
Abstract
Pial collaterals provide protection in stroke. Evidence suggests their formation late during gestation (collaterogenesis) is driven by reduced oxygen levels in the cerebral watersheds. The purpose of this study was to determine if collaterogenesis can be re-activated in the adult to induce formation of additional collaterals (“neo-collateral formation”, NCF). Mice were gradually acclimated to reduced inspired oxygen (FIO2) and maintained at 12, 10, 8.5 or 7% for two-to-eight weeks. Hypoxemia induced “dose”-dependent NCF and remodeling of native collaterals, and decreased infarct volume after permanent MCA occlusion. In contrast, no formation occurred of addition collateral-like intra-tree anastomoses, PComs, or branches within the MCA tree. Hypoxic NCF, remodeling and infarct protection were durable, i.e. retained for at least six weeks after return to normoxia. Hypoxia increased expression of Hif2α, Vegfa, Rabep2, Angpt2, Tie2 and Cxcr4. Neo-collateral formation was abolished in mice lacking Rabep2, a novel gene involved in VEGFA→Flk1 signaling and required for formation of collaterals during development, and inhibited by knockdown of Vegfa, Flk1 and Cxcr4. Rabep2-dependent NCF was also induced by permanent MCA occlusion. This is the first report that hypoxia induces new pial collaterals to form. Hypoxia- and occlusion-induced neo-collateral formation provide models to study collaterogenesis in the adult.
Introduction
Collaterals within the microcirculation, i.e. arteriole-to-arteriole anastomoses that cross-connect a small fraction of the outer branches of adjacent arterial trees, provide an alternative source of blood flow. Their number and diameter present before obstruction (i.e. native collaterals) can significantly limit the severity of injury if the trunk or branch of one of the trees becomes obstructed, for example in ischemic stroke.1–4 Studies in different strains of mice have found that the abundance of native collaterals in brain and peripheral tissues of an individual of a given strain varies widely, resulting in large differences in tissue injury following arterial ligation.5–7 Recent studies have begun to identify the genetic loci and gene polymorphisms involved in the process of their formation, termed collaterogenesis, that underlie the large strain-specific differences present in the adult.8–10 Collaterogenesis occurs during embryonic development in the pial watershed regions between the crowns of adjacent arterial trees where arterial oxygen levels are lower than levels in similarly sized arterioles. 11 The pathway that drives collaterogenesis involves vascular endothelial growth factor-A (VEGF-A) and other oxygen-sensitive genes. 12 Although genetic differences are the primary cause of differences in abundance of native collaterals in mice, vascular risk factors such as aging, hypertension and metabolic syndrome cause a decline in their number and diameter in brain and other tissues.13–17
It has long been regarded that while native collaterals can enlarge (remodel) following arterial occlusion, additional new collaterals do not form in the adult. However, recent studies in mice using high-resolution angiography have shown otherwise. Permanent occlusion of the middle cerebral artery (pMCAO) induced new pial collaterals to form, a process termed neo-collateral formation (NCF). 5 The same also occurred in skeletal muscle 18 , 19 and heart 20 following arterial ligation. In addition, several weeks of exposure to reduced inspired oxygen stimulated NCF in heart and a reduction in infarct volume following coronary artery ligation. 21 , 22 The purpose of this study was to determine if systemic hypoxemia (“hypoxia”) also induces NCF in the brain and to examine the underlying mechanisms. Understanding mechanisms of collateral development and growth, which has lagged other areas of vascular biology, has gained importance following recent reports that collateral blood flow varies widely in patients with acute ischemic stroke.1–4 The latter, which may be due in part from differences in number and/or diameter of native collaterals, is a major determinant of early infarct volume, infarct progression, response to treatment with thrombolytics and thrombectomy, and final functional outcome.1–4
Methods
See Supplement for details. C57BL/6J (B6), BALB/cByJ and B6.Rabep2−/−10 mice were from laboratory colonies. Global inducible knockdown mice were made by crossing heterozygous B6.Cg-Tg mice (CAG-cre/Esr1*) 5Amc/J (CAG-CreERT) to B6.Cxcr4fl/fl (B6;129P2-Cxcr4™ 2 Yzo /J) or B6.Vegfafl/fl to create B6.CAG-CreERT+/−;Cxcr4fl/fl and B6.CAG-CreERT−/−;Cxcr4fl/fl littermates or B6.CAG-CreERT+/−;Vegfafl/fl and B6.CAG-CreERT−/−;Vegfafl/fl littermates. Endothelial cell (EC)-specific Flk1 knockdown was achieved by crossing B6.Cdh5(PAC)-Cre-ERT2 mice to B6;CD1-Flk1fl/fl to create B6.Cdh5(PAC)-Cre-ERT2+/−;Flk1fl/fl and B6.Cdh5(PAC)-Cre-ERT2−/−;Flk1fl/fl littermates. Angiography was performed after perfusion-fixation at maximal dilation and filling of the pre-capillary vessels with viscosity-adjusted Microfil®. 17 Lumen diameters of all MCA-ACA pial collaterals in both hemispheres were determined at midpoint and averaged for each animal. pMCAO was by occlusion of the M1-MCA just distal to the lenticulostriate branches. 17 All values are mean ± SD and n-sizes and statistical tests are given in the figures and legends. Data for the 17 groups in the 7 Figures and 6 Supplemental Figures in this study (∼500 mice total), where appropriate, were subjected to D’Agostino-Pearson normality tests (Graphpad Prism; see Supplemental Methods). The hypotheses/questions that were tested are stated in Results and/or in Supplemental Methods. All procedures were approved by the University of North Carolina Institutional Animal Care and Use Committee (IACUC# 18-123.0-A, 04/2019) and were conducted in compliance with the NIH Guide for the Care and Use of Laboratory Animals and the ARRIVE and STAIR guidelines.
