
Abstract
Activin A plays an essential role in ischemic stroke as a well-known neuroprotective factor. We previously reported that Activin A could promote white matter remyelination. However, the exact molecular mechanism of Activin A in neuronal protection post-stroke is still unclear. In this study, the middle cerebral artery occlusion/reperfusion (MCAO/R)-induced ischemic stroke mouse model and oxygen-glucose deprivation/reoxygenation (OGD/R)-treated primary neurons were used to explore the molecular mechanism of Activin A-mediated neuroprotection against ischemic injuries. We found that Activin A significantly inhibits cGAS-STING-mediated excessive autophagy through the PI3K-PKB pathway, but not mTOR-dependent autophagy. Consequently, Activin A protected neurons against OGD/R-induced ischemic injury and improved cell survival in a dose-dependent manner. In addition, Activin A improved neurological functions and reduced infarct size of mice with MCAO/R-induced ischemic stroke by inhibiting autophagy. Furthermore, Activin A depended on ACVR1C receptor to exert neuroprotective effects in 1 h MCAO/R treated mice. Our findings showed that Activin A alleviated neuronal ischemic injury through inhibiting cGAS-STING-mediated excessive autophagy in mice with ischemic stroke, which may suggest a potential therapeutic target for ischemic stroke.
Introduction
Stroke is a major public health concern and affects more than 13.7 million people each year. Ischemic stroke, which accounts for approximately 70% of cerebrovascular accidents, is due to brain vessel occlusion and blockage. 1 Although recanalization is the most important treatment with a narrow therapeutic time window, many ischemic patients miss the opportunity for thrombolytic treatment.2,3 Finding new treatments for ischemic stroke is, therefore, of vital importance. Ischemic injury promotes autophagosome formation and the activation of autophagy to play neuroprotective role.4–6 However, excessive neuronal autophagy may cause axonal degeneration, ubiquitination, protein aggregation 7 and even neuronal cell death.8,9
Activin A, a member of the transforming growth factor β (TGF-β) superfamily of growth and differentiation factors, promotes and maintains the survival of cortical neurons and protects neurons from neurotoxicity.10–12 In a mouse model of ischemic stroke, exogenous Activin A, administered prior to transient focal ischemia or after reperfusion, reduces infarct volume and improves neurological functions. 13 The canonical autophagy regulatory pathway is phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB),14,15 and Activin A could regulate protein synthesis by modulating autophagic activity. 16 However, the molecular mechanism that Activin A protects neurons against ischemia/reperfusion injury through regulating autophagy is unclear.
Cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS)-adaptor protein stimulator of interferon genes (STING)-mediated induction of autophagy is the primordial function of the cGAS-STING pathway. 17 cGAS-STING pathway is also a potential therapeutic target for protection against neuronal cell death following hypoxia-tenderness in mice.18,19 In this study, we used the models of middle cerebral artery occlusion/reperfusion (MCAO/R)-induced ischemic stroke and oxygen-glucose deprivation/reperfusion (OGD/R)-induced neuronal ischemic injury to understand whether Activin A could alleviate ischemic injury through inhibiting cGAS-STING–mediated excessive autophagy in mice with ischemic stroke.
Materials and methods
Animals and ethical approval
Adult male C57BL/6J mice (at age of 6–8-weeks old; weighing 20–24 g) were purchased from the Jackson Laboratory (Bar Harbor, Maine 04609, USA). The mice were housed at the Experimental Animal Center of Capital Medical University (PR China) under appropriate rooms. All procedures were conducted according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Experimental Animal Ethics Committee of Capital Medical University (AEEI-2020-144). All experiments were reported in compliance with the ARRIVE guidelines 2.0. 20
Mouse model of MCAO/R-induced ischemic stroke, drug administration and experimental groups
The mouse model of MCAO/R was performed as previously described.21,22 Briefly, mice were anesthetized with sodium pentobarbital (70 mg/kg, i.p.). The left common carotid artery (CCA), the external carotid artery (ECA) and the internal carotid artery (ICA) were surgically separated and exposed via a ventral midline incision. The ECA and the CCA were ligated, and the ICA was clamped using microvascular clips. Next, a silicone-coated nylon monofilament (0.23 ± 0.02 mm in diameter, 3.0 cm in length, RWD Life Science, China) was inserted into the ICA through a small incision into the ECA about 11.0-12.0 mm. One hour occlusion later, the suture was carefully withdrawn; the ECA was ligated to loosen the temporary ligation of the CCA to restore blood flow. The PeriFlux 5000 System (Perimed, Stockholm, Sweden) was used to monitor cerebral blood flow during surgery. The local cerebral blood flow (CBF) was reduced to less than 20% in intraoperative mice after MCAO; CBF was completely recovered 1 h after the occlusion. The sham mice underwent a similar procedure, except that nylon monofilaments were not inserted.
The recombinant mouse (rm) Activin A (2.5 μg/kg) or vehicle (phosphate-buffered solution, PBS) and NU7441 (5 μM) were administered 6 h after MCAO/R through intracerebroventricular (ICV) injection as previously described. 13 Briefly, mice were anesthetized and stereotaxically injected with rmActivin A according to the following coordinates: 0.5 mm posterior, 1.0 mm lateral to the anterior fontanelle and 3.2 mm below the craniofacial surface.
Measurement of infarct volume
To determine the postoperative infarct volume of mice with ischemic stroke, the brain slices were observed at different time points post-reperfusion. Brain samples were separated on ice and 1.0 mm-thick coronal sections were prepared. The brain slices were dyed in triphenyl tetrazolium chloride (TTC, Sigma-Aldrich, St. Louis, MO 63105, USA) solution for 30 min. Brain slices were then removed and neatly placed on slides for scanning and imaging. According to previous reports, 21,22 the infarct volumes were determined as follows: brain edema rate (S) = (∑LT − ∑RT)/(∑LT + ∑RT) × 100%, ∑LT on behalf of the left hemisphere (ischemic hemisphere), ∑RT on behalf of the right hemisphere (non-ischemic hemisphere) volume; the background (B) = TTC unstained white matter/total brain volume in the sham group × 100%. Considering the influence of edema and background, the infarct volume is expressed as a percentage according to the following formula: I = ∑SIN(1−S)/(∑LT + ∑RT)(1−B) × 100%, where ∑SIN(1−S) represents the total infarct volume minus the edema rate.
Determination of neurological function
To determine the effects of rmActivin A on neurological outcomes of mice with ischemic stroke, we evaluated the neurological function as our previous reports.22,23 The 1 h MCAO/R 24 h post reperfusion mice were tested for neurobehavioral score according to the neurological disability status scale reported by Rodriguez et al. 24 The scoring rules are as follows: 0 points: no neurological dysfunction; 2 points: mild motor dysfunction, showing a reduction in mild to moderate motor movement, kyphosis or dragging gait; 4 points: moderate motor dysfunction, manifested as hair standing up, decreased body tension, and decreased muscle strength; 6 points: significant motor dysfunction, which may be manifested as hovering, tremor, convulsion or forelimb flexion; 8 points: severe dyskinesia, which may be complete inability to move, with or without respiratory distress; and 10 points: death. The mice were scored three times at each time point with an interval of five minutes, and the average of the three times was the final score.
Mice were trained on the balance beam prior to MCAO/R surgery. After 1 h MCAO/R 24 h, the animals were pushed to traverse on a balance beam 120 cm long, 0.7 cm wide and 40 cm away from the ground. By gently pinching the tail tip, mice were slowly pushed to traverse the balance beam. The balance beam test was repeated at different time points of reperfusion. This test was performed three times daily, and the average score from those three times was taken as the final score. The scoring criteria were 0 points: the mouse could not stay on the balance beam; 1 points: the mouse was motionless on the balance beam; 2 points: mice can walk on the balance beam but fall in the middle; 3 points: the mice can pass the balance beam test, but the number of times sliding down the balance beam on the opposite forelimb was more than 50%; 4 points: the mice could pass the balance beam test, but the sliding times were less than 50% of the steps taken; 5 points: the mouse could pass the balance beam, but it slipped once; and 6 points: no sliding.
