
Abstract
We sought to determine whether reactive oxygen species (ROS) derived from cyclooxygenase-2 (COX-2) are involved in ischemic brain injury. Focal cerebral ischemia was induced by transient middle cerebral artery occlusion in C57BL/6 mice. The time course of neocortical ROS production was assessed in vivo using hydroethidine as a marker. The same brain sections were used for infarct volume measurements. Transient middle cerebral artery occlusion led to a biphasic increase in ROS production with peaks 2 and 72 h after reperfusion. The COX-2 inhibitor NS398 (10 mg/kg) attenuated the production of COX-2-derived prostaglandin E2 and reduced brain injury, but did not affect ROS production at 2 and 72 h. Similarly, ROS production was not reduced in COX-2-null mice. In contrast, ROS production and brain injury were reduced in mice lacking the nox2 subunit of the superoxide-producing enzyme nicotinamide adenine dinucleotide phosphate (reduced form) oxidase. The data suggest that COX-2 is not a major source of oxygen radicals after cerebral ischemia and raise the possibility that other COX-2 reaction products, including prostanoids or nonoxygen-based radicals, mediate the COX-2-dependent component of the injury.
Introduction
Cyclooxygenase-2 (COX-2), the rate-limiting enzyme for prostanoid synthesis, has been implicated in the pathogenesis of a wide variety of neurologic diseases, including ischemic stroke (Iadecola and Gorelick, 2005; Minghetti, 2004). Cyclooxygenase-2 catalyzes the conversion of arachidonic acid to prostaglandin (PG) H2 (Simmons et al, 2004). Prostaglandin H2 is then converted into five prostanoids (PGE2, PGI2, PGD2, PGF2α, and thromboxane A2) by cell-specific synthases (Hata and Breyer, 2004). In addition to prostanoids, COX-2 also generates free radical species (Armstead et al, 1988; Chan and Fishman, 1980; Jiang et al, 2004; Kontos et al, 1980; Torres et al, 2004). Pharmacological inhibition or genetic inactivation of COX-2 attenuates the brain damage resulting for focal and global cerebral ischemia (Iadecola et al, 2001a; Nakayama et al, 1998; Nogawa et al, 1997; Sasaki et al, 2004). Furthermore, mice overexpressing COX-2 in the brain are more susceptible to the brain damage produced by focal cerebral ischemia (Dore et al, 2003). These observations have raised the possibility that COX-2 inhibitors could be valuable tools in the treatment of ischemic brain injury (Minghetti, 2004).
The therapeutic potential of COX-2 inhibitors has been recently challenged by clinical reports that long-term treatment with these agents increases the incidence of cardiovascular events, including myocardial infarction and stroke (Topol, 2004). The mechanisms underlying these complications have not been elucidated, but it has been suggested that COX-2 inhibitors downregulate PGI2 (prostacyclin) and upregulate thromboxane A2 resulting in a prothrombotic state (FitzGerald, 2003). Therefore, COX-2 activity is not uniformly deleterious and treatments based on inhibiting the COX-2 pathway have to focus on the mediators responsible for the deleterious effects of the enzyme sparing those mediating the beneficial actions (Iadecola and Gorelick, 2005; Rocca, 2006).
Unfortunately, the COX-2 reaction products mediating the neurotoxicity of COX-2 have not been defined. Although COX-2-derived prostanoids could mediate brain injury, COX-2-derived radicals can lead to oxidative stress, a major pathogenic factor in ischemic brain injury (Armstead et al, 1988; Chan, 2001; Torres et al, 2004). Recent evidence suggests that in models of glutamate neurotoxicity prostanoids, not oxidative stress, mediate the deleterious effects of COX-2 (Carlson, 2003; Kawano et al, 2006; Manabe et al, 2004). However, the mediators responsible for COX-2-dependent injury in models of cerebral ischemia have not been defined. In particular, COX-2 is markedly upregulated in inflammatory cells invading the postischemic brain, which are a major source of oxidative stress (Iadecola et al, 1999; Traystman et al, 1991). Therefore, COX-2-derived radical species could contribute to the toxicity exerted by postischemic inflammation.