Results
Hypoxia stimulates formation of new collaterals that is abolished in Rabep2 knockout mice
Mice were gradually acclimated to reduced FIO2, maintained at the specified level for two to eight weeks, and gradually returned (“recovery”) to normoxia (0.21 or “21%”) (Figure 1(a)). Acclimation was done because previous studies found that direct exposure to 8% FIO2 causes inflammation and a decrease in blood–brain barrier function associated with lung alveolar macrophage activation. 23 , 24 Although we did not determine if exposure to 7% hypoxia (results presented below) caused neuronal injury, the same protocol shown in Figure 1(a) did not cause loss of body weight in a study of hypoxia-induced NCF in mouse heart. 22 This does not prove absence of neuronal injury, but if it occurred it is unlikely to have altered the vascular effects and conclusions presented below. We also did not observe any overt deficits, seizures or differences in behavior over time during the 7% hypoxia experiment in the wild-type or Rabep2− / − mice (presented below), including during regular changes in bedding and CO2 and water vapor scrubbers when such behaviors would likely have been evident (see Supplemental Methods). In addition, neo-collateral formation was also induced by less severe reductions in FIO2 (see below). Since CO2 accumulation in the hypoxia chamber could alter physiological mechanisms, in a preliminary experiment, we continuously measured CO2 levels during 10% hypoxia. Levels averaged ∼0.4% (Supplemental Figure I), which is below levels that caused mild respiratory stimulation when inspired for more than one month (≥1.5%), lessened intracerebral angiogenesis induced by four weeks of 10% hypoxia (6.5%),25–27 and modified expression of angiogenic genes (2.5% FICO2 at 50% FIO2,). 28
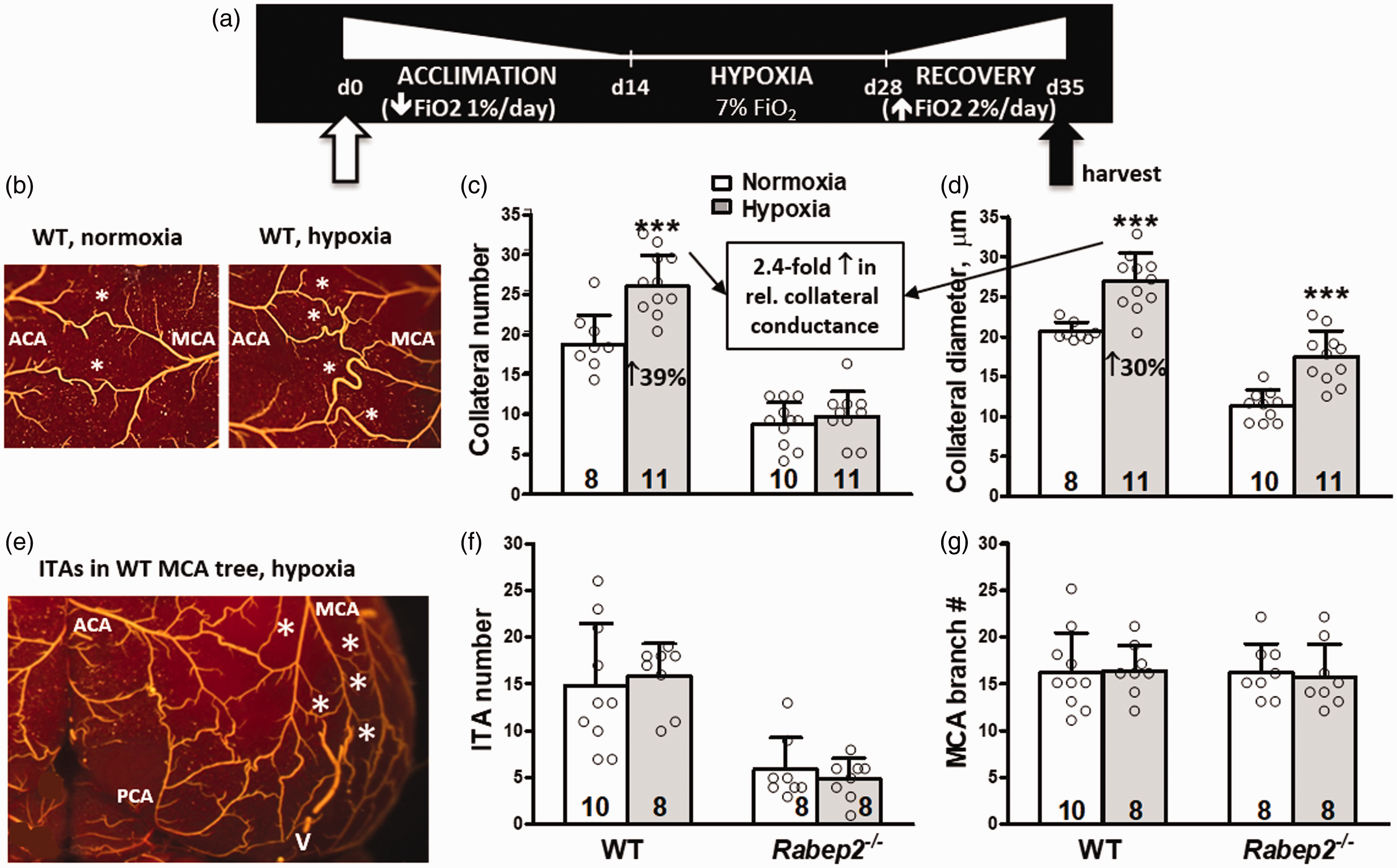
Systemic hypoxia induces new collateral formation (NCF) that is abolished in B6.Rabep2-KO mice. (a) Hypoxia (Hx) protocol. (b) Portion of the watershed zone between MCA and ACA trees (maximally dilated, fixed, filled with MicrofilR, counterstained) showing pial collaterals (∗). (c,d) Rabep2 deletion abolished Hx-induced NCF while collateral remodeling was unaffected. (e–g) Hypoxia did not affect number of intra-tree anastomoses (*, ITAs; V, partially filled venule) measured in MCA tree, or number of pial branches and branchlets along the largest second-order branch of the MCA tree extending from its bifurcation from the M1-MCA to the watershed zone. FIO2, fractional inspired oxygen content; hypoxia, 7% FIO2; d, days. Hematocrits on day-28 were 49±1.9, 80±1.6, 48±1.9, 79±6.0 for, respectively, wildtype (WT)-normoxia (Nx), WT-Hx, Rabep2−/−-Nx, Rabep2−/−-Hx. Relative conductance of the MCA-to-ACA collateral network = ((r 4 • n) • 1/hematocrit); r, collateral radius; n, collateral number. In this and subsequent figures and Supplemental figures unless indicated otherwise: values are mean ± SD; mice are C57BL/6 strain; number of mice given at base of bars; number and average diameter of MCA-to-ACA collaterals determined for both hemispheres. * , ** , ***p < 0.05, 0.01, 0.001, pre-specified one-sided t-test.
We first examined 7% FIO2. Hypoxia induced additional collaterals to form (i.e. neo-collateral formation, NCF), resulting in a 39% increase over baseline (Figure 1(b) and (c)). Hypoxia also increased lumen diameter by 30% (Figure 1(d)), a process termed collateral remodeling that is driven by increased fluid shear stress. 2 Increased shear stress is favored since hypoxia increased hematocrit by 64% when measured on day-29: 49±0.82 vs. 80±1.6 (p < 0.001), 48±1.9 vs. 79±0.6 (p < 0.001) for, respectively, wildtype (WT)-normoxia, WT-hypoxia, Rabep2−/−-normoxia, and Rabep2−/−-hypoxia groups in Figure 1. Hypoxia increased relative conductance of the ACA-to-MCA collateral network by 2.4-fold based on the formula: conductance ∝ (r 4 • n)/hematocrit, where r is collateral radius and n is collateral number. Hypoxic NCF was abolished in B6.Rabep2−/− mice, whereas collateral remodeling was unaffected (Figure 1(c) and (d)). The latter is consistent with absence of effect of Rabep2 deletion on hematocrit (see above) and flow-induced remodeling. 9 Collateral number and diameter at baseline (normoxia) in Rabep2−/− mice were smaller than WT mice, reflecting the prominent role of Rabep2 in collaterogenesis during development. 10
Stimulation of NCF by hypoxia was specific for collaterals, i.e. no increase occurred in the number of branches along the largest second-order branch of the MCA tree, including small branchlets that descend into the cortex, or collateral-like intra-tree anastomoses (ITAs) that occasionally interconnect second- and third-order branches (Figure 1(e) to (g)). Rabep2−/− mice had fewer ITAs at baseline, which indicates that like collaterals, Rabep2 is involved in their formation. Collaterogenesis and formation of ITAs occur at the same time during development, and Rabep2’s contribution to ITA formation was suggested from findings of reduced ITA formation in BALB/cBy embryos 11 that were subsequently shown to harbor a deficient variant of Rabep2. 10
To further explore the seemingly selective effect of hypoxia to induce NCF, we examined whether hypoxia increased the number of posterior communicating collateral arteries (PComs), since the majority of C57BL/6 mice lack one or both PComs at baseline. 29 Hypoxia had no effect on PCom number in WT or Rabep2−/− mice (nor did PCom number differ between WT and Rabep2−/− normoxia mice, confirming a previous study 29 ) (Figure 2(a)). Hypoxia also did not induced remodeling of the PComs or primary intracranial arteries, with the exception of the basilar artery, or branches of the MCA tree—although a trend was evident (Figure 2(b) and (c))—despite the large increase in hematocrit/shear stress. Interestingly, in Rabep2−/− mice, which had reduced pial collaterals at baseline and evidenced no hypoxic NCF, hypoxia now induced remodeling of the cerebral arteries, PComs and proximal branch-levels of the MCA tree.