Longa score was used to assess the neurological deficits of mice subjected to 1 h MCAO/R 24 h. The criteria were as follows: 0 points: no neurological dysfunction, manifested as no symptoms; 1 points: the contralateral forelimb cannot be fully extended; 2 points: unable to extend the contralateral forelimb; 3 points: slightly rotate the body to the opposite side; 4 points: severe body rotation to the opposite side; and 5 points: fall on the opposite side. Mice were scored three times at each time point, with an interval of five minutes. The average score from those three times was taken as the final score.
To evaluate the motor coordination, we trained mice for three days before MCAO/R. The 1 h MCAO/R 24 h-treated mice were placed on an automatic accelerating rod (LE8200, Panlab Harvard Apparatus, Holliston, Massachusett, 01746, USA) with an initial speed of 4 rpm that was then accelerated to 30 rpm for a maximum of 300 s. The mice were trained three times daily, with an interval of at least five minutes. The average time spent on the rod was recorded.
Primary cortical neuron culture
Suckling C57BL/6J mice, born within 24 h, were decapitated under aseptic conditions. Following immersion in 75% ethanol, the whole brain was dissected, and the cortex was cut into 0.2 cm3 fragments after the pia mater and blood vessels were removed, and 0.25% Trypsin (25200-056, Gibco, Grand Island, NY 14072, USA) was added to digest the tissue. Finally, dulbecco's modified eagle medium (DMEM, 11966025, Gibco) containing 10% horse serum (16050-122, Gibco), 10% fetal bovine serum (10099-141, Gibco), 1% penicillin-streptomycin (15070063, Life Technologies, Carlsbad, ON, Canada) and 0.25% L-glutamine (25030-081, Gibco) were used to grow neurons in 6-well plates at a density of 1 × 106 cells/well. Four hours later, the medium was changed to Neurobasal medium containing B27 (17504-044, Gibco). The medium was then changed every three days. The corresponding experimental treatments were delivered on day-in-vitro (DIV)-7 when the neurons were in optimal condition. Related reagents were as follows: Recombinant mouse Activin A (338-AC, R&D System, Minneapolis, Minnesota, 55401, USA), NU7441, GSK690693 and RU.521 (MedChemExpress, Monmouth Junction, NJ 08852, USA), 2′,3′-cGAMP (Invitrogen, Waltham, MA 02454, USA).
OGD/R treatment of primary cortical neurons
The oxygen-glucose deprivation/reoxygenation (OGD/R) model was established by using primary cultured neurons on day 7 in vitro (DIV-7). The cells were treated with glucose-free DMEM, and then were immediately placed in anoxic incubators containing the gas mixture of 5% CO2/2% O2/93% N2 for 1 h (27310, Stemcell, Canada) to simulate ischemia in vitro. To simulate reperfusion therapy, Neurobasal medium containing 2% B27 was added to cells in the presence of the following mixture of gases: 5% CO2/21% O2/74% N2.
Cell viability test
CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI 53703, USA) contains MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] and PES (phenazine ethosulfate) were used to assess the cell viability. The test was performed according to the manufacturer's instructions. Absorbance was read at a wavelength of 490 nm on a microplate reader (23225, Pierce Company, Rockford, IL 61101, USA).
Green fluorescent protein (GFP)-fused LC3 (GFP-LC3) lentivirus transfection
Primarily cultured cortical neurons were pipetted onto poly-l-lysine-coated 24-well plates at a density of 1 × 105. On DIV-3, the neurons were treated with a Lentivirus-CMV-GFP-LC3B-IRES-Puro (Virus Titer: 6.2E + 08 TU/ml) purchased from ObiO Technology (PR China). One day after transfection (MOI = 30), the cell medium was replaced with Neurobasal medium containing B27 supplement. DIV-7, the number of green fluorescent cells spots was calculated using confocal microscopy.
Western blot analysis
Western blot analysis was performed as described previously.21,25 Cells or peri-infarct region of mouse cortex were lysed by using buffer C to obtain the protein samples, the striatum tissue was not examined. The buffer C included 50 mM Tris-HCl, pH 7.5, 2 mM EDTA, 2 mM EGTA, 50 nM okadaic acid, 5 mM sodium pyrophosphate, 100 μM sodium vanadate, 1 mM DTT, 50 mM KF, 2% sodium dodecyl sulfate (SDS) and a protease inhibitor mixture. And then the protein concentration was determined with a BCA kit (Thermo Fisher Scientific, Rockford, IL 61108, USA) using albumin as a standard. Equal amounts (30 µg) of protein samples were separated on 8%, 10% and 12% SDS-polyacrylamide gels (PAGE), and then the proteins were electrically transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 10% milk and incubated with primary antibodies at 4°C overnight. Primary antibodies: Activin A (1:500, ab89307) and p62 (1:1,000, ab155686) from Abcam (Boston, MA 02101, USA); LC3 A/B (1:1000, 12741), mammalian target of rapamycin (mTOR, 1:1,000, 2983), P-Ser2448-mTOR (1:1,000, 5536), P-Ser473-PKB (1:1,000, 4060), PKB (1:1,000, 9272), P-p85 (Tyr458)/p55 (Tyr199)-PI3K (1:1,000, 4228), PI3K (1:1,000, 4257), P-Ser366-STING (1:1,000, 50907) and STING (1:1,000, 13647) from Cell Signaling Technology (Danvers, MA 01923, USA); and β-actin (1:10000, 60008-1-Ig) from Proteintech (Rosemont, IL 60018, USA).
The membranes were incubated with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG (1:4,000, 31460, Thermo Fisher) as secondary antibody for 1 h at room temperature. The signals in a specific band were detected with the enhanced chemiluminescent reagent (chemiluminescent HRP substrate, Millipore Corporation, Billerica, MA 01821, USA). The quantitative analysis was performed by using Fusion Capt 16.15 software (Fusion FX6 XT, Vilber Lourmat, Collegien, France).
ELISA assay
Cells or mouse cortex were lysed by using M-PERTM and liquid supernatant were collected. According to the manufacturer's instructions, the 2′,3′-cGAMP (Cat. #501700, Cayman Chemical, Ann Arbor, MI 48109, USA) were detected by using an enzyme-linked immunosorbent assay (ELISA).
Transmission electron microscopy (TEM)
Transmission electron microscopy was used to observe the number of autophagosomes. 26 After MCAO/R operation, the brains of mice were fixed by using cardiac perfusion with 2.5% glutaraldehyde, the peri-infarct region of brain was obtained. Analogously, after OGD/R treatment, primary cortical neurons were digested into clumps and collected for cell samples. Brain and cell samples were kept overnight in 25% glutaraldehyde at 4°C, and then fixed by 1% osmic acid for 1 h in 4°C. Samples were dehydrated in a gradient of ethanol solutions ranging from 30% to 100% ethanol (30%, 50%, 70%, and 90% for ten min; 100% for five min 3 times). Following treated with osmotic embedding and polymerization embedding, samples were subjected to double staining with 1% uranium acetate and lead citrate. 60 nm-thick ultra-thin sections were prepared. Images were acquired using a JEM-1400PLUS TEM (JEOL, Japan) at an acceleration voltage of 80 kV. The double-membranous vesicle with cytoplasmic material was defined as an autophagosome.
Statistical analysis
GraphPad Prism software version 8.0 (GraphPad Software, La Jolla, CA, 92093, USA) was used for statistical analysis. Shapiro-Wilk test and generation of QQ-plots were used to assess data distribution. The measurement data were expressed as the mean ± SD. In the anatomical and biochemical studies, one-way or two-way ANOVA was used for comparisons among multiple groups. Bonferroni's post-hoc analysis was used to determine where those differences occurred. As the attributes data, the categorical functional outcome data such as Neurological score, Longa score and Balance beam were expressed as quartile, and the Mann-Whitney U test was used for group comparisons. p < 0.05 was considered a statistically significant difference.