In the present study, we used a mouse model of transient focal cerebral ischemia to examine the contribution of COX-2-derived radicals in the early and late phases of ischemic brain damage. The data indicate that although reactive oxygen species (ROS) play a pathogenic role in ischemic injury, COX-2 does not contribute to postischemic oxidative stress. Rather, a nox2-containing nicotinamide adenine dinucleotide phosphate (reduced form) (NADPH) oxidase is a major source of ROS produced in the late stage of cerebral ischemia.
Materials and methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Experiments were performed in C57BL/6 mice, COX-2-null mice, and mice lacking the gp91phox subunit of NADPH oxidase (nox2) (Morham et al, 1995; Pollock et al, 1995). All mice (weight 20 to 22 g) were male and were obtained from in-house colonies (Iadecola et al, 2001a; Park et al, 2004b). Cyclooxygenase-2 and nox2-null mice were congenic with the C57BL/6 strain. Wild-type littermates were used as controls in all experiments.
Transient Middle Cerebral Artery Occlusion
Procedures for middle cerebral artery (MCA) occlusion were identical to those described previously (Cho et al, 2005; Park et al, 2004a) and are only summarized. Mice were anesthetized with a mixture of isoflurane (2% to 2.5%), oxygen, and nitrogen. For continuous monitoring of cerebral blood flow in the center of the ischemic territory, a fiber optic probe was glued to the parietal bone (2 mm posterior and 5 mm lateral to the bregma) and connected to a laser-Doppler flowmeter (Periflux System 5010; Perimed, Järfälla, Sweden). For MCA occlusion, a heat-blunted 6-0 monofilament surgical thread was inserted into the exposed external carotid artery, advanced into the internal carotid artery and wedged into the Circle of Willis at the origin of the MCA. Obstruction of the MCA was confirmed by an immediate cerebral blood flow reduction. The monofilament was left in place for 25 mins and then withdrawn. Only mice with a cerebral blood flow reduction by >85% during MCA occlusion and a recovery to at least 80% of baseline cerebral blood flow within 10 mins of reperfusion were included in our study. This procedure leads to reproducible cortical and striatal infarcts similar in size to those reported by others using transient MCA occlusion of comparable duration (Borsello et al, 2003; Boutin et al, 2001; Plesnila et al, 2001). Rectal temperature was kept constant (37.0°C ±0.5°C) during the surgical procedure and in the recovery period until the animals regained consciousness.
Determination of Reactive Oxygen Species Production
Reactive oxygen species production was determined using in vivo hydroethidine microfluorography (Kondo et al, 1997), as previously described (Cho et al, 2005; Manabe et al, 2004). Hydroethidine is a cell-permeable dye that is oxidized to ethidium by superoxide (Bindokas et al, 1996). Ethidium is trapped intracellularly by intercalating with DNA (Rothe and Valet, 1990). The fluorescence signal attributable to ethidium reflects cumulative ROS production during the period between administration of hydroethidine and killing of the animal. Hydroethidine (10 mg/kg) was injected into the jugular vein in isoflurane-anesthetized mice 1 h before the animal is killed. Brains were removed, frozen, and cut in a cryostat (thickness 20 μm), collected at 600-μm intervals. The sections were analyzed with a Nikon E800 fluorescence microscope (Melville, NY, USA) equipped with a custom filter set (Chroma Technology, Rockingham, VT, USA). Images were acquired by a computer-controlled digital monochrome camera (Coolsnap; Roper Scientific, Trenton, NJ, USA) attached to the microscope. The analysis of ROS production was performed in a masked manner using IPLab software package (Scantalytics, Fairfax, VA, USA) (Cho et al, 2005; Manabe et al, 2004). After subtracting the camera dark current, pixel intensities of ethidium signals were assessed in the ischemic territory. Fluorescence intensities were measured in five serial sections per animal (rostrocaudal levels +1.6, +1.0, +0.4, −0.2, −0.8 mm from bregma). The sum of the fluorescence intensity for each region was divided by the total number of pixels analyzed and expressed as relative fluorescence units (Cho et al, 2005; Manabe et al, 2004). Reactive oxygen species production was assessed 1, 2, 4, 8, 24, 48, and 72 h after MCA occlusion (Figure 1A).