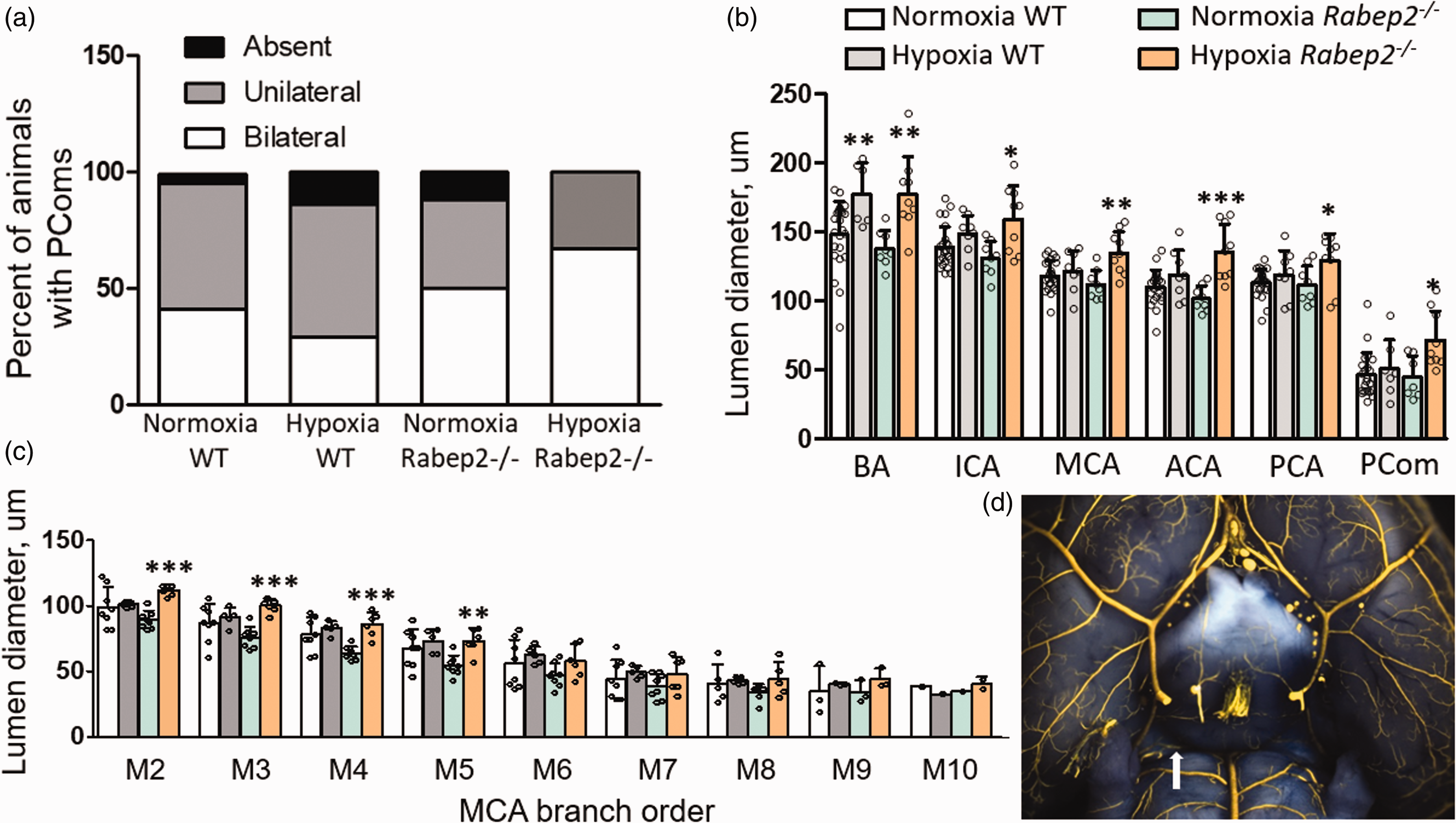
Effect of hypoxia on diameter of intracranial arteries. (a) PCom number (posterior communicating collateral artery) in WT and Rabep2−/− mice was not significantly altered by hypoxia, or different in normoxic WT vs. Rabep2−/− mice (χ2 tests). (b) Diameter of BA, ICA, MCA, ACA, PCA and PCom arteries (basilar, internal carotid, middle cerebral, anterior cerebral, posterior cerebral); values are averages from for both hemispheres. Hypoxia increased diameter of BA in WT and all arteries in Rabep2−/− mice. (c) Diameter of branches along largest second-order branch of right or left MCA tree (M2–M10), extending from its bifurcation from M1-MCA to watershed zone. Hypoxia increased diameters of M2–M5 branches in Rabep2−/− mice. (d) Representative normoxia WT mouse with unilateral PCom (star). N-sizes, 5–9 (excepting M9 and M10 in panel (c) differ from Figure 1 due to inadequate filling or damage during brain removal for imaging the circle of Willis. * , ** , ***p < 0.05, 0.01, 0.001, pre-specified two-sided t-test compared to immediately preceding bar.
Hypoxia induced neo-collateral formation, collateral remodeling, and protection against infarct volume are durable effects not lost on return to normoxia
We next examined less severe and longer durations of hypoxia. Two and four weeks of 10% hypoxia induced duration-dependent NCF and collateral remodeling (Figure 3(a) and (b)). Polycythemia had already increased maximally by two weeks (Figure 3(c)), in agreement with a previous study showing that the maximal increase during 10% FIO2 occurred within seven days. 27 Collateral remodeling also evidenced a hypoxia “dose”-dependent increase following two weeks of reduced FIO2 (Figure 3(a) and (b): 10%, 8.5%, 7% FIO2; p = 0.04 and p = 0.08 by ANOVA, respectively) that was accompanied by an expected dose-dependent increase in hematocrit (Figure 3(c)). Eight weeks of 12% hypoxia increased collateral number by an amount similar to the above groups (Figure 3(a)). Increases in collateral number and diameter exhibited maximal responses of approximately 40 and 30%, respectively, irrespective of level of hypoxia. These findings of “duration-dose effect” likely reflect an interaction between the strength and duration of activation of the collaterogenesis and remodeling pathways, as well as the time required to form new collaterals and remodel the native ones. No statistically significant remodeling occurred at 12% FIO2, in association with the smaller increase in hematocrit (Figure 3(b) and (c)).
