
Abstract
Respiratory burst activity of murine microglial cells was investigated in vitro under normoxic and hypoxic conditions with a chemoluminometric assay. Hypoxia for 24 hours reduced the release of extracellular reactive oxygen intermediates (ROIs), whereas reoxygenation increased the chemoluminescence more than sevenfold. Blockade of potassium channels inhibited the increase of oxidative burst after reoxygenation, indicating that potassium ions, which were increased in the supernatant of hypoxic microglial cells, were involved in this activation process. Also, blockade of voltage-gated calcium channels with nifedipine attenuated the increased release of ROIs. With fura-2 analysis, it was shown that the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase by potassium ions was mediated by calcium influx via voltage-gated calcium channels. Thus, influx of calcium ions through voltage-gated channels activates the NADPH oxidase in microglial cells during reoxygenation. By the increased production of ROIs, microglial cells may add to the reperfusion injury after ischemia in vivo.
Reactive oxygen metabolites have long been implicated in the development of brain lesions in reperfusion after cerebral ischemia (Siesjo et al., 1989). However, only recent advances in methodologies have allowed investigators to measure reactive oxygen metabolites directly, showing that reactive oxygen intermediates (ROIs) are released during reperfusion after cerebral ischemia (Kontos et al., 1992; Dirnagl et al., 1995) but not during ischemia. This was associated with neuronal death (Kitagawa et al., 1990) and an increased blood-brain barrier permeability after ischemia and added to the concept of reperfusion injury (Nelson et al., 1992).
A variety of enzyme systems generate reactive oxygen metabolites. In addition to the release from mitochondria or conversion of xanthine by xanthine oxidase, the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidase system on the surface of granulocytes and macrophages is the main source for ROIs in the extracellular space. Resident macrophages of the brain, the microglial cells, are representatives of the immune system in the central nervous system parenchyma (Graeber and Streit, 1990; Kreutzberg, 1996). Their most characteristic property is their ability to modify their behavior in response to diverse signals from other cells in a variety of experimental conditions and human diseases, both acute and chronic (Celada and Nathan, 1994; Banati et al., 1993). The transformation from a quiescent state into phagocytic brain macrophages is under strict control and is accompanied by the production of several secretory products. These include cytokines, excitatory amino acids, and ROIs (Spranger and Fontana, 1996; Piani et al., 1992; Colton et al., 1994b; Frei et al., 1987). The latter are released by the NADPH oxidase during the “oxidative or respiratory burst” (Rosen et al., 1995). Protein kinase C activators, such as phorbol 12-myristate 13-acetate (PMA) or opsonized yeast particles, which bind to complement receptors, increase the release of ROIs (Bastain and Hibbs, 1994; Baggiolini and Wymann, 1990). By this, they may contribute to microglial cytotoxicity in vivo under pathophysiologic conditions (Colton et al., 1994b; Thery et al., 1991). Toxicity of O2- stems mostly from its spontaneous conversion to hydrogen peroxide (H2O2), which gives rise to hydroxyl-free radicals (OH-) by the iron-catalyzed Haber—Weiss reaction. Hydroxyl radicals are known to initiate multiple cellular lesions, including membrane lipid peroxidations, DNA strand breaks, and protein alterations (Rosen et al., 1995), which may lead to cell death. Particularly in brain edema and ischemia-induced neuronal death, ROIs have been attributed a causative role (Werns and Lecches, 1990).
Therefore, we investigated the generation of ROIs and possible regulatory mechanisms in murine microglial cells in vitro after hypoxia and reoxygenation.
MATERIALS AND METHODS
Dulbecco's modified Eagle's medium, heat-inactivated fetal calf serum, and antibiotics were obtained from Life Technology (Eggenstein, Germany). All other chemicals were obtained from Sigma (Deisenhofen, Germany).