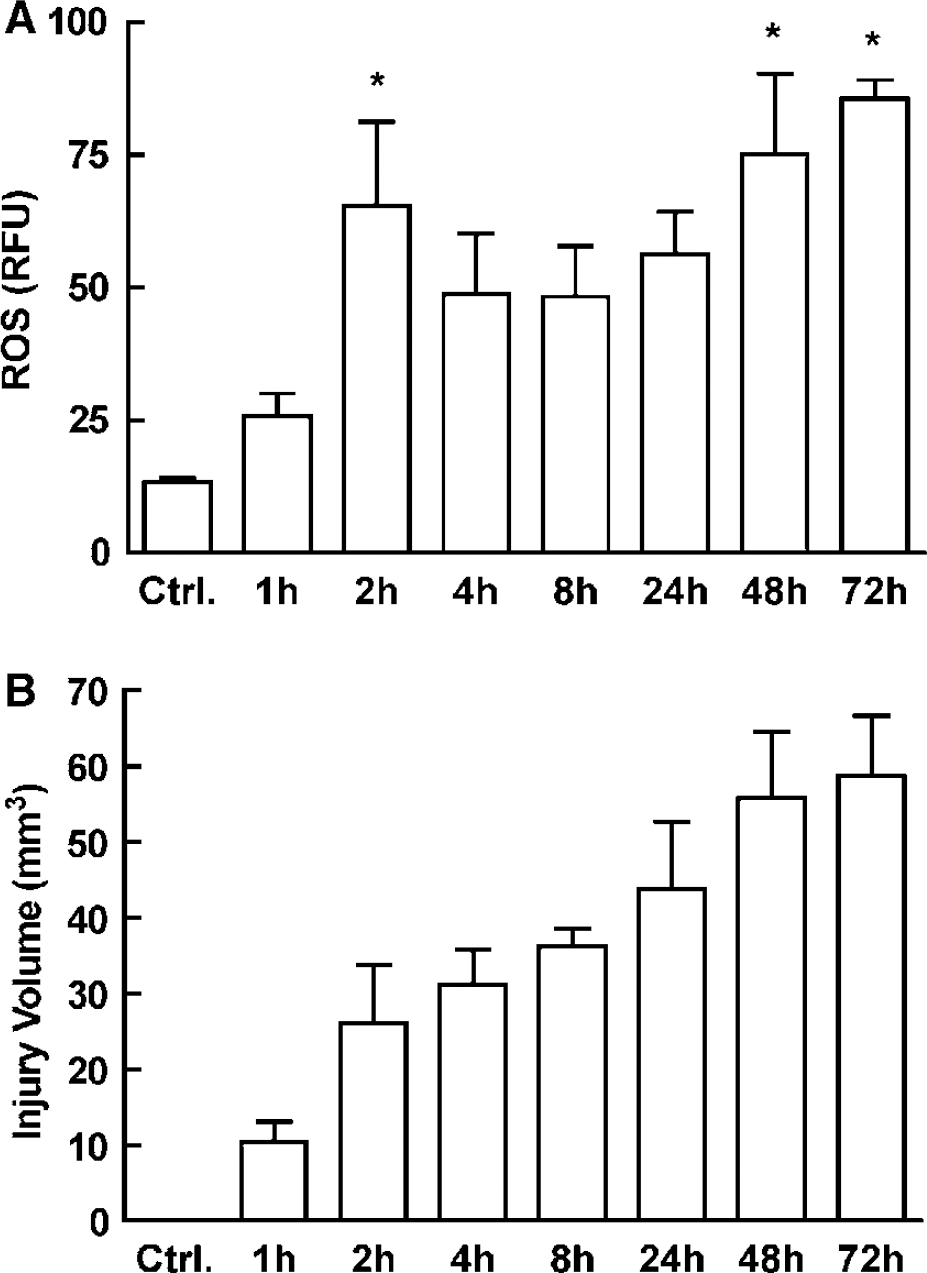
Temporal profiles of ROS production and tissue injury after MCA occlusion in C57BL/6 mice. (
Infarct Volume Measurement
Infarct volume measurements were performed using the same brain sections in which ROS production was assessed. After staining the sections with cresyl violet, the infarct volume was measured using an image analyzer (MCID; Imaging Research, St Catharines, Ontario, Canada). To eliminate the contribution of postischemic edema to the volume of injury, infarct volume measurements were corrected for swelling according to the method of Lin et al. (1993) as previously described (Zhang and Iadecola, 1994).
