
Abstract
Oxidative stress and zinc release are both known to contribute to neuronal death after hypoglycemia; however, the cause—effect relationships between these events are not established. Here we found, using a rat model of profound hypoglycemia, that the neuronal zinc release and translocation that occur immediately after hypoglycemia are prevented by the nitric oxide synthase inhibitor 7-nitroindazole but not by overexpression of superoxide dismutase-1 (SOD-1). However, overexpression of SOD-1 prevented activation of poly(ADP-ribose) polymerase-1 (PARP-1) and neuronal death, suggesting that zinc release is upstream of superoxide production. Accordingly, zinc-induced superoxide production was blocked in neuronal cultures by the NADPH oxidase inhibitor apocynin and by genetic deficiency in the p47phox subunit of NADPH oxidase. A key role for the vesicular zinc pool in this process was suggested by reduced superoxide formation and neuronal death in mice deficient in zinc transporter 3. Together, these findings suggest a series of events in which nitric oxide production triggers vesicular zinc release, which in turn activates NADPH oxidase and PARP-1. This sequence may also occur in other central nervous system disorders in which zinc, nitric oxide, and oxidative stress have been linked.
Introduction
Hypoglycemia frequently occurs in diabetic patients who attempt tight control of blood glucose levels. With profound hypoglycemia, specific subsets of neurons in the brain are injured (Auer et al, 1985). Hypoglycemic neuronal death is not a direct result of energy failure, but is instead a process consisting of several events that are triggered by glucose reperfusion (Engelsen et al, 1986; Wieloch, 1985). Several steps have been identified in this pathway that, when blocked, reduce or prevent neuronal death. These include glutamate receptor activation, NADPH oxidase activation, zinc translocation, poly(ADP-ribose) polymerase-1 (PARP-1) activation, and mitochondrial permeability transition (Ferrand-Drake et al, 1999, 2003; Suh et al, 2003, 2004). However, the temporal order and the cause—effect relationships between these events remain uncertain. In particular, it is not known if zinc release is a cause or effect of oxidative stress, and whether the earlier described effects of zinc chelation on neuronal survival are because of chelation of zinc released from synaptic vesicles or zinc released from other routes.
It is also unclear how zinc might induce production of reactive oxygen species (ROS) in neurons. It has been proposed that zinc can act on mitochondria to increase superoxide production (Sensi et al, 1999). However, recent studies have suggested that NADPH oxidase is a major source of neuronal ROS production in hypoglycemia and other conditions (Suh et al, 2007a; Abramov et al, 2007). NADPH oxidase is a multicomponent enzyme consisting of several subunits that must coalesce at the cell membrane for enzyme activation. Our previous study showed that the p47phox and p67phox subunits translocate from the cytoplasm to the plasma membrane after glucose deprivation, and that this translocation is prevented by zinc chelation (Suh et al, 2007a). Zinc-induced neuronal NADPH oxidase activation in neuronal cultures has also been described (Noh and Koh, 2000; Kim and Koh, 2002).
This study used primary cultured cortical neurons and rodent models of acute hypoglycemia to identify the temporal relationship between zinc translocation, superoxide production, and neuronal death after hypoglycemia. We found that hypoglycemia-induced vesicular zinc release was dependent on nitric oxide synthase (NOS) activity, and then subsequent zinc translocation into hippocampal neurons produced superoxide through an NADPH oxidase-mediated pathway.
Materials and methods
This study was approved by the San Francisco Veterans Affairs Medical Center animal studies committee. Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA, USA). The superoxide dismutase-1 (SOD-1) transgenic rat colony was maintained as heterozygotes, with outbreeding to wild-type rats (Chan et al, 1998). Experiments with the SOD-1 transgenic rats used nontransgenic littermates as controls. The p47phox-deficient mice and the wild-type mice used as their controls were of the C57/B16 background strain, and both were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Zinc transporter 3 (ZnT3)-deficient mice were obtained from Washington University and the wild-type mice used as their controls were from the C57/B16 background strain. Cultured cortical neurons were obtained from the CD-1 strain (Simonsen, Gilroy, CA, USA). Cell culture reagents were obtained from Mediatech (Herndon, VA, USA). All other reagents were obtained form Sigma-Aldrich (St Louis, MO, USA) except where noted.
Hypoglycemia in Rats and Mice
Insulin-induced hypoglycemia in rats was induced under isoflurane/nitrous oxide anesthesia as described previously (Suh et al, 2003). The period of hypoglycemia is defined here as the interval during which the electroencephalogram (EEG) showed an isoelectric pattern (Auer et al, 1984; Suh et al, 2003). Hypoglycemia was terminated with intravenous glucose infusion. Rats subjected to sham hypoglycemia received glucose in conjunction with insulin to maintain blood glucose levels between 5 and 10 mmol/L and were otherwise treated identically. For studies in mice, the insulin dose was reduced to 5 U/kg and isoelectricity period was extended to 45 mins before glucose reinfusion. Hexamethonium chloride was administered at 30 mg/kg intraperitoneally 15 mins before the insulin injections to reduce autonomic responses.
Cell Cultures
Glia-free neuron cultures were prepared by plating the embryonic neurons onto a plastic surface (Kauppinen and Swanson, 2005). These neurons were cultured in glia-conditioned medium and used at days 10 to 14 in vitro, at which time the cultures contained greater than 95% neurons, as assessed by immunostaining for the neuron marker microtubule-associated protein-2 and the astrocyte marker glial fibrillary acidic protein (Kauppinen and Swanson, 2005). Glucose concentration in the culture media was 5.5 mmol/L.