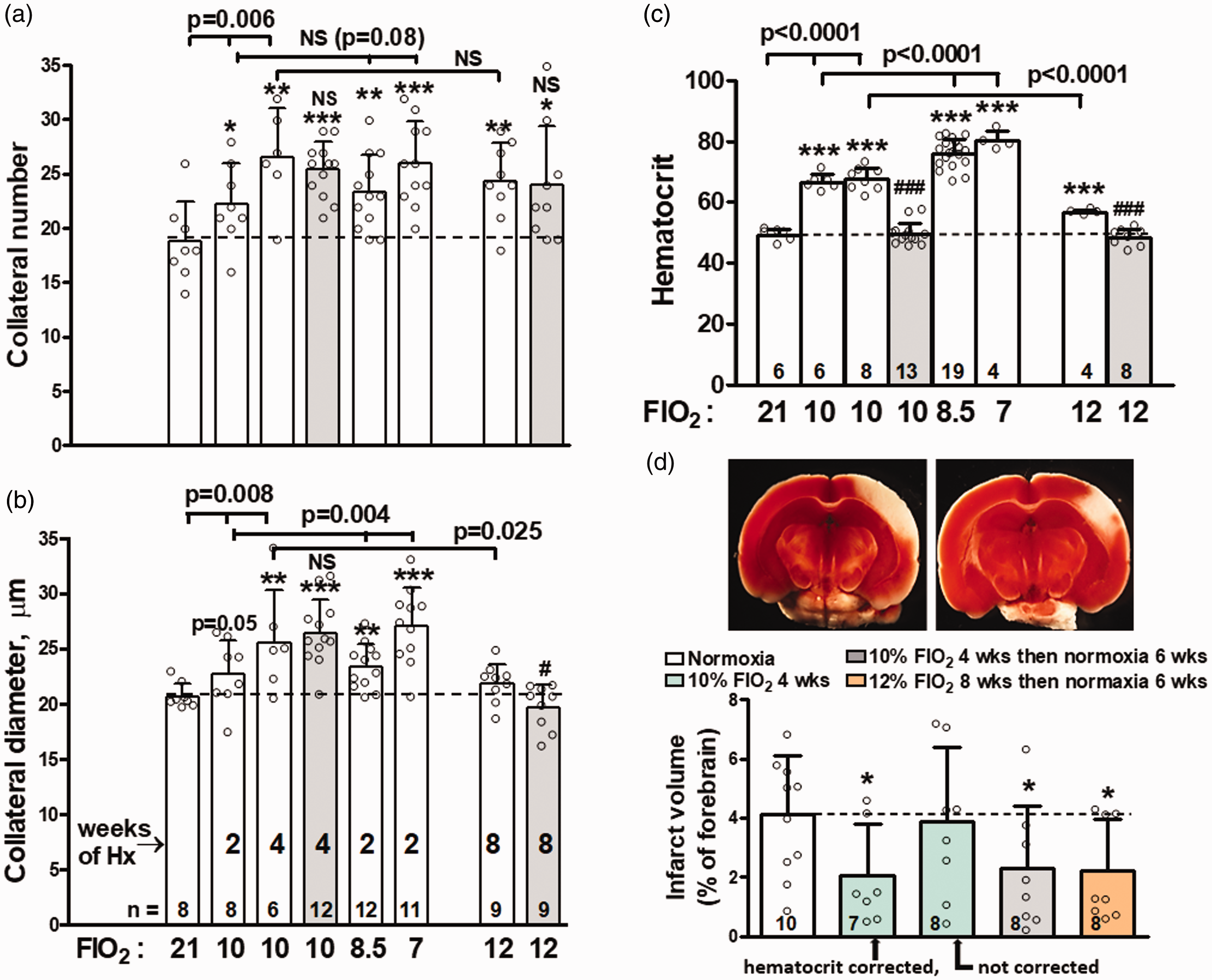
Hypoxia induces dose- and duration-dependent increases in collateral number and diameter and smaller infarct volumes that persist on return to normoxia. (a,b) Collateral number and diameter. (c) Hematocrit determined at conclusion of hypoxia (Hx) period. (d) Infarct volume (top, representative images) determined 24 h after pMCAO performed after the number of weeks (wks) indicated. Black bars, hematocrit “corrected” (or not corrected) by removal of blood and replacement with mouse plasma, to reduce hematocrit, on day-5 after return to normoxia (21% FIO2) just before pMCAO. 7% hypoxia data in a–c are from Figure 1. Gray and orange bars in a–d show that hypoxic NCF, collateral remodeling and smaller infarct volumes persist despite having returned to normoxia for six weeks and normalization of hematocrit. (a–c) bracket above bars gives ANOVA value for three comparisons indicated or t-test value for two comparisons indicated; NS: not significant (p ≥ 0.05); #,###,NS, p < 0.05, <0.001, ≥0.05 above gray bars is for t-test vs. preceding bar. * , ** , ***p < 0.05, 0.01, 0.001 vs. 21% FIO2/normoxia. All tests pre-specified as one-sided.
Hypoxic NCF was durable. There was no significant decline in collateral number six weeks after return to normoxia (Figure 3(a) to (c)). Remodeling was also sustained despite normalization of hematocrit which occurs within 7–14 days after return from 10% FIO2. 30 , 31 Neo-collateral formation was associated with a 50% decrease in infarct volume measured 24 h after pMCAO (Figure 3(d), see Supplemental Figure II for body weight and absolute and normalized forebrain volume and infarct volume). Occlusion was performed five days after return to normoxia to allow re-acclimation and hematocrit to decline naturally by neocytolysis from 67.5 ± 3.8 on day 29 (Figure 3(c)) to 60.2 ± 7.1. As expected, infarct protection only occurred if the increased hematocrit (thus increased viscosity) was normalized (“corrected”) the day of removal from hypoxia by withdrawal of blood and replacement with serum (Figure 3(d), hematocrit for bar-2 group, 51.7 ± 3.8 is not significantly different from the normoxia values of 48–49 in Figure 3(c)). Infarct volume remained reduced when pMCAO was performed six weeks after return to normoxia, well after hematocrit had naturally normalized.
Hypoxia increases expression of genes within the collaterogenesis pathway
Given the involvement of certain angiogenic genes 11 , 12 as well as requirement of the novel gene, Rabep2, in embryonic collaterogenesis but not developmental, newborn or adult angiogenesis, 10 we examined mRNAs in adult neocortex for Rabep2 and several angiogenic genes known to be driven by hypoxia→Hif signaling (Figure 4). Expression varied when examined 24 h after exposure to 10% FIO2. Thereafter, Hif2α, Vegfa, Rabep2, Angpt2, Tie2 and Cxcr4 were increased after one week of hypoxia and, with the exception of Angpt2, remained elevated at two weeks. Expression was normalized to β-actin which unlike 18 s ribosomal mRNA was not altered by hypoxia.