Treatment with NS398
Some mice were treated with the COX-2 inhibitor NS398 (10 mg/kg; ip) or vehicle (H2O, pH 12), as previously described (Sugimoto and Iadecola, 2003). Mice sacrificed 2 h after MCA occlusion received one injection of NS398 10 mins after reperfusion. Animals that survived for 72 h were treated 10 mins and 6 h after reperfusion, and thereafter twice daily at days 1 and 2, and once at day 3.
Prostaglandin Assay
PGE2 concentration in the ischemic tissue was measured 2 h and 72 h after MCA occlusion. These time points correspond to the peak of ROS production (Figure 1A). Samples of the ischemic neocortex were dissected, weighted, and frozen in liquid nitrogen. Samples were homogenized and centrifuged for 10 mins at 12,000 r.p.m. at 4°C. PGE2 concentration was measured in the supernatant using an enzyme-linked immunosorbent assay-based assay kit (Cayman Chemical Co., Ann Arbor, MI, USA) (Manabe et al, 2004; Nogawa et al, 1997). PGE2 concentration was expressed as ng/g of wet tissue. Although brain edema could lead to underestimation of the final PGE2 concentration, we have previously shown that NS398 does not influence brain edema (Nogawa et al, 1997).
Statistical Analysis
Data are presented as mean±s.e.m. Comparisons between two groups were statistically evaluated by the Student's t test. Multiple comparisons were evaluated by analysis of variance followed by Bonferroni's multiple comparison test. Differences were considered significant at P<0.05.
Results
Temporal Profile of Reactive Oxygen Species Production and Tissue Injury After Middle Cerebral Artery Occlusion
We first examined the time course of ROS production in the postischemic brain. Transient MCA occlusion increased ROS production in the affected hemisphere (Figure 1A). The increase was first observed 1 to 2 h after MCA occlusion (P<0.05). Reactive oxygen species production stabilized at a lower level between 4 and 24 h, and increased again 48 to 72 h after MCA occlusion (Figure 1A). Injury volume developed rapidly between 1 and 2 h after MCA occlusion, corresponding to the first peak of ROS production (Figures 1A and 1B). Thus, 2 h after MCA occlusion the volume of injury was 44% of the volume at 72 h (Figure 1B). Lesion volume rose steadily between 4 and 48 h after MCA occlusion, reaching a plateau at 48 to 72 h (Figure 1B).
Cyclooxygenase-2 Contribution to Reactive Oxygen Species Production 2 and 72 h After Middle Cerebral Artery Occlusion
We next sought to define the contribution of COX-2 to postischemic oxidative stress. Analyses were performed at 2 and 72 h because these times correspond to the peaks of ROS production (Figure 1A). Administration of NS398 reduced the postischemic elevation of the COX-2-derived prostanoid PGE2 (P<0.05; Figure 2A). The effect was associated with a reduction in infarct volume 72 h after MCA occlusion (P<0.05; Figure 2B). However, NS398 did not influence postischemic ROS production 2 or 72 h after MCA occlusion (Figures 2C and 2D). Similarly, infarct volume, but not ROS production, was attenuated in COX-2-null mice (Figures 2B, 2C and 2D).