Immunostaining
Immunostaining was performed as described (Kauppinen and Swanson, 2005). Briefly, cell cultures were fixed and incubated with rabbit polyclonal antibody to microtubule-associated protein-2 (Chemicon International, Temecula, CA, USA) and mouse polyclonal antibody glial fibrillary acidic protein (Chemicon International) at 1:1,000 dilution to confirm pure neuron culture. To identify PARP-1 activation, rabbit polyclonal antibody to poly(ADP-ribose) (PAR; Trevigen, Gaithersburg, MD, USA) was added at 1:500 dilution and incubated at 4°C overnight. After washing, sections were incubated with Alexa Fluor 594-conjugated goat anti-mouse or Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes, Eugene, OR, USA) at 1:500 dilution for 3 h at room temperature. Negative controls were prepared by omitting the primary antibodies. Digital images were acquired with a confocal laser-scanning microscope.
Detection of Superoxide
For in vivo studies, dihydroethidium (Molecular Probes) was prepared as a 1 mg/mL solution in 1% dimethyl sulfoxide and administered at 1 mg/kg by the femoral vein to rats and through the intraperitoneal space to mice at the onset of EEG isoelectricity. Animals were killed at the designated time points and perfusion fixed with 4% paraformaldehyde. Brains were cryostat sectioned and photographed with a confocal fluorescent microscope at excitation = 510 to 550 nm and emission > 580 nm for detection of ethidium (Et; Chan et al, 1998). Five sections were analyzed from each brain, taken at 80 μm intervals, to span the hippocampus. Ethidium signal intensity was expressed as the ratio of the mean fluorescence in neuronal perikaria to fluorescence in the stratum radiatum of hippocampal CA1. For cell culture studies, 5 μmol/L dihydroethidium (Budd et al, 1997) was added to the cultures 30 mins before zinc treatment. The cultures were photographed with a fluorescence microscope 1 h after zinc treatment and the digitized images were analyzed and expressed as percent of neurons with an Et signal at least 50% higher than background. Earlier studies have confirmed that the Et signal reflects primarily superoxide under the conditions of hypoglycemia and glucose reperfusion used here (Suh et al, 2007a).
Neuronal Death
Neuronal death after hypoglycemia in vivo was evaluated after a 7-day survival period. Brain sections were stained by the Fluoro-Jade B method (Histo-Chem, Jefferson, AR, USA) (Schmued and Hopkins, 2000; Suh et al, 2003). Five coronal sections were collected from each animal, spaced 80 μm apart and spanning the hippocampus. An observer blinded to the experimental protocol counted the total number of Fluoro-Jade B-positive neurons in each structure of interest, in both hemispheres. Data from each animal were expressed as the mean number of degenerating neurons per structure of interest.
In Vivo Zinc Detection
Vesicular zinc was imaged using the N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) method (Frederickson et al, 1987). Rats and mice were killed immediately after the designated time and freshly frozen brains were coronally sectioned with five evenly spaced sections collected through the hippocampal region of each brain. Sections were stained by immersion in a solution of 4.5 μmol/L TSQ (Molecular Probes), 140 mmol/L sodium barbital, and 140 mmol/L sodium acetate (pH 10.5 to 11) for 60 secs and then rinsed for 60 secs in 150 mmol/L NaCl. TSQ-zinc binding was imaged and photographed with a fluorescence microscope with 360 nm UV light and a 500 nm long-pass filter. The mean fluorescence intensity within the mossy fiber terminal area was measured and expressed in arbitrary intensity after subtraction of background fluorescence as measured in the lateral ventricle (Frederickson et al, 1987). Measurements from the five sections were averaged for each ‘n’. Zinc levels in postsynaptic hippocampal CA1 neurons wee evaluated in the same manner.
Statistical Analyses
Data are expressed as mean + s.e.m. Measurements of Et and TSQ fluorescence intensities were evaluated with the Kruskal—Wallis test and Dunn test for comparisons between multiple groups. All other data were assessed by one-way analysis of variance followed by either the Tukey—Kramer test for multiple comparisons between groups or the Dunnett's test for comparisons of multiple groups against a common control group.
Results
Nitric Oxide Synthase Inhibition Prevents Hypoglycemia-Induced Vesicular Zinc Release and Postsynaptic Zinc Accumulation
Earlier studies have shown that release of vesicular zinc from hippocampal mossy fibers is induced by 30 mins of hypoglycemia (HG) followed by 30 mins of glucose reperfusion (GR) (Suh et al, 2007a). Here we used the fluorescent dye TSQ, which binds free zinc (Frederickson et al, 1987), to evaluate the relationships between vesicular zinc release, nitric oxide production, and superoxide production after HG/GR in the hippocampal CA1 region. Vesicular zinc release was almost completely prevented in rats treated with the NOS inhibitor 7-nitroindazole (7-NI), whereas rats overexpressing SOD-1 showed zinc release comparable with wild-type rats (Figures 1A and 1B). TSQ staining was also used to evaluate zinc translocation into postsynaptic neurons. The postsynaptic pyramidal cell bodies lacked zinc staining under control conditions, but showed a marked increase in zinc staining 3 h after HG/GR (Figure 2), suggesting that zinc released from presynaptic vesicles is translocated into the postsynaptic neurons after hypoglycemia (Suh et al, 2004). This intracellular zinc accumulation was almost completely prevented by the NOS inhibitor 7-NI. It also prevented subsequent neuronal death when evaluated at 24 h after reperfusion (Figure 2). By contrast, rats overexpressing SOD-1 did not show attenuated zinc translocation at the 3-h time period, but showed reduced neuronal death at 24 h after reperfusion (Figure 2).