Results
Upregulated activin a in peri-infarct region protected neurons against ischemic injury by inhibiting autophagy
Illustration of the experimental timelines is showing in Figure 1(a). To investigate the role of Activin A in ischemic stroke, we first examined Activin A expressions in the brain of mice with ischemic stroke. Activin A (ActA) protein expression levels significantly increased in the peri-infarct region of mice with 1 h MCAO/R 12 h or 1 d when compared with that of Sham group; the peak reached on 1 d reperfusion (Figure 1(b) and (c)). The neuron survival rate in the 1 h OGD/R 24 h group was significantly lower than that of Normoxia group, and rmActivin A (50, 100, and 200 ng/mL) significantly increased the neuron survival rate in a dose-dependent manner (Figure 1(d)). Since LC3 is now widely used to monitor autophagy. 27 And p62 accumulates when autophagy is inhibited.28–30 To explore how Activin A protects neurons against ischemic injury, we observed that addition of rmActivin A could significantly reduce 1 h OGD/R 24 h-induced increase of LC3 II/(LC3 I + LC3 II) ratio and inhibit p62 degradation (Figure 1(e) to (g)). Similarly, the addition of rmActivin A could significantly decrease the quantity of autophagosomes (Figure 1(h) and (i)) but did not affect the cleaved-caspase3 levels in cortical neurons (Figure 1(j) and (k)). To further clarify the effect of Activin A on autophagy, we observed LC3 conversion by using autophagy inhibitor Baf A1, the results showed that on the basis of Baf A1 treatment, LC3 accumulation were decreased upon Activin A addition in 1 h OGD/R 24 h neurons (Figure S2).These results suggested that Activin A inhibits autophagy but has no impact on cell apoptosis in ischemic neurons.
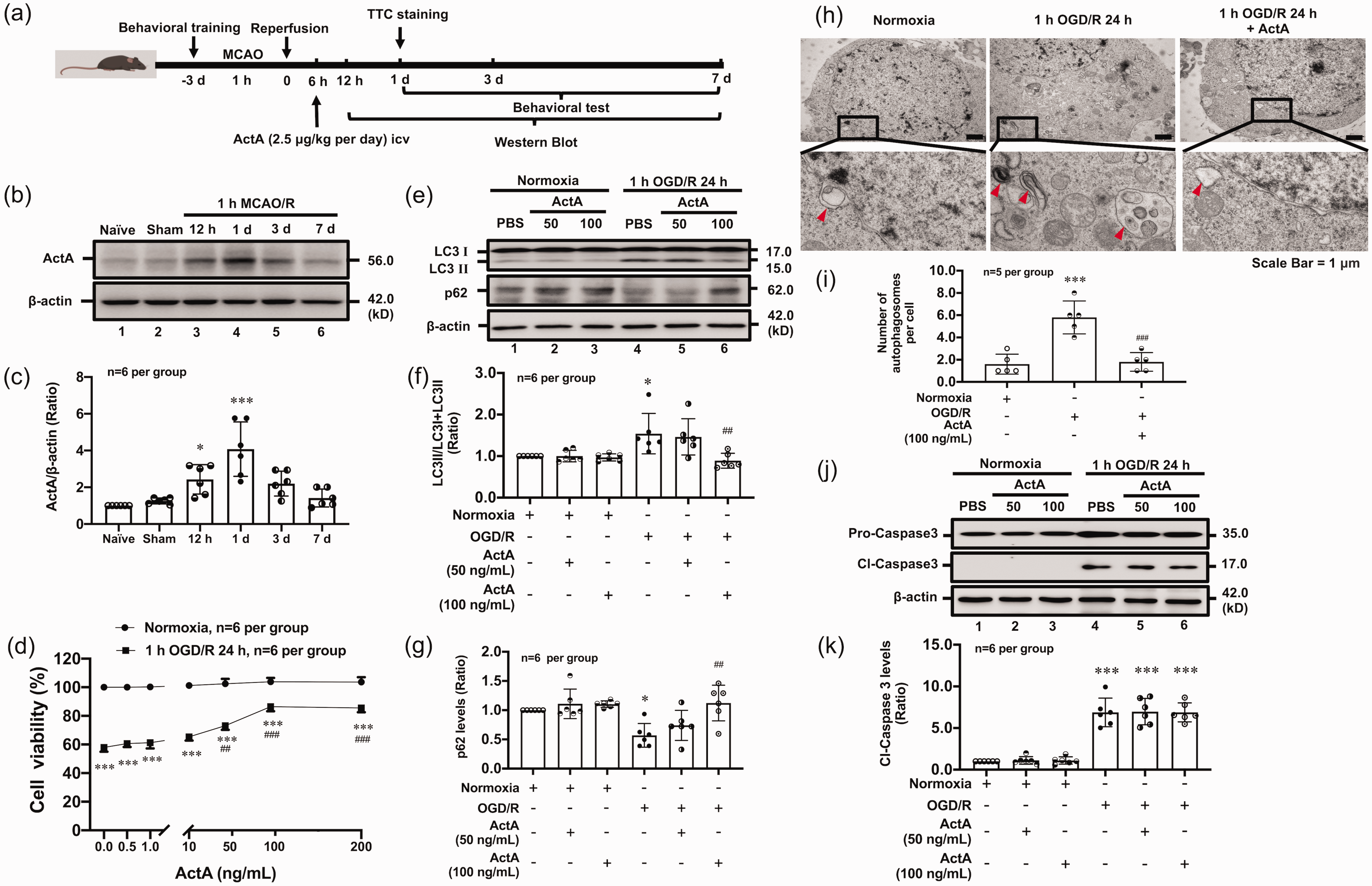
Activin A protein levels were upregulated in the brain of mice with ischemic stroke and protected neurons against ischemic injury by inhibiting autophagy. (a) shows the illustration of animal experimental timelines. The typical image and quantitative analysis results of Western blot (b, c) showed that Activin A protein levels were upregulated in peri-infarct region of mice with ischemic stroke. The MTS assay results showed that the addition of rmActivin A could significantly increase the cell viabilities of cortical neurons after 1 h OGD/R 24 h treatment (d). The typical image and quantitative analysis results of Western blot demonstrated that rmActivin A addition reduced the LC3 II/LC3 (I+II) ratio (e, f) and inhibited p62 degradation (e, g), but did not affect the cleavage levels of pro-caspase-3 (j, k) in primary cortical neurons subjected to 1 h OGD/R 24 h. The typical image (h) and quantitative analysis (i) results of TEM showed that rmActivin A decreased the quantity of autophagosomes.The red arrows, autophagosomes.*p < 0.05, ***p < 0.001 vs Sham or Normoxia group; ##p < 0.01, ###p < 0.001 vs 1 h OGD/R 24 h group, n = 6 per group (c, d, f, g, j, k), n = 5 per group (h, i).
Activin a inhibited PI3K-PKB-activated autophagy in primarily cultured neurons after OGD/R
As a canonical pathway in the regulation of autophagy, PI3K-PKB is modulated by Activin A. In response to 1 h OGD/R 24 h, the levels of P-PI3K and P-PKB were significantly lower than that of the Normoxia group. However, the addition of rmActivin A significantly increased P-PI3K and P-PKB levels (Figure 2(a) to (d)); and the PI3K inhibitor NU7441 could reverse the effect of Activin A on LC3 conversion (Figure 2(e) and (f)) and p62 degradation (Figure 2(g) and (h)). Consistent with the Western blot results, rmActivin A could significantly reduce the numbers of LC3 positive puncta that were reversed by PI3K inhibitor NU7441 (Figure 2(i) and (j)) in 1 h OGD/R 24 h-treated neurons. These results suggested that Activin A inhibits ischemic neurons' autophagy by activating the PI3K-PKB pathway.