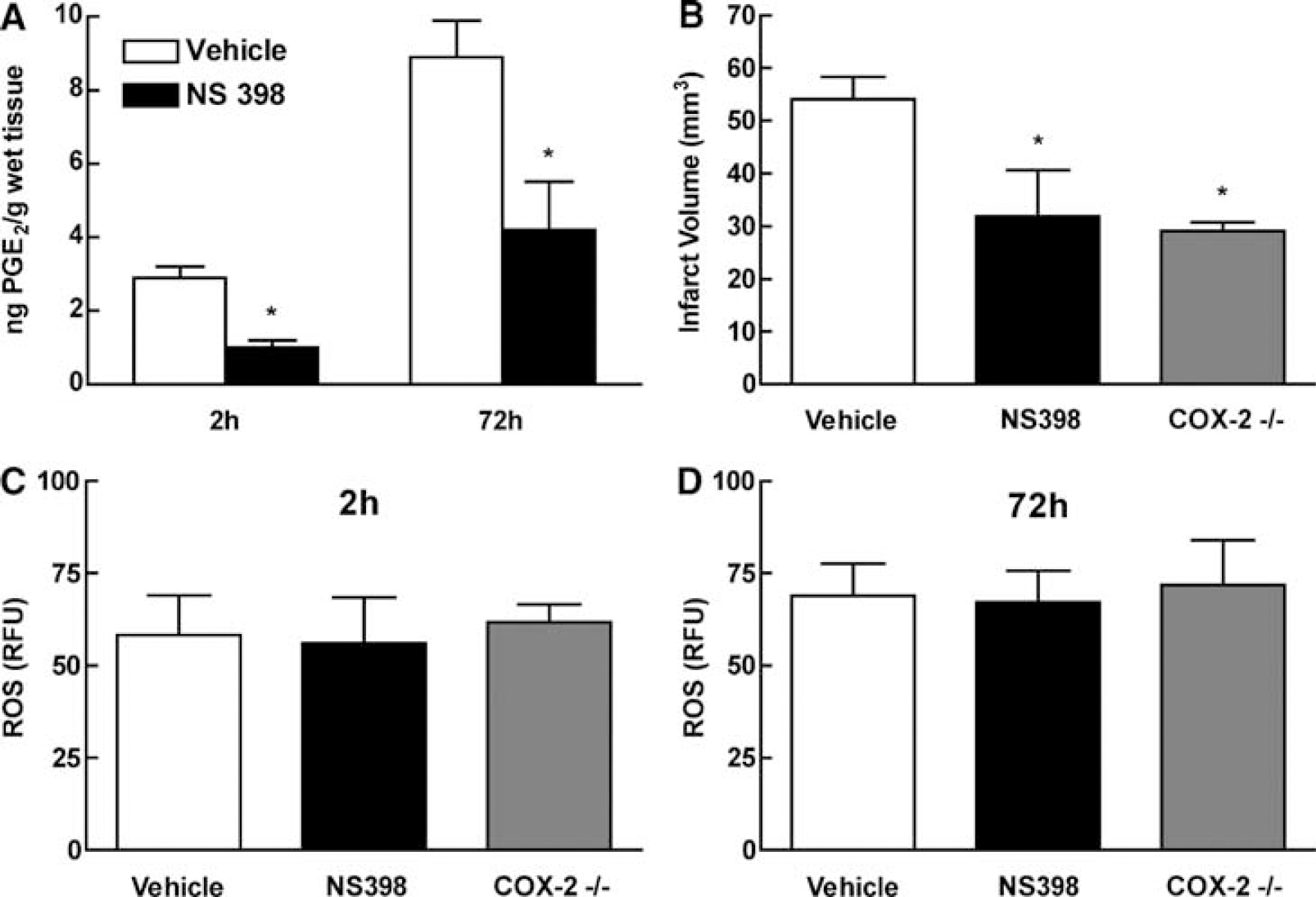
Cyclooxygenase-2 contribution to ROS production 2 and 72 h after MCA occlusion. (
Reactive Oxygen Species Production and Ischemic Brain Injury in nox2-null Mice
Nicotinamide adenine dinucleotide phosphate (reduced form) oxidase is a potential source of ROS in the late phase of cerebral ischemia (Park et al, 2004a; Vallet et al, 2005) and contributes to ischemic injury (Walder et al, 1997). Therefore, we used mice lacking nox2 to assess the contribution of NADPH oxidase to ROS production and ischemic injury 72 h after MCA occlusion. Nox2-null mice exhibited a 44% reduction in ROS (P<0.05) in the postischemic brain at 72 h compared with wild-type mice (Figure 3A). In addition, the infarct volume at 72 h in nox2-null mice was 61% (P<0.05) smaller than in wild-type mice (Figure 3B).