Cell culture
Cultures of microglial cells were prepared from newborn NMRI mice brain, as described previously (Piani et al., 1992). After mechanical dissociation of the tissue in Dulbecco's modified Eagle's medium and 20% fetal calf serum, primary cultures were kept in 75-cm2 culture flasks at 5% carbon dioxide (CO2) and 37°C for 2 weeks. Cells growing on top of a confluent cell layer were removed by vigorous shaking (800 rpm for 4–6 hours) and replated. Based on immunofluorescence studies using anti-C3b receptor antibodies (Mac-1, Sera Laboratory, Crawley Down, Great Britain), less than 95% of the cells in microglia cultures were of the macrophage lineage. The purified cells (106/mL) were incubated for 24 hours at 37°C and 5% CO2 in phenol-red free Dulbecco's modified Eagle's medium supplemented with 1% fetal calf serum and antibiotics. Hypoxic conditions were achieved by incubating the cells in a hypoxic incubator (Labotec, Göltingen, Germany) with 95% N2 and 5% CO2, which reduced the O2 concentration in the incubator to 1%. Microelectrode measurements revealed that the pericellular steady state of oxygen pressure (PO2) was reduced to 15 mm Hg as compared with 143 mm Hg in the normoxic control cells (Metzen et al., 1995). To obtain reoxygenation, the hypoxic medium was replaced by oxygenated medium. Replacement with medium that previously had been placed in a hypoxic atmosphere for 24 hours served as control to exclude an influence of the medium change per se. To investigate whether the chemoluminescence (CL) was caused by an intra- or extracellular ROI production, the cell cultures were treated with superoxide dismutase (50 ng/mL) and catalase (200 units/mL), which are unable to penetrate the cell membrane. To investigate the involvement of potassium and calcium ions in the activation of NADPH oxidase, potassium chloride (KC1; 25 mmol/L) was added for 5 minutes to microglial cell cultures. Blockade of potassium and calcium channels was assayed by adding tetraethyl ammonium and nifedipine at concentrations of 10 mmol/L and 1 mmol/L, respectively (all reagents from Sigma, Deisenhofen, Germany). Reactive oxygen intermediate-scavenging enzymes and ion channel blockers were added to the cells 10 minutes before reoxygenation.
Quantification of reactive oxygen intermediate generation
Activation of the microglial oxidative burst was measured as luminol-enhanced CL. The CL response was monitored with a chemoluminometer (Lumat LB 9501, Bertholt, Bad Wildbad, Germany) in the presence of 50 mmol/L luminol (Sigma, Deisenhofen, Germany). After baseline CL was recorded, the respiratory burst was triggered with PMA (1 mmol/L). At 1-minute intervals up to 30 minutes, the integrated counts were recorded. The peak counts per minute of microglial cells that were not pretreated was set as 100%.
Potassium and calcium measurement
Potassium content in the supernatant was measured electro-chemically (Chiron Diagnostic System, Schlebusch et al., 1994). For measurement of intracellular calcium, microglia were plated at low density 10 times 103/cm2 on glass coverslips. After loading the cells with Fura 2 acetoxymethylester (1 mmol, Boehringer Mannheim, Mannheim, Germany) added to the medium for 30 minutes at 37°C, they were washed twice in a buffer containing 137.6 mmol sodium chloride (NaCl), 1 mmol/L sodium bicarbonate (NaHCO3), 0.34 mmol/L Na2HPO4, 0.44 mmol KH2PO4, 5.36 mmol/L KC1, 1.25 mmol/L calcium chloride (CaCl2), 5 mmol/L Hepes, 22.2 mmol/L glucose, pH 7.4. The coverslips were placed in a superfusion chamber and challenged by addition of 25 mmol/L KC1 with or without nifedipine (1 mmol/L), a blocker of voltage-gated calcium channels. Fluorescence was monitored as previously described (Nobiling and Bührle, 1989) using a microspectrofluorimeter (Ionoquant, Heidelberg, Germany). Signals were taken from single cells that were excited alternately at 340- and 380-nm wavelengths. Emission at 517 nm was registered by a photomultiplier and simultaneously recorded digitally. Changes in intracellular calcium are shown as the ratio of photon counts for the excitation wavelengths 340 nm to 380 nm.
Statistical analysis
Data are displayed as mean values±standard deviation. The statistical comparison of differently treated cells were performed by means of the Mann—Whitney U nonparametric statistic. The difference was considered significant if corresponding P values were less than 0.05.
RESULTS
Microglial cells that were kept in a hypoxic atmosphere for 24 hours released significantly less ROIs as measured by luminol-enhanced CL compared with normoxic control cultures (Fig. 1). However, when these cells were reoxygenated by the replacement of oxygenated medium, the CL measured after induction of the NADPH oxidase with PMA was increased more than sevenfold. This was not caused by the mere change of medium, which may activate microglial cells, because the replacement with hypoxic medium did not increase the oxidative burst. The measured CL was caused by an increase in extracellular ROIs because superoxide dismutase and catalase almost completely abolished the CL (Fig. 1).