Glucose reperfusion (GR) increased vesicular zinc release in the hippocampus after hypoglycemia (HG). (
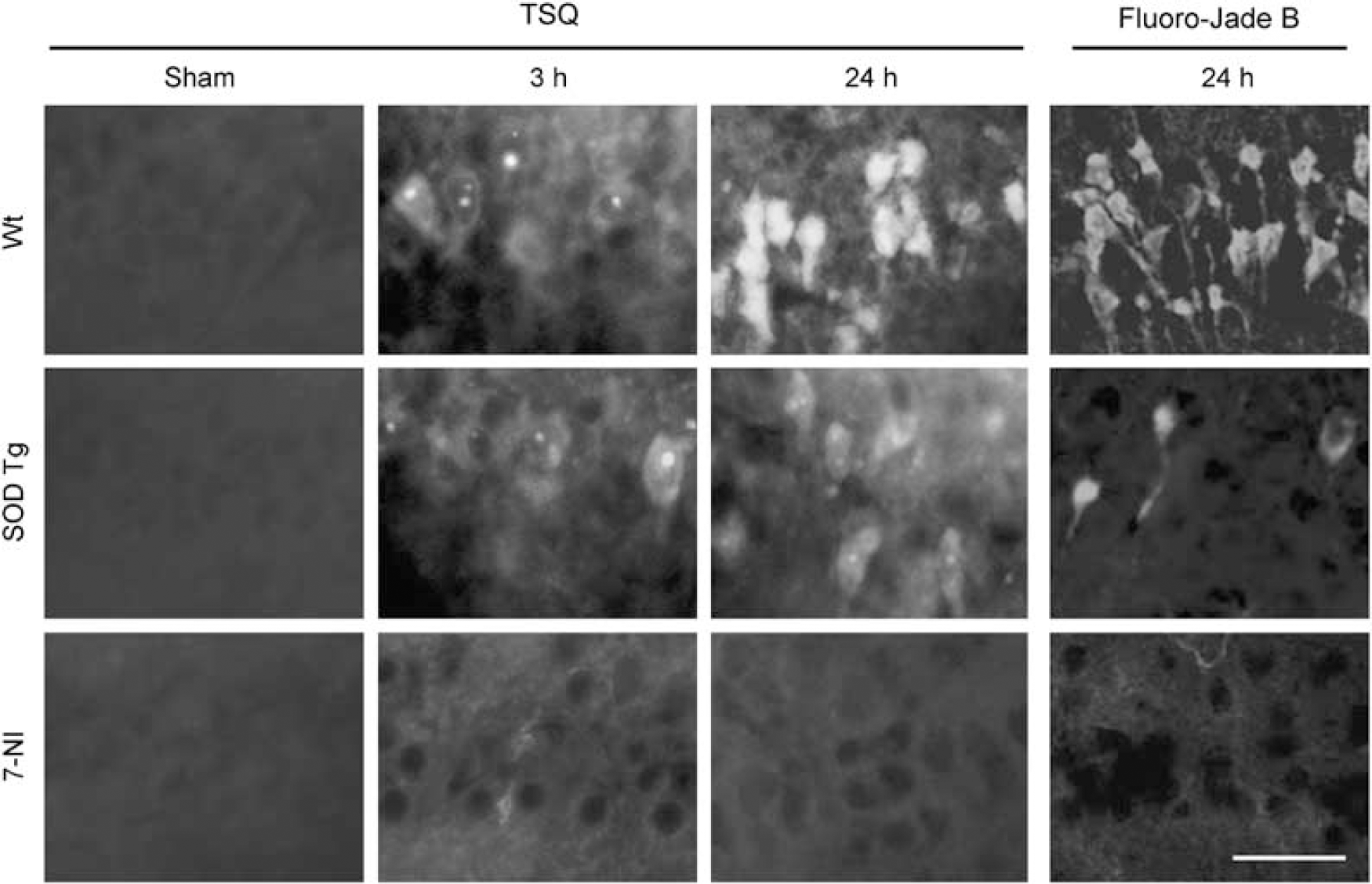
Intracellular zinc accumulation and neuronal death after hypoglycemia. Images show neuronal zinc accumulation at 3 or 24 h after HG/GR and neuronal death at 24 h after HG/GR. The images in the upper panel are representative of wild-type rat hippocampal CA1. The intensity of TSQ in CA1 pyramidal neurons is increased compared with sham-operated rats by 3 h after hypoglycemia and further increased at 24 h. CA1 pyramidal neurons show Fluoro-Jade B staining (green) at 24 h. The images in the middle panel are from SOD-1 transgenic rats. The intensity of TSQ is not elevated at 3 h, but is increased to a degree comparable with wild-type rats at 24 h. The images in the lower panel are from 7-NI-treated rats. The increase in TSQ intensity and neuron degeneration is blocked by 7-NI at both time points. Scale bar = 200 μm. n = 3 to 4.
Overexpression of Superoxide Dismutase-1 Prevents Poly(ADP-ribose) Accumulation in Neurons After Hypoglycemia/Glucose Reperfusion
The activation of PARP-1 is known to be induced by oxidative stress (Watson et al, 1995; Hwang et al, 2002), and it is a crucial event in hypoglycemia-induced neuronal death (Suh et al, 2003). We used the SOD-1 transgenic rats to test whether superoxide production is upstream of PARP-1 activation after HG/GR. In wild-type rats, robust PARP-1 activation is induced in cortical and hippocampal neurons 3 h after HG/GR, as evidenced by the accumulation of PAR. By contrast, PAR accumulation was minimal in the SOD-1 transgenic rats (Figure 3).