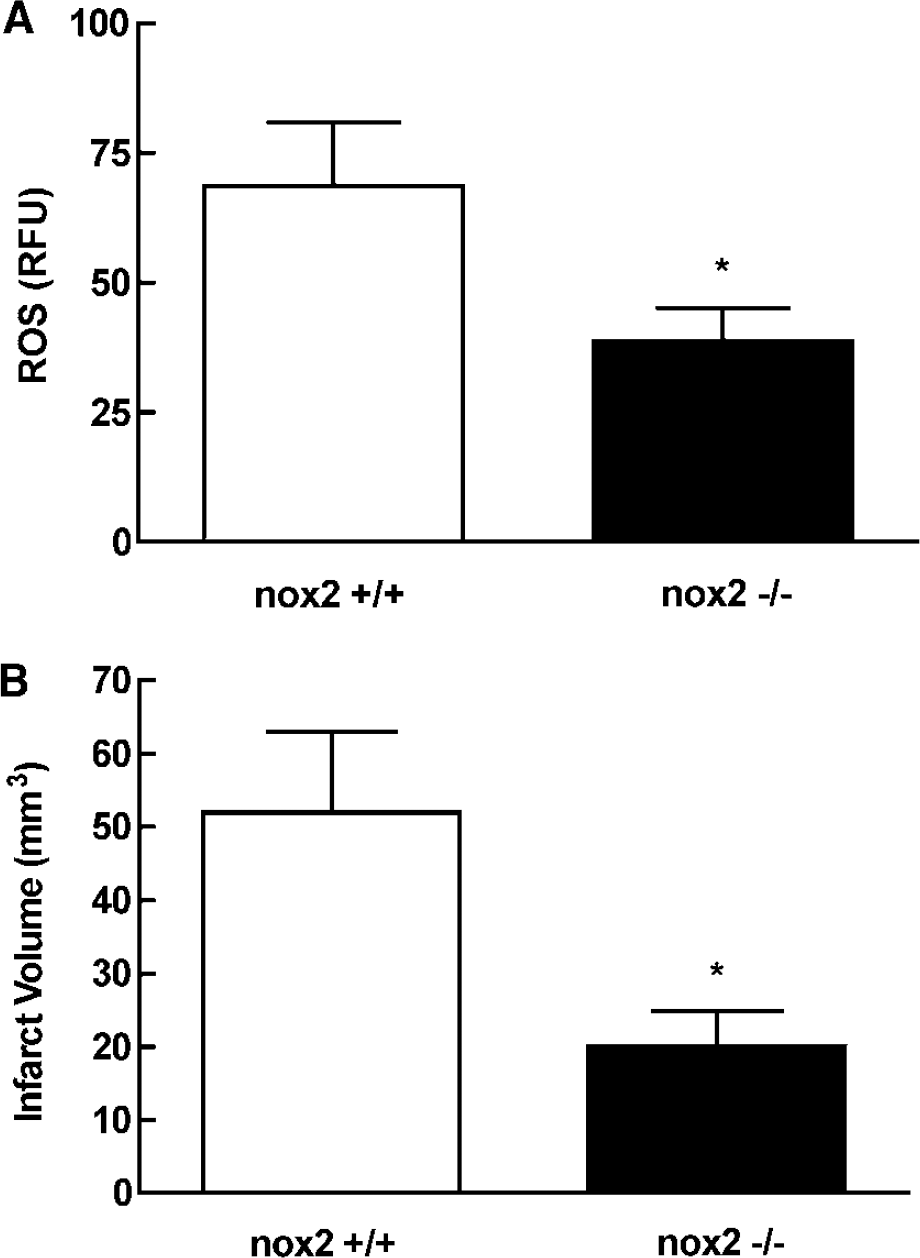
Reactive oxygen species production and ischemic brain injury in nox2-null mice 72 h after MCA occlusion. (
Discussion
It is well established that ROS are produced after cerebral ischemia and play a critical role in the resulting tissue damage (Chan, 2001). However, the sources of postischemic ROS have not been elucidated. Cyclooxygenase-2 plays a deleterious role in ischemic brain injury and is also a potential source of oxidative stress (Armstead et al, 1988; Chan and Fishman, 1980; Kontos et al, 1980; Minghetti, 2004). Therefore, we sought to determine whether COX-2-derived ROS contribute to the brain damage induced by transient focal ischemia. We found that MCA occlusion produced a marked increase in ROS production, which peaked 2 and 72 h after ischemia. Cyclooxygenase-2 inhibition with NS398 attenuated PGE2 production and reduced injury volume, but did not attenuate postischemic ROS production. Similarly, ROS production was not altered in COX-2-null mice. In contrast, ROS production and brain injury were reduced in mice lacking nox2. These data, collectively, demonstrate that COX-2 does not play a role in postischemic ROS production. In particular, COX-2 is not involved in the oxidative stress produced by inflammatory cells in the late stages of cerebral ischemia. Furthermore, the evidence suggests that a nox2-containing NADPH oxidase is a significant source of ROS in the late postischemic period.
The fact that ROS production was unchanged after treatment with NS398 cannot be attributed to insufficient inhibition of COX-2, because PGE2 production was attenuated and injury volume was reduced. Moreover, postischemic ROS production was not affected in null mice lacking COX-2. It is also unlikely that the increased fluorescent signal observed in the postischemic brain reflects oxidation of hydroethidine by factors other than ROS, because ROS production was attenuated in mice lacking nox2, in