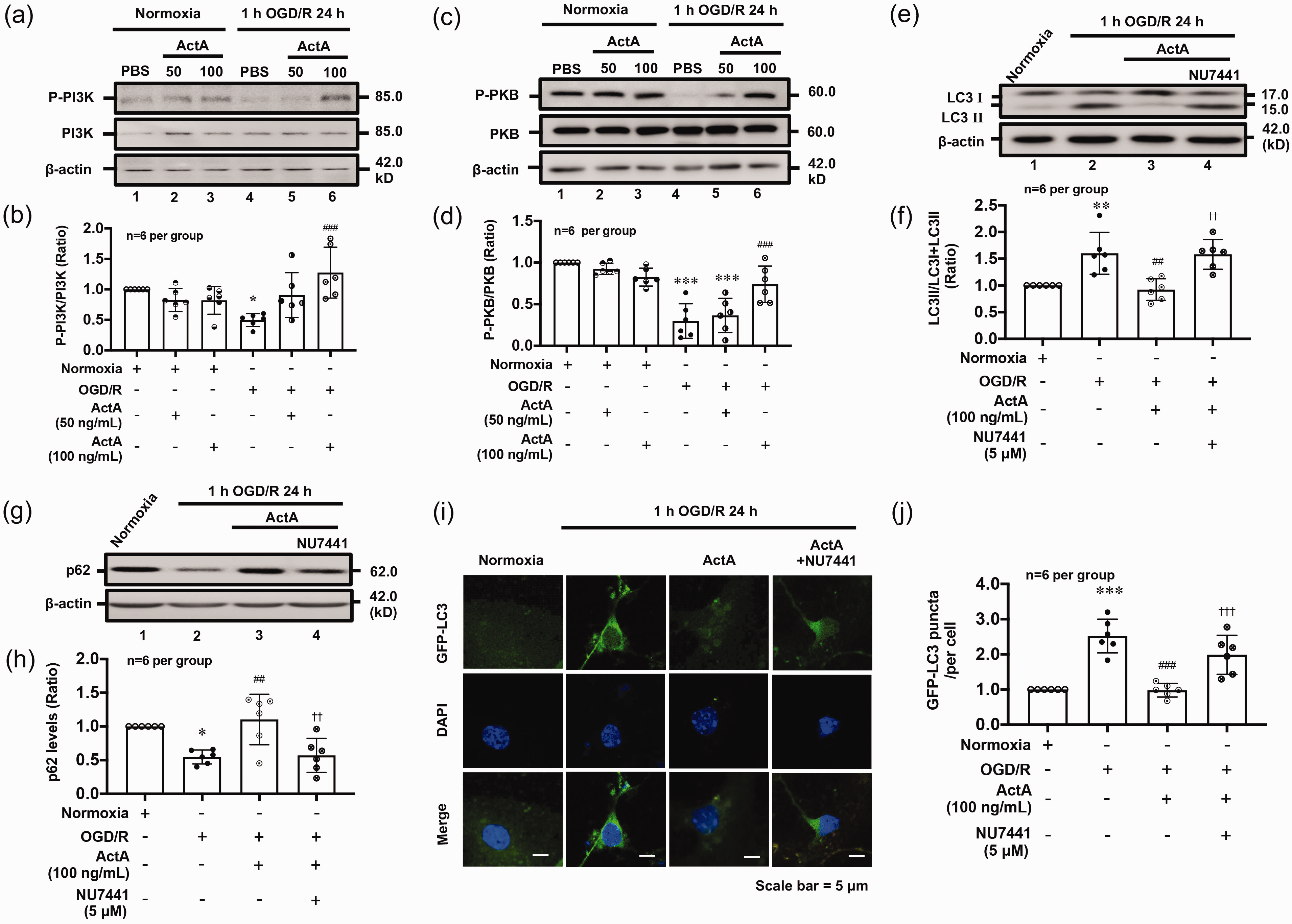
Activin A inhibited PI3K-PKB-mediated autophagy in OGD/R-treated cortical neurons. The Western blot and fluorescent staining results showed that rmActivin A addition could significantly increase the levels of P-PI3K (a, b) and P-PKB (c, d). However, PI3K inhibitor NU7441 could reverse the inhibitory effects of Activin A on LC3 conversion (e, f), p62 degradation (g, h) and number of LC3 positive puncta and (i, j) in 1 h OGD/R 24 h-treated cortical neurons. *p < 0.05, **p < 0.01, ***p < 0.001 vs Normoxia group; ##p < 0.01, ###p < 0.001 vs 1 h OGD/R 24 h group; ††p < 0.01, †††p < 0.001 vs 1 h OGD/R 24 h + rmActivin A group, n = 6 per group.
Activin a regulated the cGAS-STING pathway by activating PI3K-PKB signaling to inhibit autophagy of neurons
PI3K/PKB regulates downstream molecules of autophagy including mTOR and cGAS. To investigate the mechanism by which Activin A regulates autophagy following OGD/R, we observed the levels of P-mTOR and activities of cGAS-STING. Results showed that rmActivin A did not affect the P-mTOR levels after OGD/R (Figure 3(a) and (b)). However, rmActivin A could significantly reduce 2′,3′-cGAMP generation, representing cGAS activity (Figure 3(c)), and P-STING levels (Figure 3(d) and (e)) in 1 h OGD/R 24 h-treated neurons; STING agonist 2′,3′-cGAMP reversed the effect of rmActivin A to LC3 positive puncta and p62 degradation (Figure 3(f) and (i)). Consistent with Western blot data, 2′,3′-cGAMP reversed the effect of Activin A on LC3 positive puncta (Figure 3(j) and (k)). Furthermore, cGAS inhibitor RU.521 increased the viability of primary cultured cortical neurons compared with that of 1 h OGD/R 24 h group, but the addition of 2′,3′-cGAMP reversed (p < 0.001) the effect of rmActivin A (Figure 3(l)). These results indicated that Activin A suppresses excessive autophagy via the cGAS-STING pathway in primary cultured cortical neurons after OGD/R. PKB regulates cGAS phosphorylation as an upstream signal of cGAS-STING in mice and humans. 31 To determine the effects of Activin A on the PI3K-PKB and cGAS-STING signaling pathways after OGD/R in neurons, we investigated cGAS and STING activities after blocking PI3K activation. PI3K suppression increased 2′,3′-cGAMP generation (Figure S1 (a)) and P-STING (Figure S1 (b) and (c)); these were reduced by rmActivin A following OGD/R. cGAMP, however, did not influence the levels of P-PKB (Supplementary data 1(d) and (e)). These data suggested that cGAS-STING, locate downstream of the PI3K-PKB pathway, was regulated by Activin A.