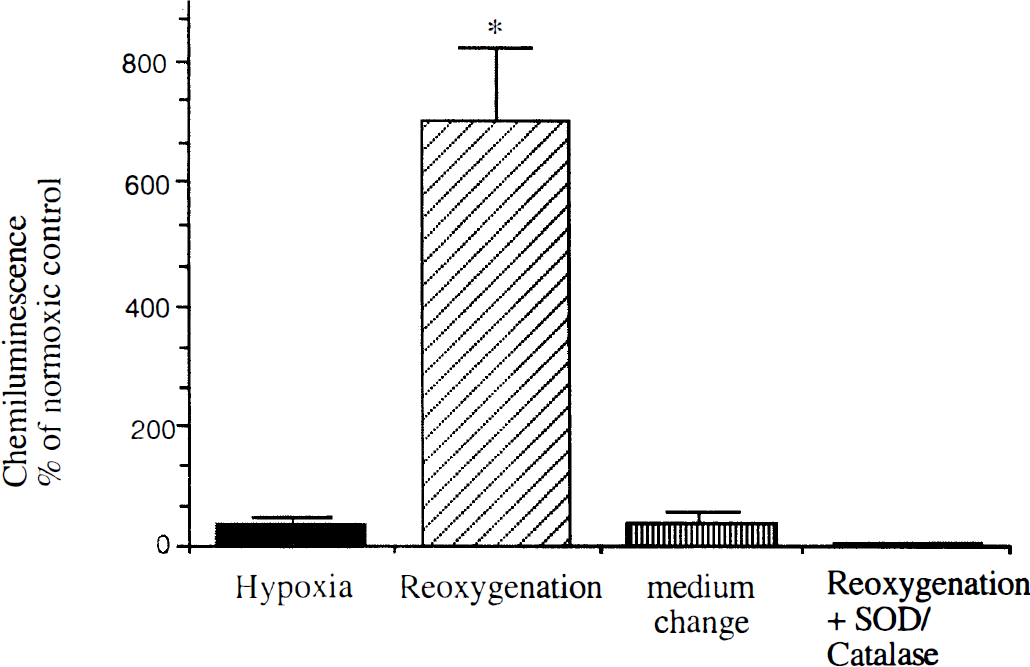
Reoxygenation increases the release of reactive oxygen intermediates (ROIs) from murine microglial cells. After incubating the cells for 24 hours in an atmosphere with 1% oxygen (O2), they were reoxygenated by the change to oxygenated medium. Oxidative burst was induced by 1 mmol phorbol 12-myristate 13-acetate (PMA), and luminol-enhanced chemoluminescence was measured. Hypoxia without change to reoxygenated medium or change to hypoxic medium rather inhibited the release of ROIs. Chemoluminescence was blocked almost completely by extracellular superoxide dismutase (50 ng/mL) and catalase (200 units/mL), which indicated the presence of extracellular ROIs after reoxygenation. The data represent the arithmetical mean and standard deviation of the increase in percent compared with untreated control cells under normoxic conditions in at least five independent experiments.* P < 0.002 of reoxygenated cells versus control.
We further investigated possible pathways by which the NADPH oxidase was activated during reoxygenation. In our in vitro model of hypoxia/reoxygenation, we observed elevated levels of potassium ions in the supernatant of microglial cells after 24 hours of hypoxia (16.38±0.11 mmol vs. 5.51±0.05 mmol in normoxic parallel cultures). Furthermore, KC1 (25 mmol/L) added to microglial cells cultivated under normoxic conditions increased CL more than twofold over untreated control cells (Fig. 2). Blockade of potassium channels with tetraethyl ammonium inhibited the induction of the NADPH oxidase by increased extracellular potassium and by reoxygenation.