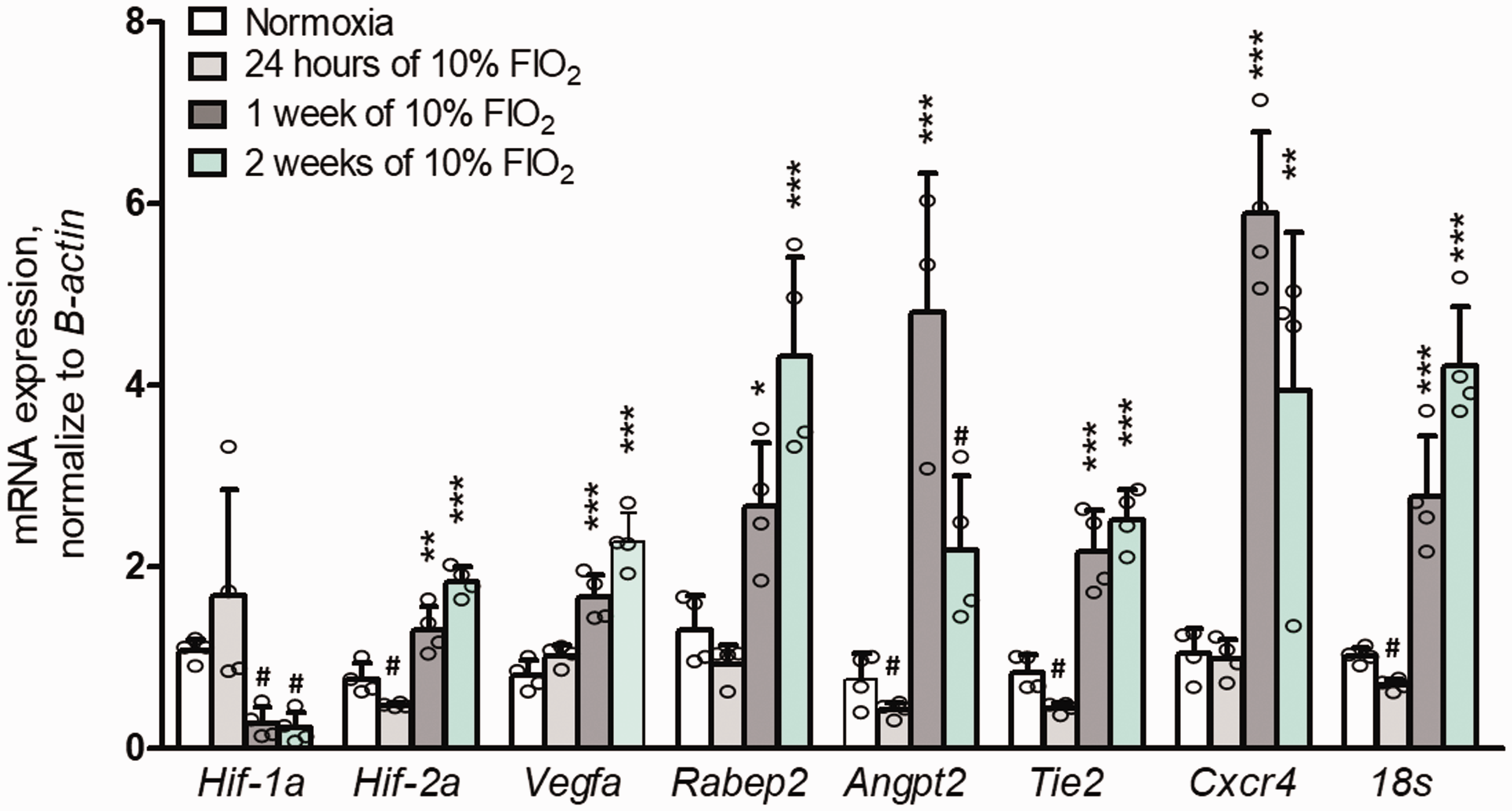
Hypoxia increases expression of angiogenic genes. Three days of acclimation (16%, 12%, 12%) preceded indicated durations of 10% FIO2. Neo-cortex frozen immediately after removal from hypoxia chamber. n = 4 mice per group. * , ** , ***p < 0,05, 0.01, 0.001, ANOVA followed by Bonferroni two-sided t-test for three pre-planned comparisons to normoxia. #p < 0.05 by two-sided t-test vs. normoxia. Bars 2–4 were each significantly different from normoxia (p < 0.05) by two-sided t-tests for all genes except bar-2 of Hif1a, Vegfa (p = 0.07), Rabep2 (p = 0.13) and Cxcr4.
Hypoxic neo-collateral formation is inhibited by knock-down of Vegfa, Flk1 and Cxcr4
Previous studies found that collaterogenesis in the embryo and collateral remodeling in the adult following arterial occlusion are reduced in mice deficient in either VEGF-A or Flk1. 12 As well, hypoxia increased Cxcr 4 expression (Figure 4) and induced SDF1 expression, 32 , 33 and MCAO stimulated SDF1 release from a number of cell types that recruit immune and endothelial progenitor cells to regions undergoing neovascularization.34–38 Furthermore, SDF1 and CXCR4 are required for collateral formation after occlusion of the dorsal aorta in zebrafish embryos. 39 Given the above and the assumption that pathways that drive collaterogenesis and NCF likely have certain factors in common, we generated mice with deficiencies in Vegfa, Flk1 and Cxcr 4 . Hypoxic NCF was abolished by conditional knockdown of Vegfa and Cxcr4, and reduced by conditional knockdown of endothelial cell (EC)-specific Flk1 (Figure 5). Remodeling was not significantly reduced. Efficiency of knockdown in neocortex for Vegfa, Flk1 and Cxcr4 averaged 73, 81 and 64%, respectively (Supplemental Figure II).
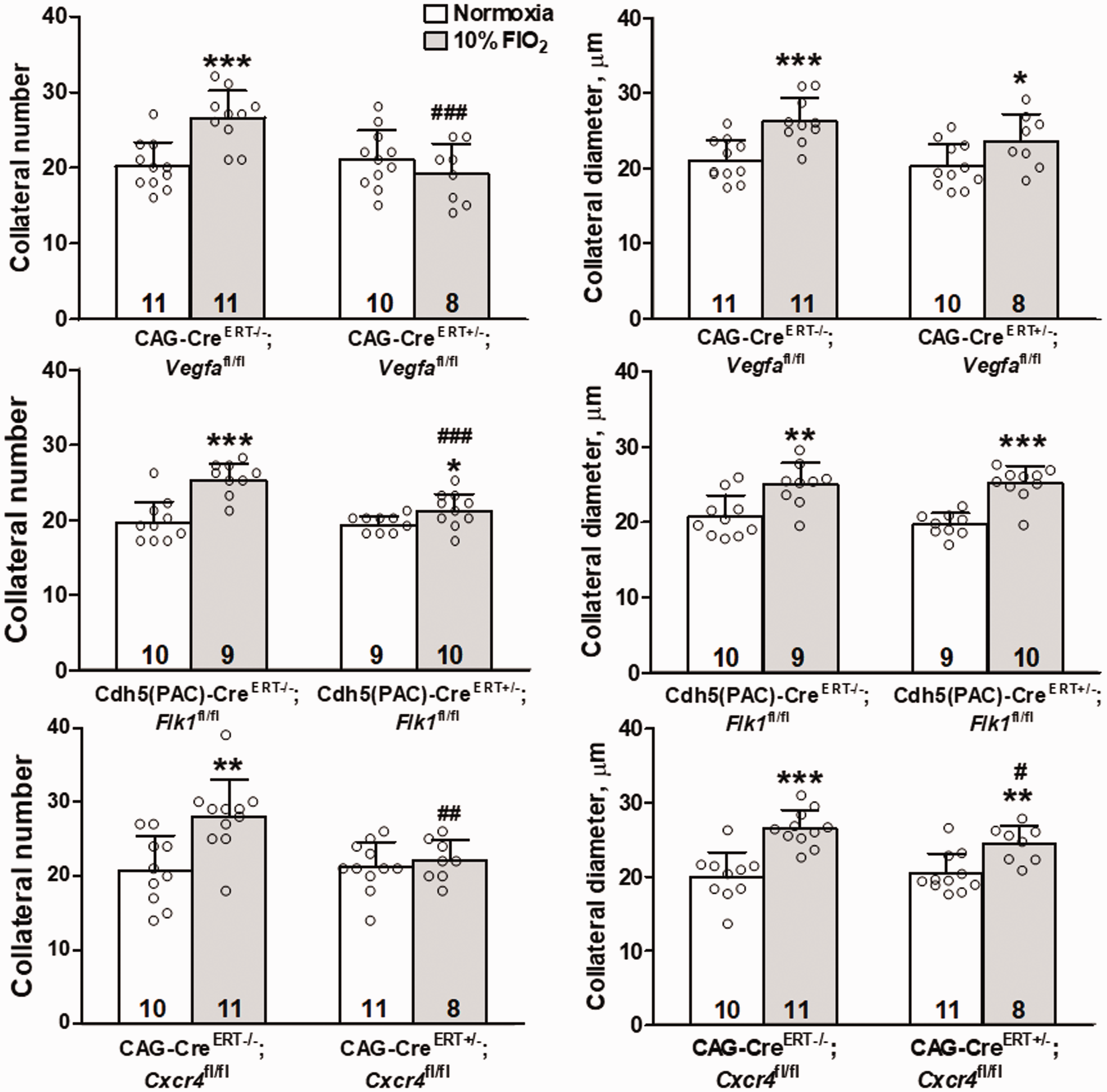
Hypoxic neo-collateral formation is inhibited by knockdown of VEGF-A, Flk1 and CXCR4. Data are for MCA-to-ACA collaterals. Four weeks exposure to 10% FIO2. * , ** , ***p < 0,05, 0.01, 0.001, pre-planned one-sided t-test vs. normoxia. #,##,###p < 0,05, 0.01, 0.001, pre-planned one-sided t-test vs. Cre − / − group. Efficiency of knockdown in neocortex for Vegfa, Flk1 and Cxcr4 averaged 73, 81 and 64%, respectively (Supplemental Figure II).
