1995). However, the signaling pathways responsible for these opposing effects have not been elucidated. Peroxynitrite activates numerous signaling cascades, including stress pathways involved in both cell death and survival (Klotz et al, 2002). Although concentration-dependent activation of beneficial or deleterious pathways could underlie its protective and destructive effects, these interactions need to be explored further.
We used NMDA injection into the cortex as a model of injury, thereby focusing predominantly on the mechanisms of tolerance to excitotoxic neuronal death. However, this model is not suitable to explore potential vasoprotective effects of iNOS derived NO. Several reports suggest that improved cerebral perfusion contributes to the protective effect of preconditioning in models of focal cerebral ischemia (Dawson et al, 1999; Furuya et al, 2005; Zhao and Nowak, 2006) raising the possibility that the mechanisms of the tolerance include both vasoprotective and neuroprotective components (Gidday, 2006). Ongoing experiments are addressing whether, in models of focal cerebral ischemia, the preconditioning effects of iNOS depend on improved CBF.
In conclusion, we found that iNOS-derived NO is critical for the tolerance to NMDA lesions afforded by BCCAO or LPS. The mechanisms of the tolerance do not involve the NO second messenger cGMP, but require superoxide derived from a nox2 containing NADPH oxidase. The preconditioning is associated with upregulation of the peroxynitrite marker 3-NT and is blocked by a peroxynitrite decomposition catalyst. These observations, collectively, provide evidence that peroxynitrite, formed from iNOS-derived NO and nox2-derived superoxide, is a key factor in the mechanisms of the tolerance. Thus, peroxynitrite, in addition to its well-known deleterious effects, can also be beneficial. Modulation of peroxynitrite production may be an important factor in balancing neuronal death and survival in the setting of brain injury.
Footnotes
Acknowledgements
CI is the recipient of a Javits award from NIH/NINDS.