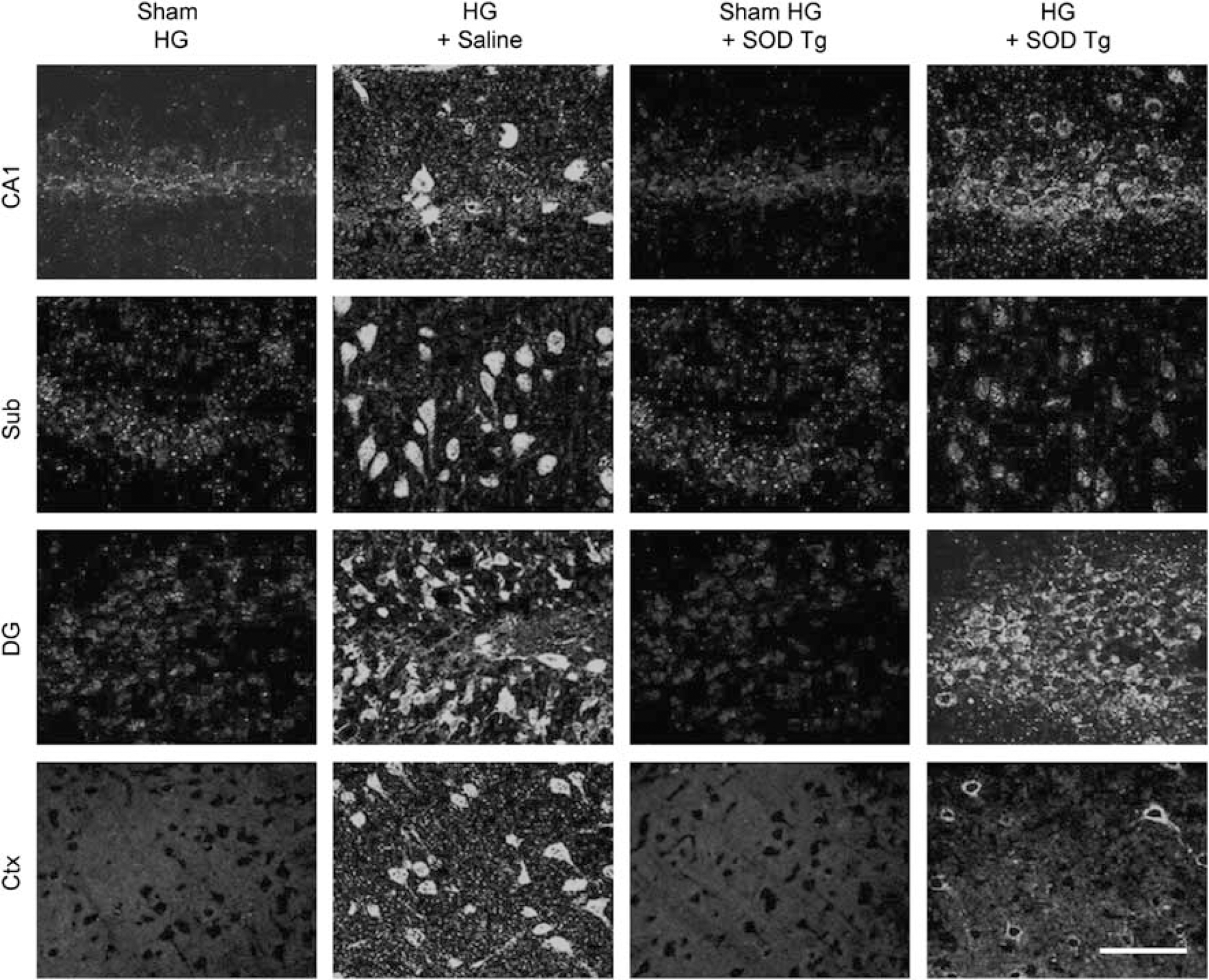
Overexpression of SOD-1 gene prevents PAR accumulation in the hippocampal and cortical neurons. Confocal images of PAR immunoreactivity in the hippocampus and cortex are shown. Rats were exposed to 30 mins of HG/GR and 3 h later the brain was collected. Immunoreactivity for PAR (green) is evident in hippocampal CA1, dentate gyrus (DG), subiculum (Sub), and perirhinal cortex neurons (Ctx). Immunoreactivity for PAR was not detected in the brain of rats subjected to sham hypoglycemia. The results are representative of three rats in each group. Scale bar = 100 μm.
Mice Deficient in Zinc Transporter 3 (ZnT3−/−) Show Reduced Reactive Oxygen Species Formation and Neuronal Death After Hypoglycemia
To further evaluate the events leading from HG/GR to neuronal death, we adapted this experimental model for use in mice deficient in ZnT3 (ZnT3−/−). Intraperitoneal injection of 5 U/kg of regular human insulin induced a gradual reduction in blood glucose concentrations and a concomitant change in the EEG pattern culminating in an isoelectric signal (Figure 4), similar to that previously reported in rats (Suh et al, 2003). Blood glucose level was below 19 mg/dL (< 1 mmol/L) during EEG isoelectricity. Neuronal death was evaluated 7 days after hypoglycemia. Forty-five minutes of isoelectric EEG induced substantial neuronal death in the hippocampus and cortex 7 days later (Figure 4).