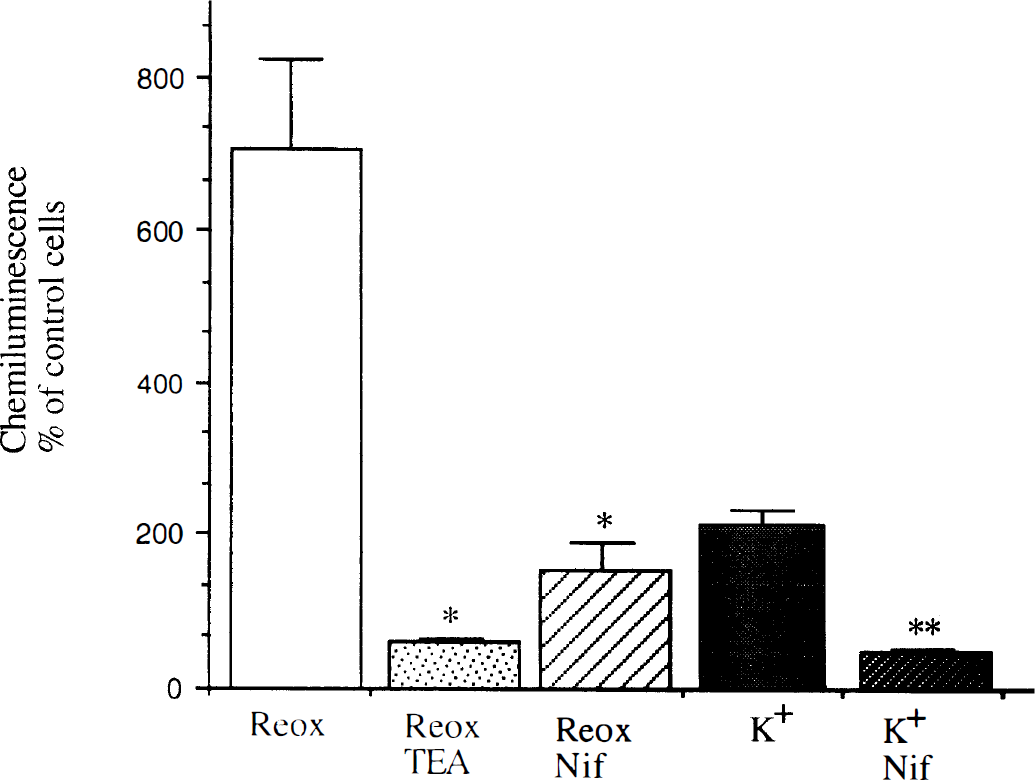
Potassium and calcium ion channels are involved in the triggering of the oxidative burst by reoxygenation in murine microglial cells. The potassium channel blocker tetraethyl ammonium (10 mmol) and nifedipine (Nif; 1 mmol), a blocker of voltage-dependent calcium ion channels, reduced the induction of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase by reoxygenation. Treating the cells with 25 mmol potassium chloride (KCI) for 5 minutes under normoxic conditions increased the oxidative burst after activation with 1 mmol phorbol 12-myristate 13-acetate (PMA) more than twofold, which could be inhibited by the presence of nifedipine. The data represent the arithmetical mean and standard deviation of the increase in percent compared with untreated control cells under normoxic conditions in at least three independent experiments. * P < 0.02 of reoxygenated cells treated with tetraethyl ammonium or nifedipine vs. untreated reoxygenated cells; ** P < 0.025 of cells treated with potassium and nifedipine versus cells treated with potassium alone.
After loading microglial cells with Fura 2, we observed an increase in intracellular calcium after stimulating the cells with potassium, which could be blocked by nifedipine (Fig. 3). This correlated well with the inhibition of the potassium-induced induction of respiratory burst by nifedipine (Fig. 2). The functional role of voltage-dependent calcium channels in the activation process of the NADPH oxidase during reoxygenation was indicated by the finding that nifedipine also attenuated the increased release of ROIs after reoxygenation (Fig. 2).
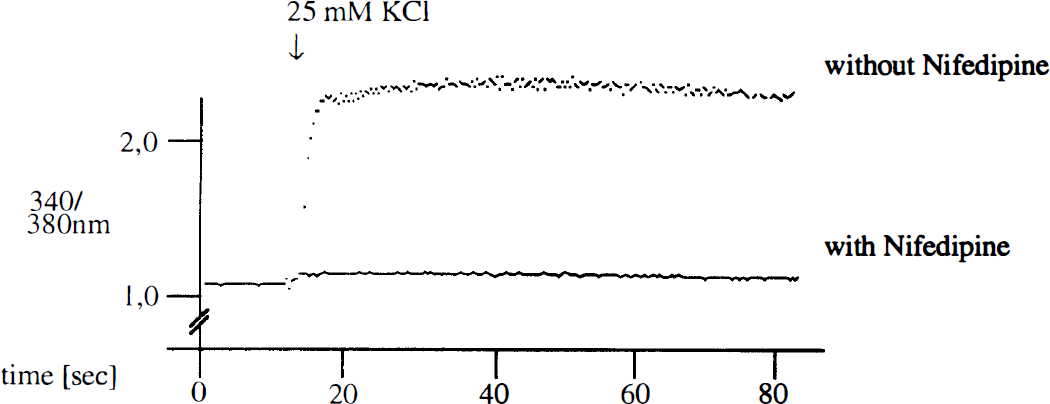
After loading murine microglial cells with 1 mmol Fura-2/acetoxymethylester, they were treated with 25 mmol/L potassium chloride (KCI) in the presence or absence of 1 mmol nifedipine, and intracellular free calcium was measured in a magnesium-free buffer. Intracellular calcium is given as the ratio of light emission at the excitation wave length of calcium-dye complex and free dye. The figure shows representative curves of seven independent experiments, which indicate that the increase of intracellular calcium after potassium treatment could be blocked by nifedipine.