which NADPH oxidase-dependent ROS production is reduced (Park et al, 2004b). Cytochrome c released from mitochondria as a result of brain injury could also oxidate hydroethidine (Papapostolou et al, 2004). However, this is unlikely to be the case in our model because COX-2 inhibition or gene inactivation reduced the tissue damage, and presumably cytochrome c release, but did not affect the fluorescent signal. Therefore, the findings of the present study cannot be attributed to confounding effects related to the experimental preparation or the method used to assess ROS production.
The identity of the radical species produced by COX-2 has not been established. Studies in vivo suggest that COX activity can generate oxygen radicals, such as superoxide (Armstead et al, 1988; Chan and Fishman, 1980; Kontos et al, 1980; Torres et al, 2004). However, in vitro investigations have provided evidence that COX-2 activity is coupled to generation of carbon centered radicals that leads to lipid peroxidation (Jiang et al, 2004). Although our data suggest that COX-2 is not a source of postischemic oxygen radicals, we cannot rule out the participation of other radical species derived from COX-2.
To our knowledge, this is the first study to correlate the time course of postischemic ROS production with the maturation of cerebral infarction. Analysis of the temporal profile of postischemic ROS production revealed a biphasic pattern with an early peak at 2 h and a late increase beginning 48 h after MCA occlusion. The fact that the ROS peak at 2 h in parallel with rapid expansion of the lesion suggests a contribution of this early ROS burst to the evolution of ischemic brain injury. The slightly lower ROS production from 4 to 24 h could be related to the cell death resulting from the early ROS burst, whereas the second peak of ROS activity could result from the inflammatory reaction that involves the ischemic brain at this time (Iadecola et al, 2004).