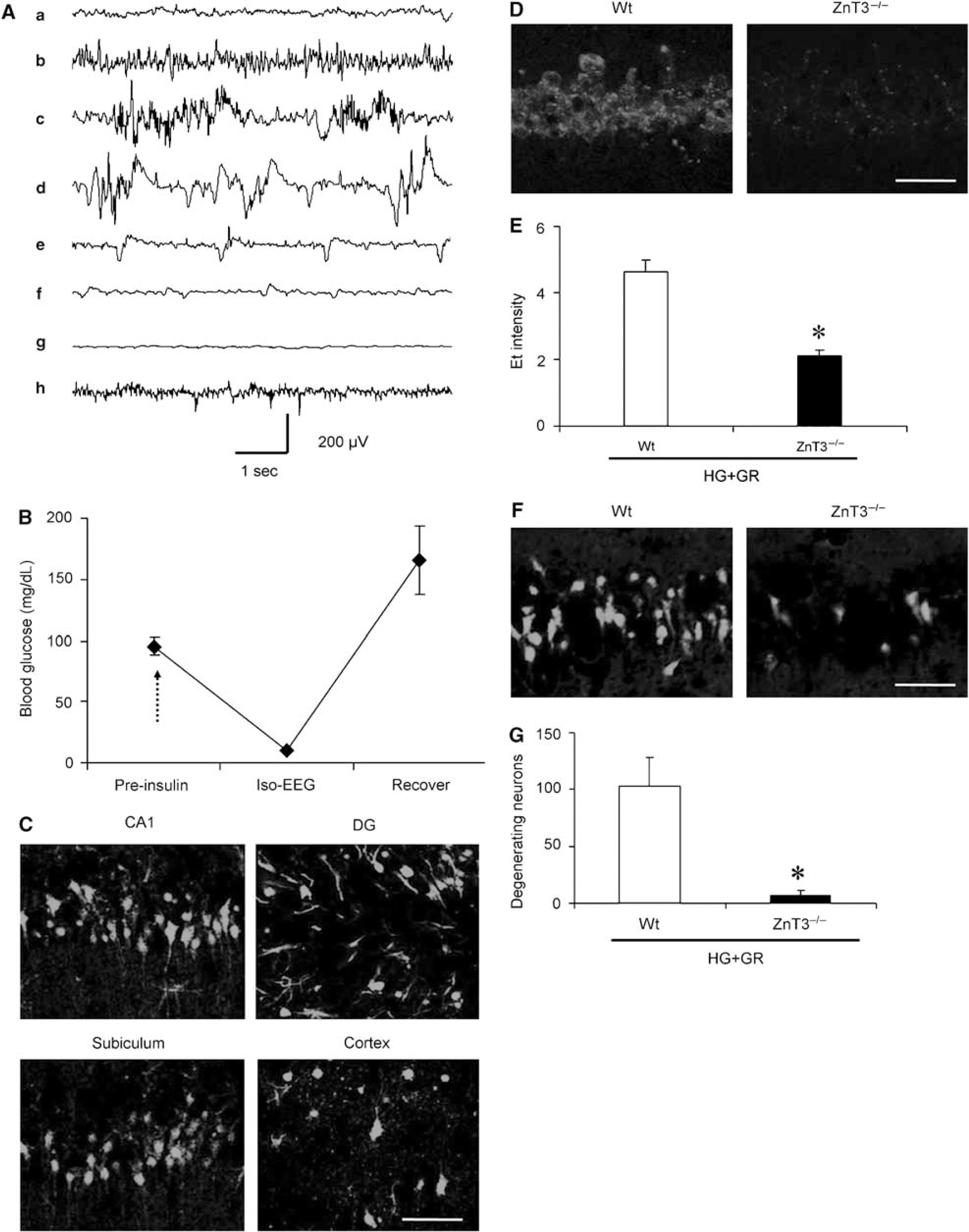
Hypoglycemic neuronal injury in mice. (
To confirm that vesicular zinc release contributes to superoxide production and neuronal death after hypoglycemia, we examined Et fluorescent activity in hippocampal CA1 neurons of ZnT3-deficient mice. The ZnT3−/− mice are devoid of vesicular zinc (Palmiter et al, 1996). These mice showed a much smaller increase in Et fluorescence in hippocampal CA1 neurons after HG/GR, relative to wild-type mice of the same C57/B16 background strain (Figures 4D and 4E). The ZnT3−/− mice also showed reduced neuronal death after HG/GR (Figures 4F and 4G).
Zinc-Induced Reactive Oxygen Species Production is Mediated by NADPH Oxidase Activation
The mechanism by which NADPH oxidase is activated in nonphagocytic cells, such as neurons, is not well understood, but zinc has been identified as both an inducer of neuronal NADPH oxidase activity (Kim and Koh, 2002) and a contributor to hypoglycemic neuronal death (Suh et al, 2007a). To confirm that NADPH oxidase is the source of superoxide production after zinc exposure, we prepared mouse cortical neuronal cultures. Neurons exposed to 100 μmol/L zinc for 1 h increased the number of Et (+) neurons by 86.9% ± 6.8%. This increase was reduced to 32.4%+ 11.1% in cultures treated with 500 μmol/L apocynin, an NADPH oxidase assembly inhibitor, during zinc exposure (Figures 5A and 5B), and similarly reduced in neurons from p47phox−/− mice (Figures 5C and 5D).
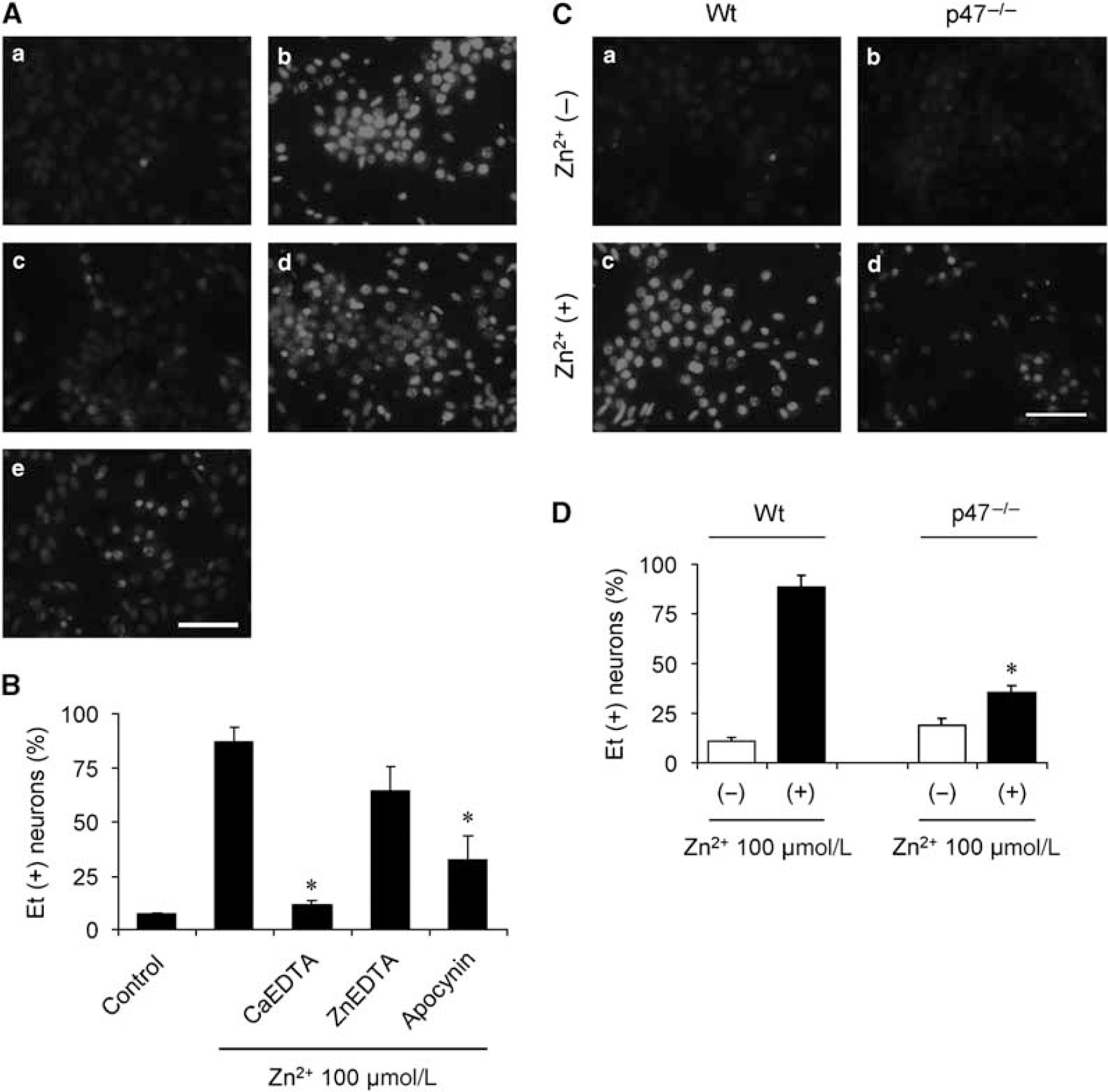
Inhibition of NADPH oxidase prevents zinc-induced ROS production in cultured cortical neurons. (A) Production of ROS in neurons detected by Et fluorescence. (
Superoxide can also be generated from several sites in mitochondria (Loschen et al, 1974; Starkov et al, 2004); however, neuronal superoxide production after zinc exposure was unaffected by inhibiting glucose-derived substrate delivery to mitochondria with 500 μmol/L of the glycolysis inhibitor iodoacetate. NADPH oxidase requires glucose for regeneration of NADPH by the hexose monophosphate shunt (Decoursey and Ligeti, 2005). In contrast to iodoacetate, incubation with 500 μmol/L 6-aminonicotinamide, an inhibitor of the hexose monophosphate shunt (Gupte et al, 2005), significantly blocked the superoxide production induced by zinc in cortical cell cultures (Figure 6).
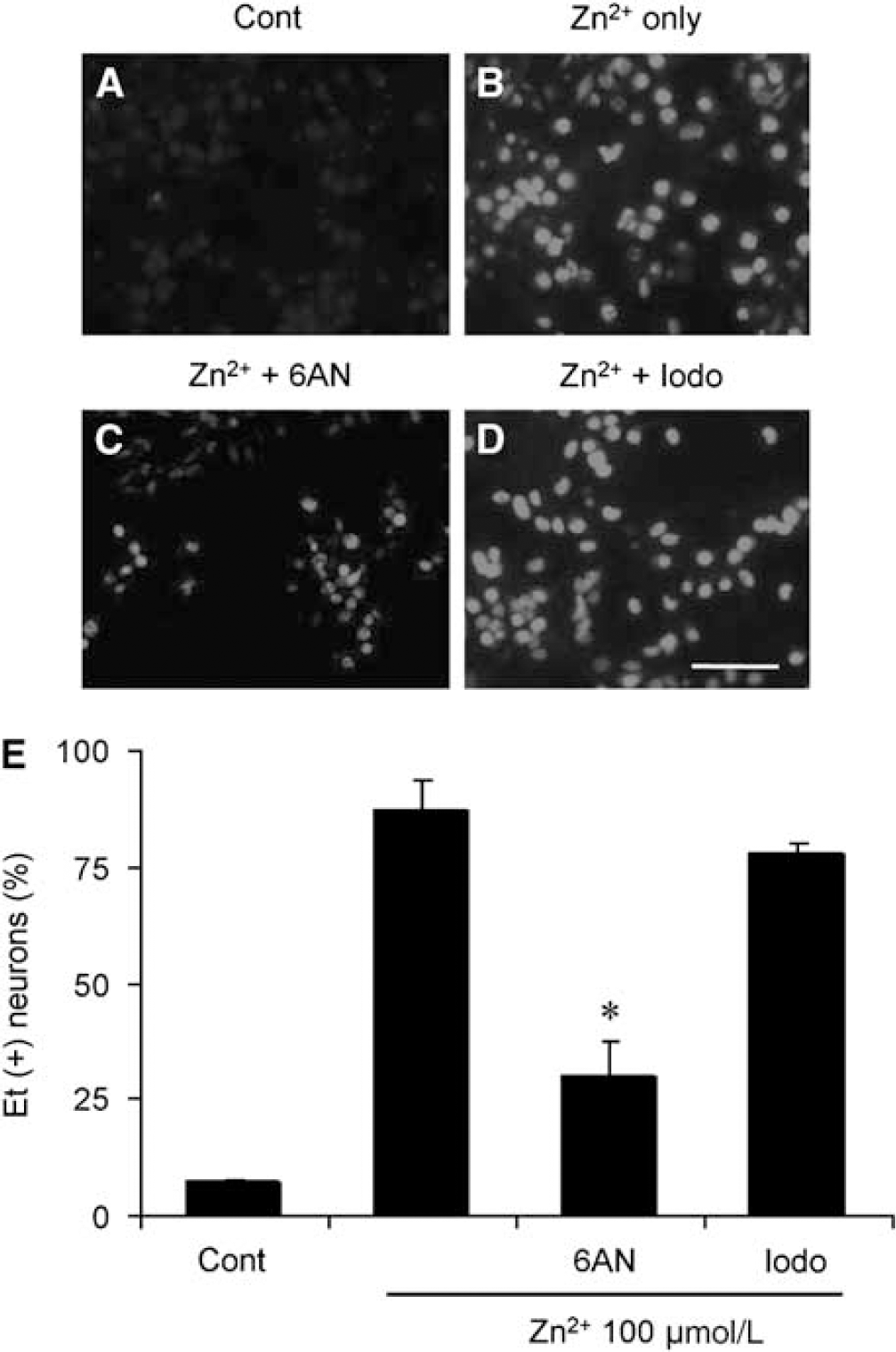
Production of ROS by cortical neurons was unaffected by the glycolytic inhibitor iodoacetate (lodo) but blocked by 6-aminonicotinamide (6AN). (