DISCUSSION
The release of ROIs by murine microglial cells was reduced under hypoxic conditions and increased by reoxygenation. The activation of NADPH oxidase was shown to be responsible for the increased release of superoxide anion after reoxygenation in a cell-free system and in a number of cell types, including endothelial cells, hepatic Kupffer cells, and polymorphonuclear neutrophils (Zulueta et al., 1995; Gyenes and deGroot, 1993; Simms et al., 1996; Derevianko et al., 1996). Furthermore, the generation of superoxide anions in leukocytes was associated with other indicators of cellular activation, such as upregulation of cytokine receptors (Simms et al, 1996) and increased production of cytokines (Simms and D'Amico, 1996). This increased production of ROIs and proinflammatory cytokines from endothelial cells and granulocytes may account for the destruction of the blood—brain barrier. Microglial cells are derived from perinatally invading monocytes and form a network in the parenchymal white and gray matter of the adult brain (Graeber and Streit, 1990). On a variety of lesions, including ischemia, these resting cells are reactivated and are believed to play a major role in the pathogenesis of both acute and degenerative neurologic diseases (Spranger and Fontana, 1996; Kreutzberg, 1996; Banati et al., 1993) The release of ROIs after reoxygenation may be responsible for the neuronal death in reperfusion injury.
Because extracellular potassium levels are known to rise rapidly in ischemic brain tissue because of the failure of the energy consuming adenosine triphosphate-dependent sodium—potassium pump, we examined the possibility that extracellular potassium mediates changes of microglial function. In pilot experiments, we found elevated potassium concentrations in our hypoxic tissue cultures and confirmed previous reports that extracellular potassium increases microglial superoxide production (Colton et al., 1994b). From whole cell patch clamp experiments, it is well known that microglial cells possess inwardly rectifying potassium ion channels (Kettenmann et al., 1993). In a human lung adenocarcinoma cell line, it was shown that these potassium ion channels are involved in the activation of the NADPH oxidase (Koong et al., 1993). In this report, we could demonstrate that tetraethyl ammonium-sensitive delayed rectifier K+ channels also are involved in the activation of the NADPH oxidase by reoxygenation.
The electrophysiologic behavior of microglial cells is distinct from other cells because they express an inward rectifying K+ channel but no outward currents (Kettenmann et al., 1990). Thus, even small inward currents lead to a large membrane depolarization because K+ outward currents are not activated with depolarization. Kettenmann and coworkers (1993) showed that this depolarization triggers calcium influx. Intracellular calcium can be increased by liberation from intracellular stores or by transmembrane influx through voltage-dependent or ligand-regulated calcium channels. Several groups reported the presence of voltage-dependent calcium channels on microglial cells (Norenberg et al., 1994, Colton et al., 1994a). In keeping with these observations, we detected an increase of intracellular calcium in microglial cells after challenge with high extracellular potassium. Additionally, we showed that influx of calcium ions triggered the activation of the NADPH oxidase in microglia during hypoxia/reoxygenation. The involvement of voltage-dependent calcium channels was indicated by the blocking effect of nifedipine. The modulation of the NADPH oxidase by intracellular calcium ion concentration was shown in another intraparenchymal derivative of monocytes, the hepatic Kupffer cells. In these cells, hypoxia/reoxygenation induced prostaglandin production and superoxide anion release, which were preceded by an increase in intracellular calcium (Gyenes and deGroot, 1993). The reduction of ROI release by blockade of voltage-dependent calcium channels also was observed in vivo. A significant decrease in oxidative metabolism, as evidenced by decreased CL response and subnormal phagocytic cell responses, was recorded in peritoneal macrophages and neutrophils from nifedipine-treated mice compared with control animals (Kalra et al., 1993). A positive correlation between the increase in calcium levels and the CL response of macrophages and neutrophils was observed.
In conclusion, we observed an increased release of ROIs by murine microglial cells after reoxygenation, which could be attenuated by inhibition of potassium channels and voltage-dependent calcium channels. This finding suggests that an elevated extracellular potassium concentration—as it occurs in ischemic brain tissue—activates voltage-dependent calcium channels in microglial cells. The following increase of intracellular calcium concentration potentiates the activation of NADPH oxidase via protein kinase C after PMA treatment. This generation of ROIs from activated microglial cells might augment reperfusion injury in vivo after an ischemic brain lesion.
Footnotes
Acknowledgment
The authors thank Carmen Walter for her expert technical assistance.