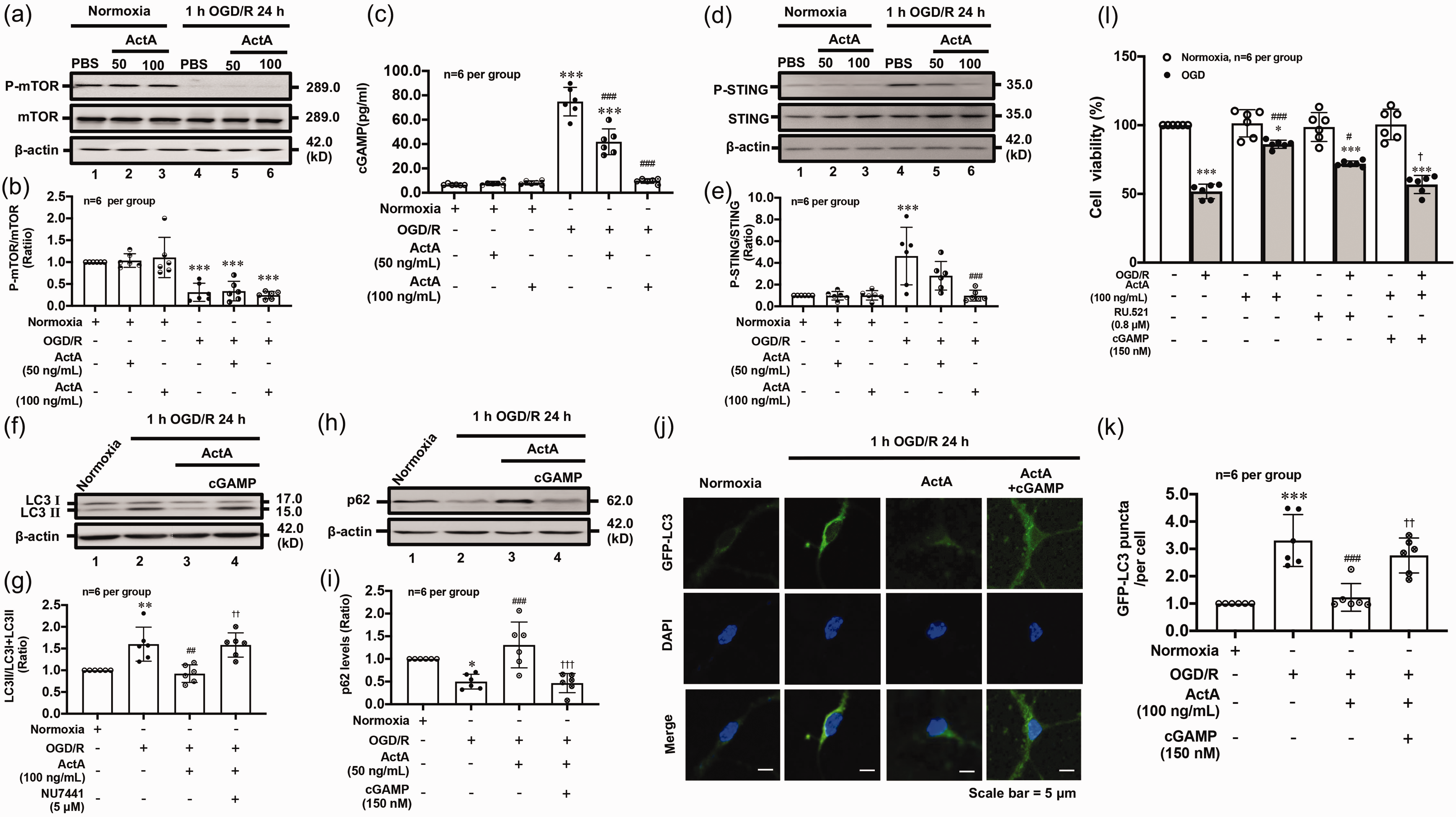
Activin A suppressed excessive autophagy via cGAS-STING pathway in OGD/R-exposed cortical neurons. The typical image and quantitative analysis results of Western blot showed that rmActivin A addition did not affect mTOR phosphorylation levels (a, b), but could significantly decrease the generation of 2′,3′-cGAMP (c) and P-STING levels (d, e) in OGD/R-treated neurons. Similarly, the STING agonist 2′,3′-cGAMP reversed the effects of rmActivin A on LC3 conversion (f, g) and p62 degradation (h, i) in cortical neurons after OGD/R; rmActivin A addition significantly inhibited OGD/R-induced increase of LC3 positive puncta numbers, which could be reversed by addition of 2′,3′-cGAMP (j, k). In addition, the cell viability test demonstrated that both cGAS inhibitor RU.521 and rmActivin A could significantly inhibit the OGD/R-induced decrease of neuronal viability, but 2′,3′-cGAMP abolished the effect of Activin A (l). *p < 0.05, **p < 0.01, ***p < 0.001 vs. Normoxia group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. 1 h OGD/R 24 h group, and †p < 0.05, ††p < 0.01, †††p < 0.001 vs. 1 h OGD/R 24 h + rmActivin A group, n = 6 per group.
rmActivin a improved neurological outcomes of mice after ischemic stroke
Next, we investigated whether Activin A improved the prognosis of ischemic stroke in vivo and the autophagy levels. Compared with the 1 h MCAO/R 24 h group, the rmActivin A-treated mice had a much smaller infarct size than mice exposed only to 1 h MCAO/R 24 h (Figure 4(a) and (b)). In addition, we performed neurobehavioral tests to determine whether Activin A improved neurological function. The neurological function score increased significantly after 1 h MCAO/R 1, 3 and 7 day, indicating neurological impairment (Figure 4(c)). Mice treated with ICV injection of rmActivin A exhibited better performance (Figure 4(c) and (e)) as per the neurological function and Longa scores. After ICV injection of rmActivin A, neurological function scores of mice were significantly lower than those of the PBS (vehicle) group, suggesting an improvement in neurological function. Mice administered exogenous rmActivin A exhibited significant improvement in the balance beam and rotating rod tests (Figure 4(d) and (f)).
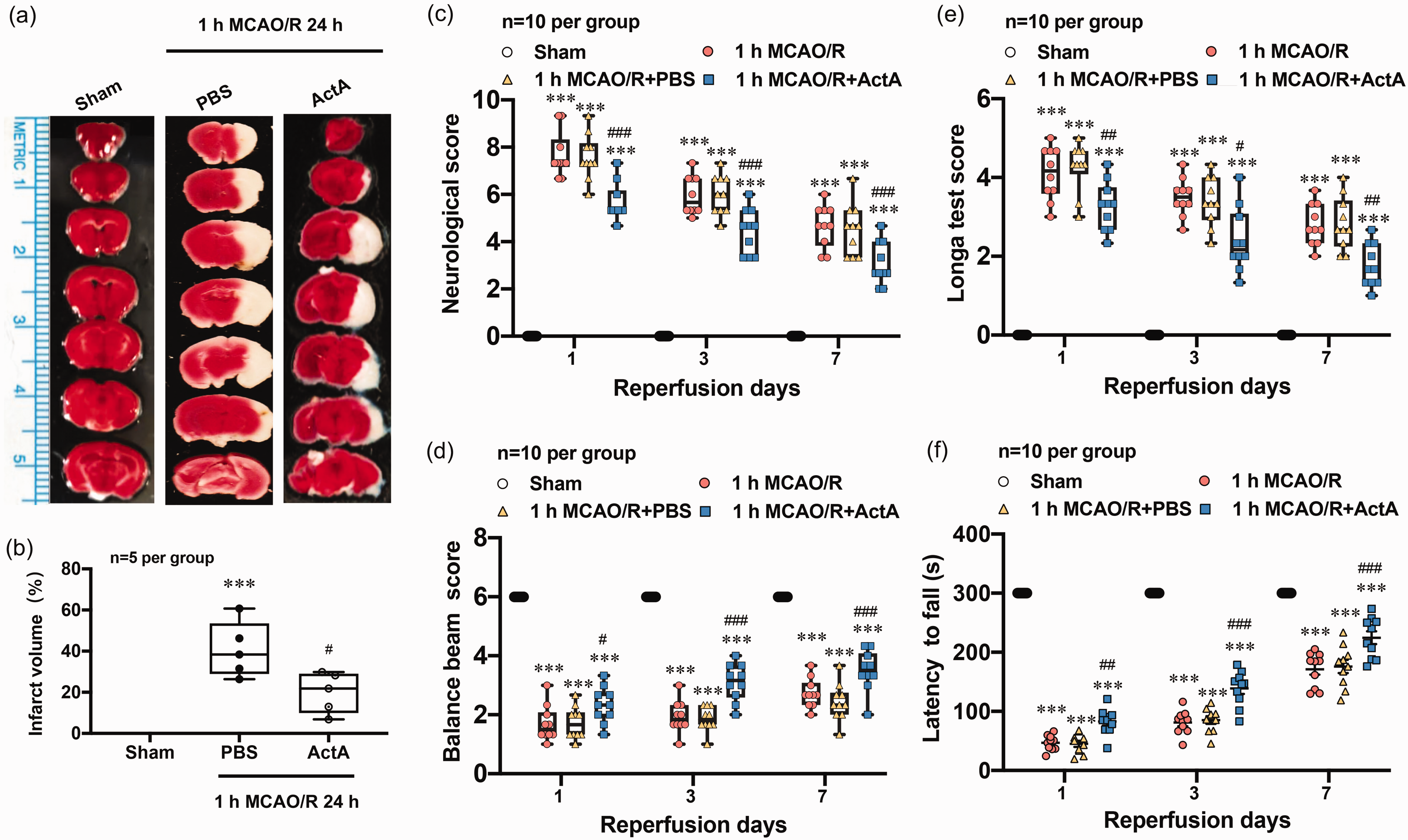
rmActivin A reduced infarction volume and improved neurological outcomes of mice with ischemic stroke. Representative image (a) and statistical analysis result (b) of triphenyl tetrazolium chloride (TTC) staining showed that the infarct size in brain of mice with 1 h MCAO/R 24 h + rmActivin A treatment was significantly smaller than that of 1 h MCAO/R 24 h + PBS group. The statistical analysis results of neurological function score (c), balance beam (d), and Longa score (e) and rotating rod (f) tests demonstrated that rmActivin A could significantly improve the neurological deficits of mice with ischemic stroke. ***p < 0.001 vs. Sham group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. 1 h MCAO/R + PBS group, n = 5 per group (a, b), n = 10 per group (c–f).