p47phox−/− Mice Have Similar Vesicular Zinc Content and Release Comparable with Wild-Type Mice, but Show Reduced Zinc Accumulation in Hippocampal CA1 Neurons 24 h After Hypoglycemia
To determine whether the reduced superoxide production in p47Phox−/− mice after hypoglycemia is because of decreased basal vesicular zinc concentration or decreased zinc release from the synaptic terminals, we compared TSQ staining in p47phox−/− and wild-type mice brains. The basal intensity of hippocampal mossy fiber zinc was almost identical in the sham model for both mice models (Figures 7A and 7B). Vesicular zinc release after hypoglycemia, as shown by loss of TSQ intensity, was also indistinguishable between these mice (Figures 7C and 7D). Three hours after hypoglycemia, TSQ intensity in the CA1 neuron was present in both mice models, as also seen in rat hypoglycemia (Figure 2). Evaluation of the TSQ intensity in the hippocampal CA1 neuron 24 h after hypoglycemia showed that the wild-type mice TSQ intensity was bright, indicating chelatable zinc fully accumulated in the neuron, whereas the p47phox−/− TSQ intensity was nearly the same as that seen at 3 h.
Discussion
This study presents three novel observations concerning hypoglycemia-induced neuronal death: vesicular zinc release is triggered by nitric oxide, vesicular zinc release activates neuronal NADPH oxidase, and activation of neuronal NADPH oxidase leads to intracellular PAPvP-1 activation, zinc accumulation, and subsequent neuronal death.
Superoxide production and zinc translocation are key steps in the sequence of events that lead to neuronal death after hypoglycemia (Suh et al, 2004, 2007a). In earlier studies of hypoglycemia in the rat, nitrotyrosine formation was detected shortly after glucose reperfusion, but not during hypoglycemia per se (Suh et al, 2003). Here, using the same rat model, the neuron-specific NOS inhibitor 7-NI was found to significantly inhibit hypoglycemia-induced vesicular zinc release from hippocampal hilus. It also prevented intracellular zinc accumulation and neuronal death at the 24 h after hypoglycemia time point. These findings suggest that nitric oxide production is upstream of vesicular zinc release and postsynaptic zinc accumulation. This observation is consistent with previous studies in which intrahippocampal injection of nitric oxide donor induced vesicular zinc release and intracellular zinc accumulation (Cuajungco and Lees, 1998; Frederickson et al, 2002).