Evidence that reduced tissue oxygen is a proximal stimulus of NCF: Permanent MCA occlusion induces NCF
We next asked whether reduced tissue oxygen stimulates NCF in a setting where inspired oxygen is normal. We previously reported 5 that pMCAO induced a 2-to-4 fold increase in MCA-ACA collaterals, i.e. NCF, in the ipsilesional hemisphere six days after pMCAO (maximum NCF occurred between three and six days post-occlusion) in BALB/cBy, SWR, AKR and A/J strains. These strains have a low number of native MCA-ACA collaterals (1–4 per hemisphere). In contrast, no increase occurred in C57BL/6 (B6) mice and 10 other strains with high native collaterals (5–11 per hemisphere). It is possible that NCF occurred in the low- but not high-collateral strains due to shared difference(s) in genetic background, given the genetic relatedness of the strains. However, it is also possible that NCF requires reduced tissue oxygen as a stimulus and that NCF can only occur, by definition, in the anatomic watershed zone between the artery trees where collaterals reside. Accordingly, tissue oxygen in the MCA side of the watershed region of the low-collateral strains would be reduced after pMCAO due to the watershed’s proximity to the strains’ large evolving infarctions; however, this would not occur in the high-collateral strains whose small infarctions are restricted to the proximal MCA tree well away from the watershed zone (Figure 7(a) and (b)). We therefore examined NCF six days after pMCAO in a population of 162 three-months-old F2 mice that we created by reciprocal mating of B6 and BALB/cBy mice. Each B6 × BALB-F2 mouse has a distinct, randomly assorted but closely related genome, thus a broad range of native collateral numbers exists among the 162 individuals. 8 In support of the above hypothesis, pMCAO stimulated NCF in mice with low- but not high-collateral number (Figure 6(a) and (b)). Figure 6(c) and (d) shows that NCF was absent in Rabep2−/− mice even though they have low native collaterals, 10 while remodeling of pre-existing collaterals was unaffected, as also seen in the data in Figures 1 and 3.
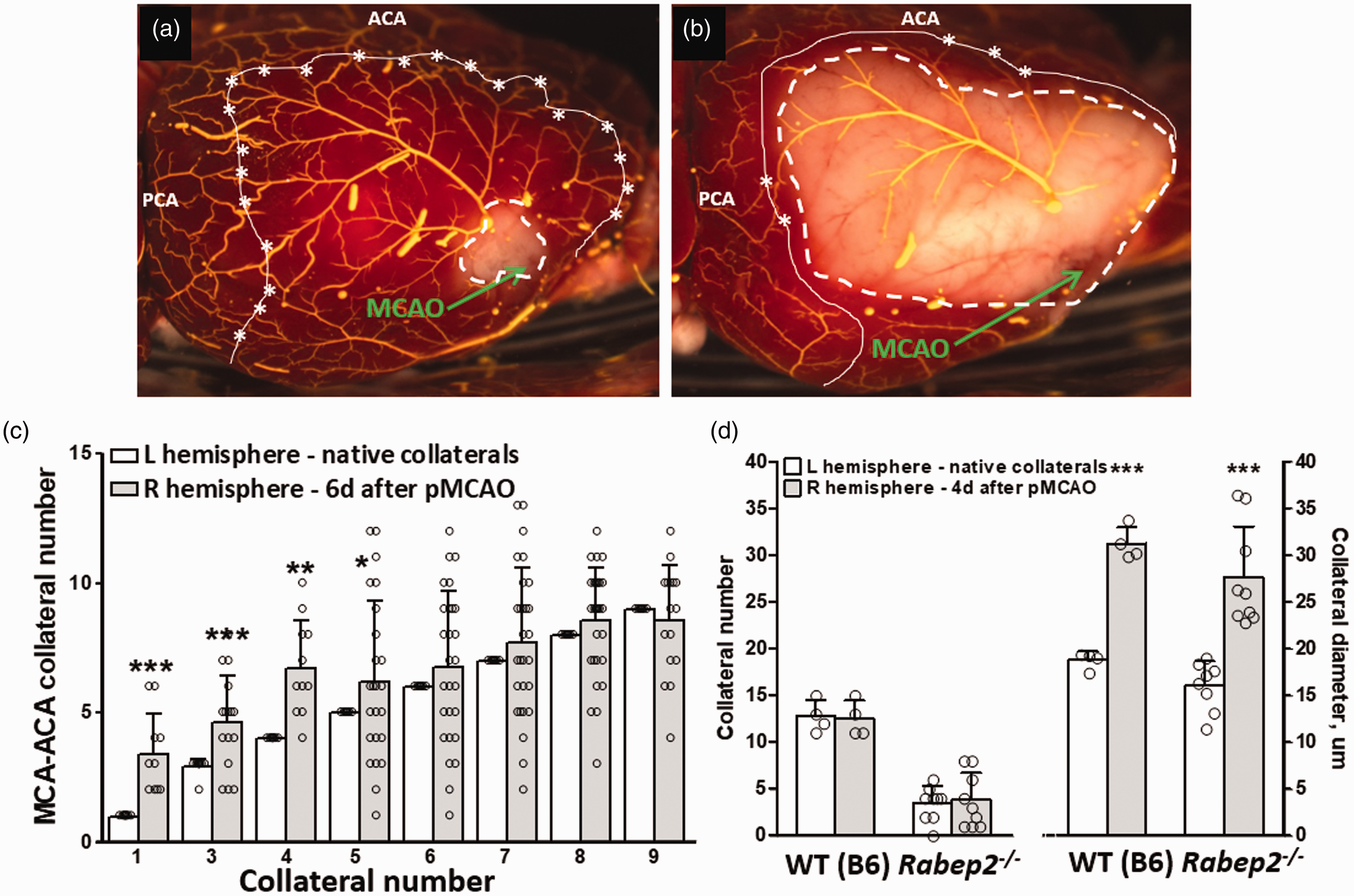
Permanent right distal M1-MCA occlusion induces neo-collateral formation in F2 mice with low but not high number of native collaterals. (a–c) 162 three-months-old F2 mice were created by reciprocal mating of C57BL/6 (B6) and BALB/cBy to produce mice with randomly assorted genomes and thus varying numbers of native collaterals. (a,b) TTC-stained brains of high- and low-collateral F2 mice six days (d) after right pMCAO. Stars and solid line, collaterals in anatomic watershed zone between MCA, ACA and PCA trees; dashed line, ∼location of functional watershed defined by extent of retrograde collateral perfusion of territory downstream of occlusion site (arrows) sufficient to prevent cell death. (c) Frequency plot of number of native collaterals (white bars) and neo-collaterals (gray bars) in the population (16–31 mice per bar-pair); e.g. mice with four native collaterals in left hemisphere (3rd white bar, n = 26) had seven collaterals in the right hemisphere six days after pMCAO (3rd gray bar). (d) MCA-ACA collateral number and diameter; n = 4 and 9 for WT and Rabep2−/−, respectively. Similar to hypoxic NCF in Figure 1, no NCF occurred post-MCAO in low-collateral B6.Rabep2−/− mice, whereas remodeling was unaffected. * , ** , ***p < 0.05, 0.01, 0.001, pre-specified one-sided t-test.