Activin a inhibited cGAS-STING-mediated autophagy by activating PI3K-PKB pathway in mice after MCAO/R
To determine the mechanism by which Activin A protects brain against ischemic stroke, we verified the effects of Activin A on the PI3K-PKB and cGAS-STING signaling pathways after MCAO/R in mice. We observed LC3II/LC3(I + II) ratio and p62 degradation significantly increased after 1 h MCAO/R 24 h treatment, compared with the Sham group, but ICV injection of rmActivin A decreased LC3II/LC3 (I + II) ratio (Figure 5(a) and (b)) and p62 degradation (Figure 5(c) and (d)). However, the PI3K inhibitor NU7441 reversed the effects of Activin A (Figure 5(a) to (d)). We further observed that phosphorylation levels of PKB significantly lowered after 1 h MCAO/R 24 h treatment, compared with the Sham group. The phosphorylation levels of PKB significantly increased after rmActivin A i.c.v administration, and PI3K inhibitor reversed the effects of Activin A (Figure 5(e) and (f)). Similarly, we observed 2′,3′-cGAMP generation and phosphorylation levels of STING elevated after 1 h MCAO/R 24 h treatment, and PI3K suppression increased 2′,3′-cGAMP generation (Figure 5(g)) and P-STING levels (Figure 5(h) and (i)) that could be reduced by rmActivin A following MCAO/R. These data revealed that Activin A regulates the cGAS-STING pathway by activating PI3K-PKB signaling to inhibit autophagy of mice with ischemic stroke.
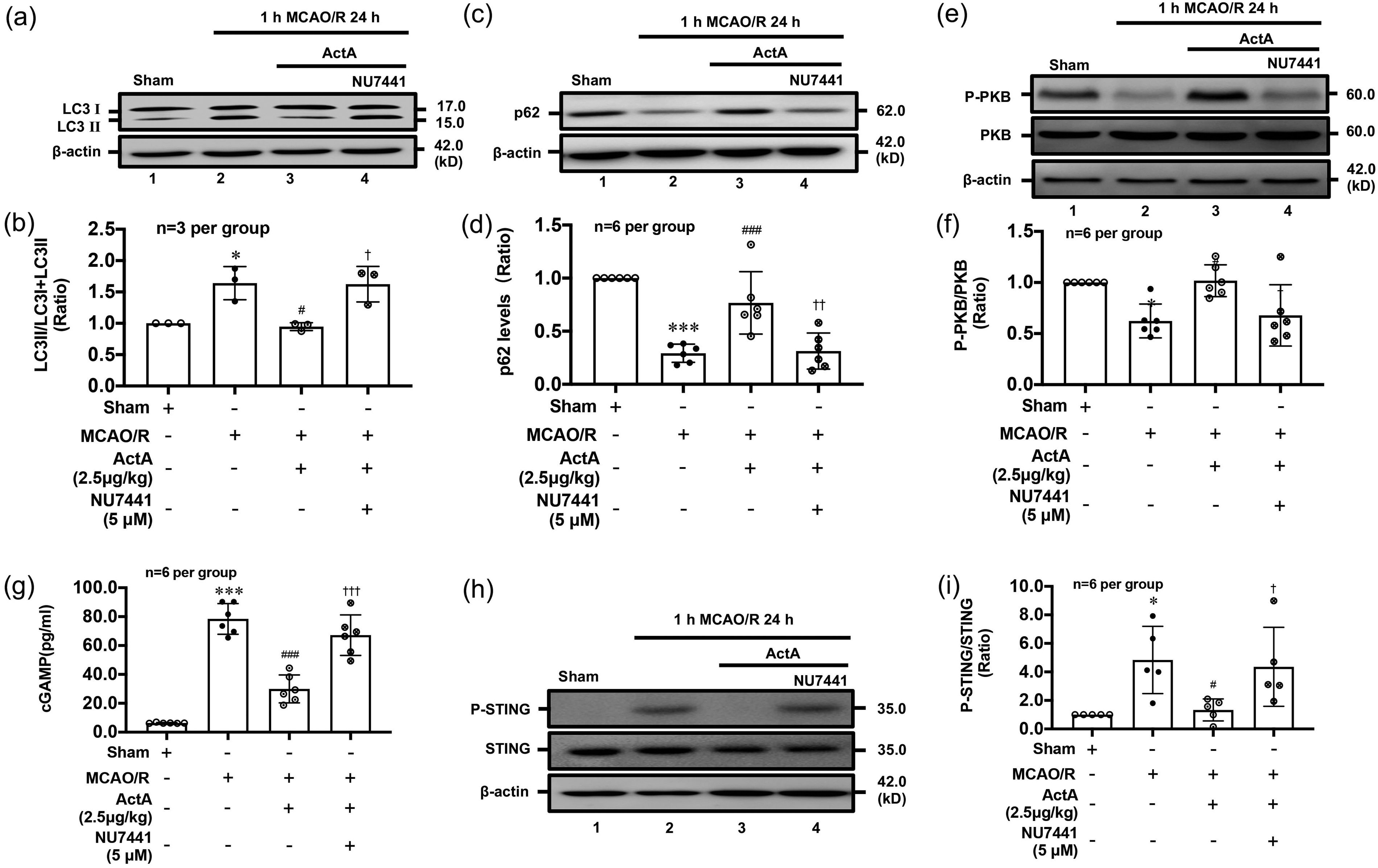
Activin A suppressed autophagy levels via PI3K-PKB and cGAS-STING pathway in mice with ischemic stroke. Western blot data showed that LC3 II/LC3 (I+II) ratio (a, b), p62 degradation (c, d), 2′,3′-cGAMP generation (g) and P-STING (h, i) were increased while P-PKB levels (e, f) decreased in 1 h MCAO/R 24 h group compared with that of Sham group. ICV injection of rmActivin A reduced the LC3 II/LC3 (I+II) ratio (a, b), p62 degradation (c, d), 2′,3′-cGAMP generation (g) and P-STING levels (h, i), and increased P-PKB levels (e, f). PI3K inhibitor NU7441 reversed the effect of rmActivin A on LC3 II/LC3 (I+II) ratio (a, b), p62 degradation (c, d), P-PKB levels (e, f), 2′,3′-cGAMP generation (g) and P-STING levels (h, i) in peri-infarct region of mice with ischemic stroke. Data are presented as the mean ± S.D.; **p < 0.01, ***p < 0.001 vs. Sham group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. 1 h MCAO/R 24 h + PBS group, and †p < 0.05, ††p < 0.01, †††p < 0.001 vs. 1 h MCAO/R 24 h + rmActivin A group, n = 3 per group (a, b), n = 6 per group (c–i).
Conditional knocking out ACVR1C receptor on neurons aggravated autophagy levels and neurological deficits in mice with ischemic stroke
Activin A, as a ligand, binds to its receptor to activate downstream signals. Activin A receptor type 1C (ACVR1C), also known as activin like kinase receptor 7 (ALK7), is mainly expressed in the central nervous system. 32 To further study the effect of endogenous Activin A in neurons on the prognosis of ischemic stroke, we established conditional Knockout (cKO) mice using Crisper/Cas9 technique. LC3 conversion (Figure 6(a) and (b)) and quantity of autophagosomes (Figure 6(c) and (d)) were increased in brain of WT and Acvr1cfl/fl; Nestincre+/− mice with ischemic stroke. In addition, the LC3 conversion and quantity of autophagosomes showed bigger increase in Acvr1cfl/fl; Nestincre+/− mice than WT mice (Figure 6(a) to (d)). We further explored the role of ACVR1C in neurological outcome of mice with ischemic stroke. The results showed that the neurological deficits were aggravated in Acvr1cfl/fl; Nestincre+/− mice than WT mice after 1 h MCAO/R day 1, 3 and 7 (Figure 6(e) to (h)). These results suggested that endogenous Activin A may improve neurological outcome of mice with ischemic stroke through ACVR1C-mediated autophagy.