As peroxynitrite is formed by the reaction of NO with superoxide (Beckman and Koppenol, 1996), we also sought to investigate the role of superoxide in presynaptic zinc release and postsynaptic zinc accumulation after hypoglycemia. Previous studies showed that overexpression of SOD-1 reduced hypoglycemia-induced neuronal death (Suh et al, 2007a). To determine whether the neuroprotective role of SOD-1 was because of reduced vesicular zinc release, SOD-1 transgenic rats were subjected to hypoglycemia. We found that SOD-1 overexpression had no effect on hypoglycemia-induced zinc release or on the initial zinc translocation into hippocampal postsynaptic neurons at 3 h, but that SOD-1 overexpression reduced neuronal death and neuronal accumulation of zinc assessed at 24 h after hypoglycemia. These results suggest that zinc release is upstream of superoxide production, but that superoxide production contributes to zinc accumulation in postsynaptic neurons.
Zinc in neurons is present both in synaptic vesicles and as a pool bound to protein sulfhydryl groups (Danscher and Rytter Norgaard, 1985). An earlier study suggests that nonvesicular zinc may be more important than vesicular zinc in promoting brain injury (Lee et al, 2000). Here we used the ZnT3−/− mouse, which has no vesicular zinc in the presynaptic terminals (Palmiter et al, 1996), to determine the role of vesicular zinc pool in hypoglycemia-induced superoxide production and neuronal death. The ZnT3−/− mice showed less superoxide production and less neuronal death after hypoglycemia. This result confirms earlier reports that zinc chelation prevents ROS production and neuronal death after hypoglycemia (Suh et al, 2004, 2007b) and strongly suggests that it is the vesicular zinc pool that contributes to neuronal demise in this setting.
The administration of PARP-1 inhibitors at the time of glucose reperfusion leads to a marked and long-lasting increase in neuronal survival (Suh et al, 2003), and zinc chelation prevents PARP-1 activation in the vulnerable hippocampal neurons after hypoglycemia (Suh et al, 2004). Evidence suggests that zinc activation of PARP-1 is mediated by ROS production (Kim et al, 1999; Kim and Koh, 2002; Suh et al, 2003; Sheline et al, 2003). In agreement with this idea, the present findings show that SOD-1 overexpression almost completely prevents hypoglycemia-induced accumulation of PAR in neurons. This suggests a sequence of events in which zinc induces superoxide production, which in turn leads to PARP-1 activation and cell death.