Cyclooxygenase-2 contributes both to the early and late phases of ischemic brain injury. In the early phase, evidence suggests that the contribution of COX-2 is related to the neurotoxicity exerted by glutamate receptors, which are critical for the initiation of ischemic brain injury (Lo et al, 2003). Thus, COX-2 plays a role in the brain damage produced by activation of glutamate receptors (Carlson, 2003; Hewett et al, 2000; Iadecola et al, 2001a; Manabe et al, 2004). Our findings that COX-2-derived ROS do not contribute to the ROS production in the early phase of cerebral ischemia is consistent with our previous demonstration that pharmacological inhibition or genetic inactivation of COX-2 does not attenuate the ROS production induced by N-methyl-
The contribution of COX-2 to the late phase of ischemic damage is likely to be related to its role in postischemic inflammation. Thus, COX-2 is expressed in infiltrating inflammatory cells and delayed treatment with COX-2 inhibitors attenuates the injury (Iadecola et al, 1999; Miettinen et al, 1997; Sugimoto and Iadecola, 2003). The present study suggests that COX-2 does not contribute to the ROS production that occurs in the late phase of ischemic injury. Rather, our data clearly indicate that a nox2-containing NADPH oxidase is a substantial source of ROS and contributes to ischemic brain injury. Although the role of NADPH oxidase in ischemic brain injury was previously established (Walder et al, 1997), nox2-dependent ROS production in the late postischemic period had not been demonstrated. However, the fact that ROS production in nox2-null mice is not completely abolished suggests additional sources of ROS, including NADPH oxidases containing other nox homologues, mitochondrial enzymes, lipoxygenases, p450 enzymes, xanthine oxidase, as well as NOS uncoupling from substrate and cofactor depletion (Chan, 2001; Gottlieb, 2003; Munzel et al, 2005; Traystman et al, 1991; Vallet et al, 2005). Although COX-1 is also a potential source of radicals, inhibition of COX-1 enlarges the infarct produced by MCA occlusion, suggesting a protective role (Iadecola et al, 2001b). Therefore, it is unlikely that COX-1-derived radicals are a major factor in the tissue damage produced by ischemia.
There is increasing evidence that COX-2 exerts its neurotoxic effect through prostanoids, PGE2 in particular (Carlson, 2003; Kawano et al, 2006; Manabe et al, 2004). It has been shown that the COX-2-dependent component of excitotoxic brain injury is mediated by PGE2 via activation of EP1 receptors (Kawano et al, 2006). Activation of EP1 receptors disrupts intracellular Ca2+ homeostasis by impairing Na+–Ca2+ exchange and contributes to excitotoxic cell death (Kawano et al, 2006). However, it is unclear whether EP1 receptor-dependent mechanisms are also involved in the inflammatory component of the damage in which COX-2 also plays a role (Hata and Breyer, 2004). Irrespective of these mechanisms, the data presented here suggest that COX-2-derived ROS do not contribute to the late inflammatory phase of the injury. However, we cannot exclude the participation of other radical species derived from COX-2, such as carbon centered radicals, which can lead to lipid peroxidation (Jiang et al, 2004).
The COX-2 pathway is an important therapeutic target for acute ischemic stroke and other neurologic diseases (FitzGerald, 2003; Minghetti, 2004). However, recent reports that longterm treatment with COX-2 inhibitors increases the incidence of stroke and myocardial infarction have led to a reevaluation of the therapeutic potential of these drugs (Iadecola and Gorelick, 2005; Topol, 2004). These vascular complications have been attributed to the inhibition of the vasoprotective effects of COX-2-derived PGI2 (FitzGerald, 2003). Thus, therapeutic interventions based on the COX-2 pathway need to target the downstream effectors of the injury sparing the beneficial vascular actions of COX-2 (Rocca, 2006). We have shown here that COX-2-derived ROS do not contribute to ischemic damage. Although the data are consistent with the hypothesis that COX-2-derived prostanoids are important mediators of injury and a potential therapeutic target (Ahmad et al, 2006; Carlson, 2003; Kawano et al, 2006), we cannot rule out a pathogenic role of COX-2-derived carbon-centered radicals.
In conclusion, we have shown that focal cerebral ischemia is associated with a biphasic pattern of ROS production that correlates with the development of tissue damage. We found that COX-2 inhibition or genetic inactivation reduces ischemic brain injury without altering postischemic ROS production. Reactive oxygen species production and ischemic injury are attenuated in mice lacking the nox2 subunit of NAPDH oxidase. The findings suggest that COX-2 is not a major source of oxygen radicals after cerebral ischemia and support the hypothesis that other COX-2 reaction products, including prostanoids or nonoxygen-based radicals, mediate the COX-2-dependent component of the injury.