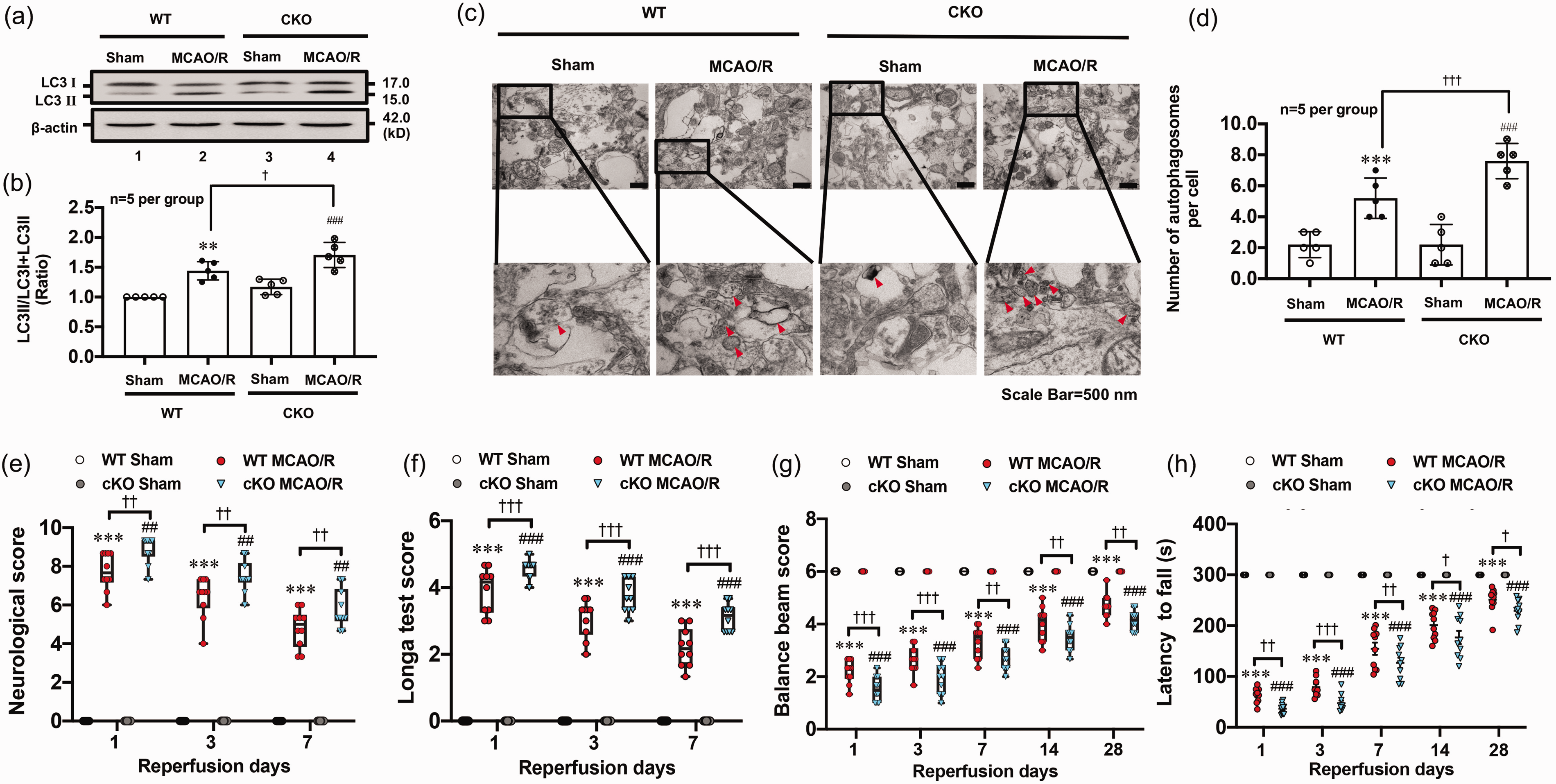
ACVR1C receptor conditional knock out enhanced autophagy levels and aggravated neurological outcomes of mice with ischemic stroke. Western blot and TEM data showed that the LC3 II/LC3 (I + II) ratio (a, b) and autophagosome number (c, d) were increased in Acvr1cfl/fl; Nestincre+/− mice after 1 h MCAO/R 24 h compared with the wild type. The statistical analysis results of neurological function score (e), balance beam (f), Longa score (g) and rotating rod tests (h) demonstrated that Acvr1cfl/fl; Nestincre+/− mice had greater neurological deficits compared with wild type mice subjected to the 1 h MCAO/R treatment. ***p < 0.05 vs. Sham group of WT mice; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. Sham group of Acvr1cfl/fl; Nestincre+/− mice; †p < 0.05, ††p < 0.01, †††p < 0.001 vs. 1 h MCAO/R group of WT mice, n = 5 per group (a–d), n = 10 per group (e–h).
Discussion
Accumulating evidence suggest that Activin A plays a protective role in ischemic stroke.11,13,33,34 Activin A mRNA levels are significantly increased during transient focal cerebral ischemia 12 and protect neurons from neurotoxicity, which plays a vital role in mitigating brain injury. 33 Consistent with these findings, our study observed the increase of Activin A protein levels in the peri-infarct region of mice with ischemic stroke. rmActivin A could improve neurological outcome, but conditional knockout ACVR1C receptor on neurons aggravated neurological deficits in mice with ischemic stroke. In addition, we found that Activin A could significantly inhibit cGAS-STING-mediated autophagy through the PI3K-PKB pathway, which in turn reduced brain infarction volume and improved the neurological functions of mice with ischemic stroke.
The association between ischemic stroke and autophagy is a major focus of research. 35 There is an increase in the number of autophagosomes, as observed in experimental models of neuronal ischemia.4,6 Autophagy, however, is a double-edged sword; moderate autophagy promotes normal neuronal physiological activity and the removal of cellular debris, whereas excessive autophagy exacerbates ischemic injury, causing neuronal death.36,37 Consistent with these results, in the present study, autophagy levels enhanced in 1 h MCAO/R 24 h-treated mice, which led to worse neurological function. Identically, autophagy levels also increased in primary cortical neurons exposed to 1 h OGD/R 24 h, these were accompanied by a decrease in the neuron survival rate. Those data indicate that MCAO/R-induced autophagy, excessive autophagy aggravated neuronal ischemic injury exposed to MCAO/R or OGD/R.
Our data showed that Activin A is neuroprotective through the autophagic process rather than apoptosis. Autophagy plays a key role in the regulation of ischemic stroke, but the specific mechanism is still unclear, among which PI3K-PKB-mTOR signaling is an important pathway that scholars pay attention to. Resveratrol provides neuroprotection by regulating the PI3K-PKB-mTOR pathway after stroke in rats. 38 Dehydrocostuslactone attenuated OGD/R-induced PC12 cell ischemic injury through autophagy inhibition by activating the PI3K-AKT-mTOR pathway. 39 In order to eliminate the interference of other factors, we explored the upstream and downstream relationship of the pathway in the cell model. In our study, Activin A attenuated autophagy levels and reduced quantity of autophagosomes. Moreover, Activin A mediated its protective action through the classical autophagy PI3K-PKB pathway and promoted PI3K and PKB phosphorylation. Interestingly, Activin A did not affect mTOR phosphorylation in 1 h OGD/R 24 h-exposed primary cortical neurons. Thus, Activin A may play a neuroprotective role by acting on autophagy pathways of other PKB downstream molecules.