Recent studies aimed at determining the source of superoxide production in the neurons suggest that NADPH oxidase is a major source of superoxide production after hypoglycemia and in a cell culture model of ischemia—reperfusion (Suh et al, 2007a; Abramov et al, 2007). If so, and if zinc release is an intermediary event in this process, then zinc alone should induce NADPH oxidase in neurons. Here we confirmed that zinc-induced superoxide production in neuron cultures was almost completely blocked in cultures from mice deficient in the p47phox subunit of NADPH oxidase and in wild-type neurons treated with the NADPH oxidase inhibitor apocynin (Stolk et al, 1994). Zinc-induced ROS production was also blocked by the hexose monophosphate shunt inhibitor 6-aminonicotinamide, which limits the rate at which NADPH substrate can be formed (Gupte et al, 2006).
To exclude the possibility that the neuroprotective effect of p47phox−/− is because of reduced vesicular zinc content or reduced presynaptic zinc release, p47phox−/− mice were compared with wild-type mice. The results of these studies showed that p47phox−/− and wild-type mice did not differ in basal vesicular zinc contents, in hypoglycemia-induced zinc release from presynaptic terminals, or in the initial zinc translocation into hippocampal postsynaptic neurons.
Taken together, the present study suggests a sequence of events that lead to neuronal death after hypoglycemia. Glucose reperfusion initiates nitric oxide production, which leads to vesicular zinc release, which in turn activates neuronal NADPH oxidase. Reactive oxygen species produced by NADPH oxidase lead to increased zinc accumulation, PARP-1 activation, and resultant cell death. Undoubtedly, this sequence is not entirely linear, and earlier work suggests that crosstalk between these events is likely (Bossy-Wetzel et al, 2004). In addition, this sequence does not exclude other contributory mechanisms, particularly in regions of brain where there is less concentration of zinc compared with hippocampus. Nevertheless, this work for the fist time delineates an ordered sequence between the several events known to contribute to hypoglycemic neuronal death. This sequence may also be operative in ischemia and other conditions in which glutamate, zinc, and oxidative stress have been implicated.
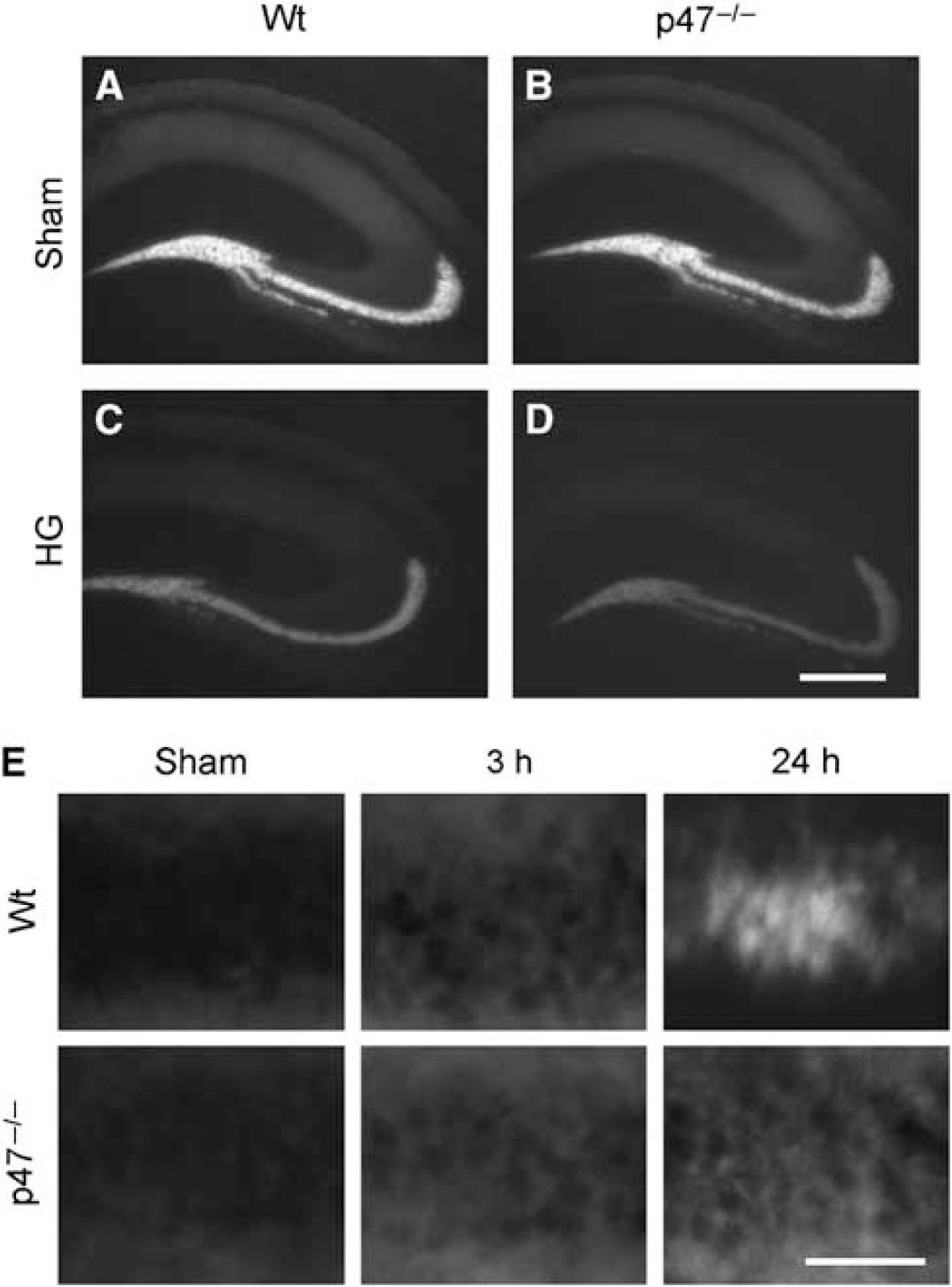
Vesicular zinc in the mouse hippocampal hilus imaged with TSQ fluorescence (white,