Figures 1 and 6(d) show that Rabep2 is required for NCF induced by hypoxia and pMCAO. A potential point of confusion arises regarding the F2 population of mice in Figure 6(c), wherein the deficient/low activity allele of Rabep2 present in the parental BALB/cBy genome contributes to the low collaterals at baseline in the low-collateral F2 progeny. 10 However, a previous study showed that two negative and one positive loci, in addition to the positive locus harboring Rabep2, contribute to variation in collateral abundance in BALB/cBy x B6 F2 mice, 8 which offers one possible explanation for why the low-collateral mice still exhibited NCF.
Differentiation of neo-collaterals from native pre-existing collaterals
Lastly, we sought to determine if neo-collaterals could be differentiated from native collaterals. Because no distinguishing molecular marker has been identified, we first asked whether neo-collaterals could be identified based on diameter, since they would be expected to have smaller diameters for some duration after their formation. We examined data from the 12% hypoxia group shown in Figure 3 because eight weeks of 12% hypoxia did not cause significant collateral remodeling, which otherwise would diminish the ability to detect two such populations. However, no bimodal distribution for collateral diameter was evident (Supplemental Figure IIIA). This presumably reflects that by eight weeks, the neo-collaterals had matured to full diameter and blended with the native collaterals. Bimodal distributions were also not evident in the 7% 2-week and 10% 4-week hypoxia groups in Figure 3 or the 7% 2-week Rabep2−/− groups in Figure 1 (Supplemental Figures IIIB-D). This was expected a priori however, given the remodeling that occurred in these groups, including rapid remodeling of the neo-collaterals and thus a thorough blending of them with the also-remodeled native collaterals.
As a second approach, we examined the hypothesis that neo-collaterals can be distinguished from native collaterals by increased proliferation. Mice were acclimated to 12% hypoxia or normal FIO2 over three days and then received EdU on days 6, 10, 18, 26, 34 and 42. Labeling was evident in the collaterals of the hypoxia group but virtually absent in the normoxia group (Figure 7); 3.5 ± 2.1 collaterals had 1 EdU+ EC and 3.5 ± 2.1 had ≥ 2 EdU+ ECs per collateral. This labeling of seven collaterals per mouse, on average, agrees with the number of neo-collaterals induced to form by 12% hypoxia (Figure 3). The small number of their ECs having undergone proliferation is consistent with findings in chick yolk sac where formation of collaterals induced by unilateral occlusion of the vitelline artery occurred almost entirely by migration and reorganization of endothelial cells recruited from the surrounding capillary-venous plexus. 40 The same also appears to underlie embryonic collaterogenesis. 12 A small number of EdU+ non-ECs—likely smooth muscle cells, pericytes, fibroblasts, and/or myeloid cells—were also associated with these presumed neo-collaterals (Figure 7). While these findings support the hypothesis that neo-collaterals and native collaterals can be distinguished by proliferation, confirmation awaits identification of a unique molecular marker of neo-collaterals.
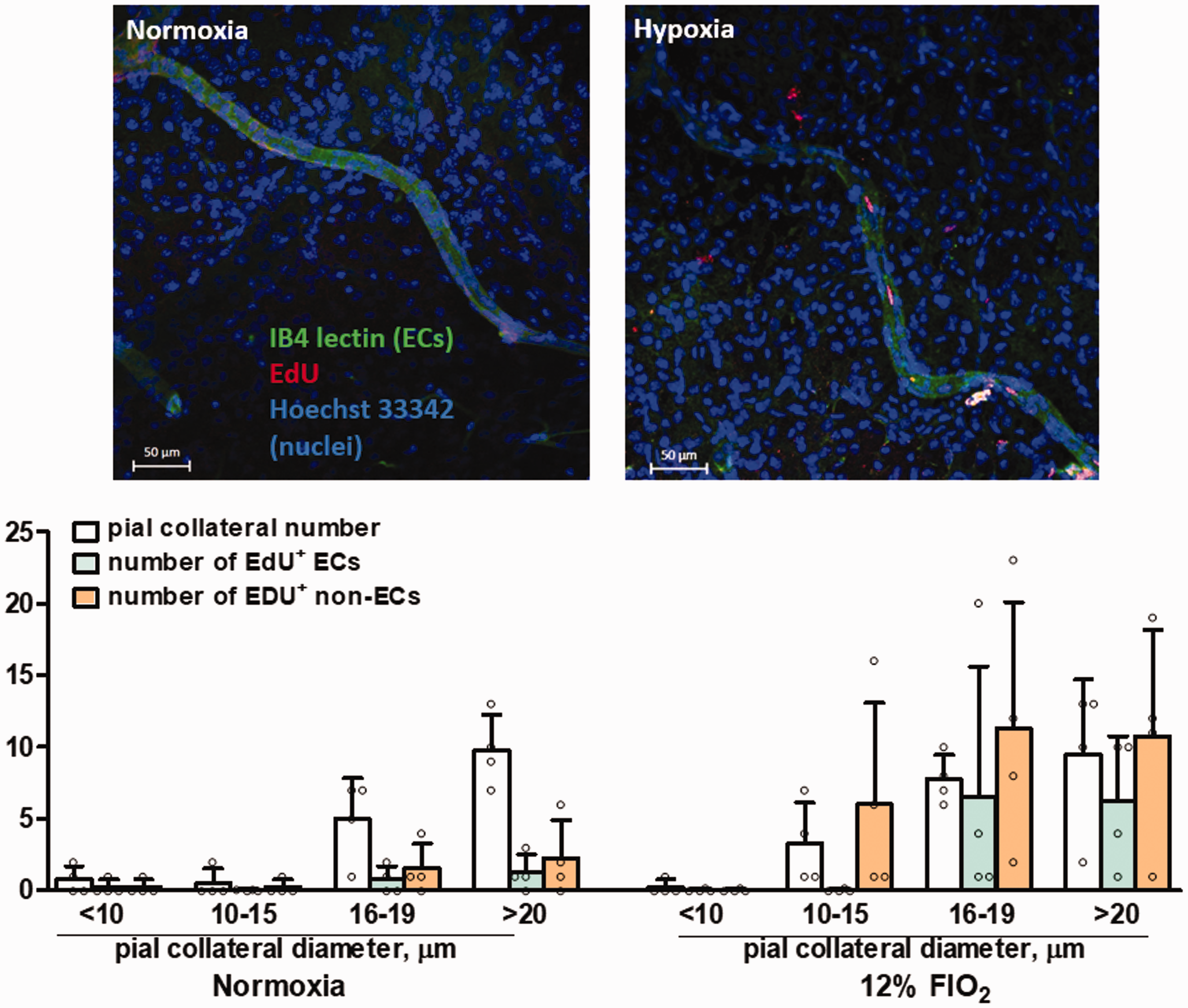
Hypoxia induces proliferation of collateral endothelial cells. 10-week-old C57BL/6 mice were acclimated to 12% hypoxia or normal FIO2 over three days, then injected with EdU (1 mg, ip) on days 6, 10, 18, 26, 34 and 42. Parasagittal whole-mounts of the MCA-ACA watershed of both hemispheres were examined on day-45. Upper panels, representative images of collaterals in normoxia and hypoxia mice (n = 4 each). Lower panels, key gives parameters determined for collaterals of four sizes. A small number of EdU+ ECs were evident in the collaterals of hypoxia mice but virtually absent in normoxia mice. 3.5 ± 1.0 collaterals had 1 EdU+ EC and 3.5 ± 1.0 had ≥ 2 EdU+ ECs per collateral. This labeling of seven collaterals per mouse agrees with the number of neo-collaterals induced to form by 12% hypoxia (Figure 3). A small number of EdU+ non-ECs, likely smooth muscle cells, pericytes, fibroblasts and/or myeloid cells, were located within or perivascular to these presumed neo-collaterals. Magnification bar, 50 μm.
Discussion