PKB (Akt) phosphorylates the S291 or S305 residue of mouse or human cGAS, respectively, 31 indicating that PKB kinase negatively regulates the cGAS pathway. The cGAS-STING pathway, widely studied in the fields of immunity, inflammation and tumor, has been shown to be important in ischemia-reperfusion injury.18,40 Moreover, cGAS-STING pathway is highly active in ischemia/reperfusion (I/R) injury, 41 and particularly in I/R-induced brain injury. 40 In accordance with these studies, cGAS-STING pathway were activated in brain of MCAO/R mice and OGD/R-treated primary neurons in this study. Some scholars have proposed that autophagy induction via STING trafficking is a primordial function of the cGAS pathway. 17 Thus, cGAS-STING pathway-mediated autophagy may provide new insights into ischemic stroke.
We determined whether Activin A regulate autophagy through cGAS-STING pathway. The results showed that the cGAS-STING pathway facilitated autophagy and rmActivin A could significantly reduce 2′,3′-cGAMP generation, representing cGAS activity and P-STING levels. Moreover, STING agonist cGAMP reversed the effect of Activin A on LC3 conversion, p62 degradation and LC3 positive puncta. Studies have shown that inhibition of cGAS reduces the expression of cGAS and neurological impairment.19,42 In keeping with these results, compared with the rmActivin A group after OGD/R, the neuron survival rate declined by further activating STING. Thus, we speculated that cGAS-STING pathway activation might induce excessive autophagy and cause neuron injury, which was suppressed by Activin A. PI3K inhibition increased 2′,3′-cGAMP generation and P-STING levels. cGAMP, however, did not affect the levels of P-PKB. We determined that cGAS-STING was located downstream of the PI3K-PKB pathway and regulated by Activin A. Interestingly, we observed that Activin A decreased LC3II increasing by Baf A1, probably the reason is, in the presence of Baf A1, which inhibits the cargo content degradation, the OGD induced LC3II increase is further up due to the block of lysosomal protein degradation, an end step of the autophagy process. These suggest that Activin A inhibits autophagy likely in the early phase. However, the limitation of this study is that we did not determine the effect of activin A on autophagy flux, the relationship among LC3, p62 and autophagy has always been a fascinating topic. These mechanism are worth exploring in future studies.
Activin A, as a ligand, binds to its receptor to activate downstream signals. Studies have shown that ACVR1C knockdown has a negative effect on cell survival.43–45 Notably, ACVR1C regulates islet β cell proliferation and apoptosis by mediating PKB phosphorylation. 46 We presumed Activin A might be play neuroprotective function through ACVR1C receptors in ischemic stroke. We established Acvr1cfl/fl; Nestincre+/− mice and observed higher autophagy levels and greater neurological deficits compared with wild type mice. And the function of Activin A in reducing infarct volume was blocked in Acvr1cfl/fl; Nestincre+/− mice with ischemic stroke (Figure S3). These results demonstrated endogenous Activin A, depending on ACVR1C receptor, has a neuroprotective effect against 1 h MCAO/R-induced neurological impairment through suppressing excessive autophagy.
In conclusion, we reported that Activin A protected neurons against ischemic injury and improved neurological outcomes by inhibiting the cGAS-STING pathway. Therefore, Activin A may play a potential therapeutic effect for ischemic stroke through reducing cGAS-STING-dependent neuronal autophagy (Figure 7).
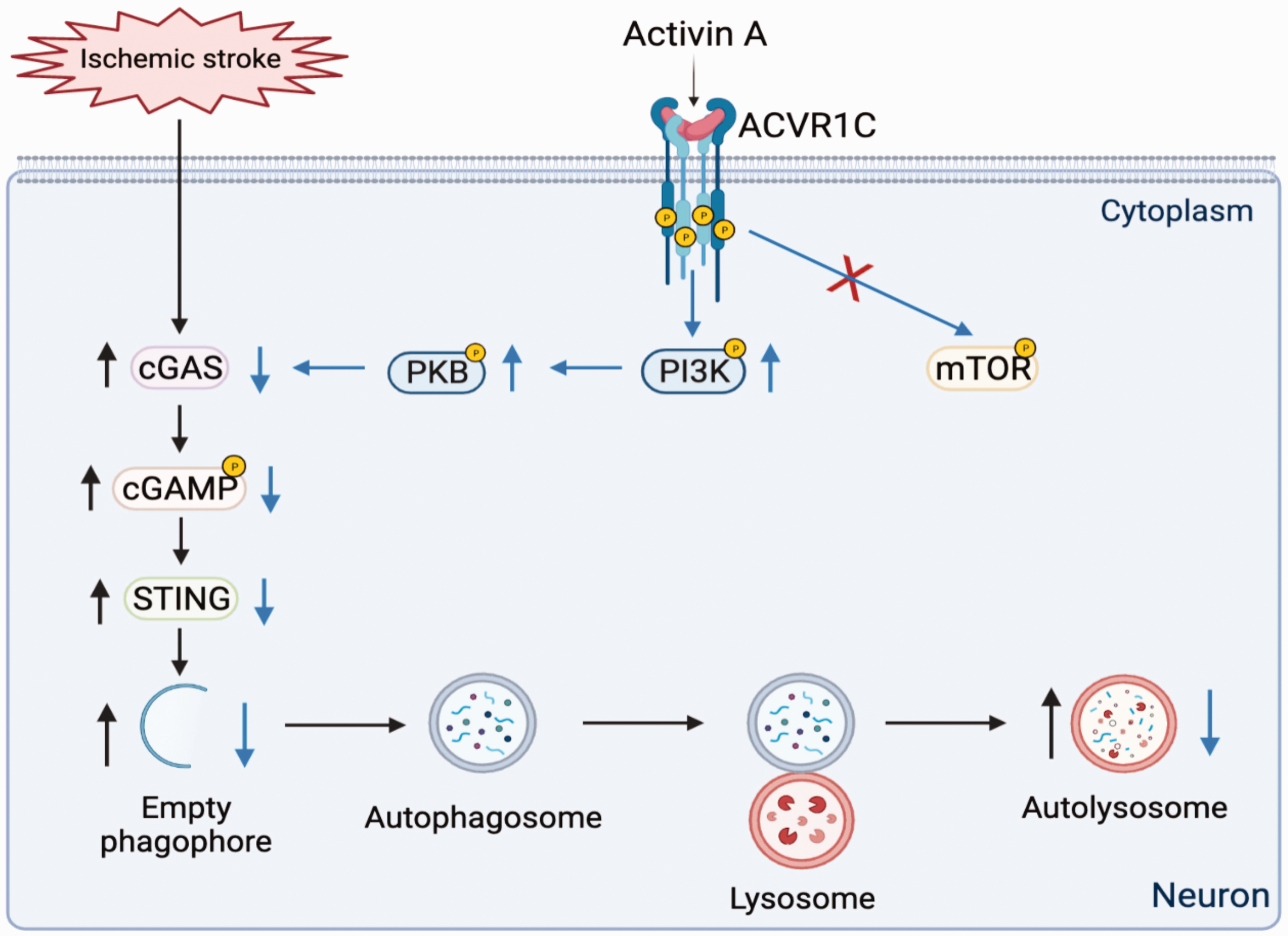
The schematic diagram showed that Activin A protects neurons against ischemic injuries through inhibiting cGAS-STING-mediated excessive autophagy depending on ACVR1C receptor.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X221147056 - Supplemental material for Activin A alleviates neuronal injury through inhibiting cGAS-STING-mediated autophagy in mice with ischemic stroke
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X221147056 for Activin A alleviates neuronal injury through inhibiting cGAS-STING-mediated autophagy in mice with ischemic stroke by Meilian Liu, Yudie Li, Song Han, Hongyu Wang and Junfa Li in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (No. 31972911 to JL, and No. 81971102 to SL).
Authors' contributions
ML and JL contributed to conception and design of the study; ML organized the database. SH, HW and YL performed the statistical analysis. ML wrote the first draft of the manuscript that was edited by JL. All authors contributed to manuscript revision, read, and approved the submitted version.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.