This study reports several notable findings. Exposure to systemic hypoxia (i.e. hypoxemia) stimulated additional collaterals to form and outward remodeling of those present before exposure. These effects were accompanied by a 50% decrease in infarct volume following pMCAO. Neo-collateral formation and remodeling were specific for pial collaterals: no formation occurred of additional branches off the MCA tree, intra-tree collateral-like anastomoses (ITAs) or PCom collateral arteries, nor did remodeling occur of primary intracranial or pial arteries/arterioles. Intriguingly, the new collaterals that formed, remodeling of native collaterals, and reduction of infarct volume were durable, evidencing no regression when examined six weeks after return to normoxia. These findings stand in contrast to other adaptations to hypoxia, such as polycythemia, pulmonary hypertension, metabolic changes, 41 and increase in cerebral capillary density. 24 , 26 , 28 ,42–44 For example, the latter accompanied two to three weeks of 10% hypoxia, reversed over the same time-frame on return to normoxia, evidenced a threshold of seven days of 12–13% FIO2, and exhibited no further increase on exposure to 8 or 7%. 28 , 42 , 44 These findings of angiogenesis, apart from the reversal, mirror our findings for NCF. Hypoxia increased expression of Rabep2, which is involved in VEGF-A→Flk1 signaling 10 , 45 and is a key element in the collaterogenesis pathway. 10 Neo-collateral formation was abolished in mice lacking Rabep2 or having undergone knockdown of Vegfa or Cxcr4 and was reduced after knockdown of Flk1. Thus hypoxia in the adult appears to re-activate the embryonic collaterogenesis pathway or a closely related one. Permanent MCAO also induced NCF. However, unlike hypoxic NCF that occurred in mice with abundant collaterals at baseline, NCF only occurred in mice with low-to-intermediate numbers of native collaterals. This plus our finding that hypoxemia, alone, elicited NCF suggests that a trifecta of tissue hypoxia, increased shear stress, and their occurrence within the watershed region between adjacent arterial trees are required for NCF.
Neo-collateral formation induced by hypoxia and MCA occlusion
It had long been regarded until recently5,18–22 that arterial obstruction in brain, heart and other tissues, while stimulating native collaterals to remodel, does not induce new ones to form.46–48 In brain, this conclusion was based on finding no increase in collateral number after pMCAO, e.g. in Wistar rats and CD1 mice.49–52 However, these and other rat strains examined previously have abundant pial collaterals at baseline. Likewise, our current and previous study 5 observed no NCF after pMCAO in 11 strains of mice with abundant native collaterals. Of note, a study employing perfusion contrast imaging reported that transient MCAO in Wistar rats induced formation of new pial arteriole anastomoses. 53 However, it is not clear whether these were either native collaterals or ITAs, both having diameters too small for detection before occlusion, that subsequently underwent dilation or remodeling. Interestingly, a presumed hypoxia-driven type of NCF occurs in patients with pMCAO caused by steno-occlusive moyamoya disease or following indirect surgical revascularization to relieve chronic ischemia in the MCA territory.54–58 Also of note, Marushima et al. 59 reported that indirect bypass surgery using implantation of the temporalis muscle with myoblasts expressing VEGFA improved outcome after pMCAO in mice. Our finding that systemic hypoxia and pMCAO induce NCF is consistent with preliminary reports in adult mouse heart for systemic hypoxia 21 , 22 and following coronary artery ligation, 20 with the latter recently being confirmed in neonatal mice. 60 Thus, new collaterals can be induced to form in adults in brain, heart and other tissues.
Hypoxia-induced remodeling
Remodeling of arteries and arterioles in peripheral tissues accompanies systemic hypoxia. 61 , 62 This is due to an increase in fluid shear stress arising from the vasodilation, increased cerebral blood flow (CBF) and increased viscosity (hematocrit) that accompany hypoxia,24,26,42,43,62 which are known to stimulate endothelial and smooth muscle cell proliferation. We propose that the same mechanism underlies hypoxic remodeling of collaterals, which to our knowledge has not be reported previously, with the caveat that it begins from a different starting point: In the absence of obstruction, flow in collaterals slowly oscillates to and fro and averages zero, at least in anesthetized mice. 11 , 63 We postulate that hypoxic collateral remodeling is stimulated by an increase in the magnitude of this oscillatory shear stress caused by cerebral vasodilation and increased viscosity. In the case of pMCAO, the situation is simpler, i.e. remodeling is well-known to arise from the sudden sustained induction of unidirectional flow/shear stress across collaterals. 47 , 62 Unlike NCF, collateral remodeling induced by hypoxia and pMCAO was unaffected in Rabep2−/− mice. This agrees with previous findings for occlusion-induced remodeling of pial and hindlimb collaterals, 9 and supports other evidence that collaterogenesis and collateral remodeling depend on different signaling pathways. 5 , 6 , 8 Remodeling of pial collaterals following pMCAO was stronger (∼200% increase in diameter, Figure 6(d) and Zhang et al. 5 ) than the ∼30% increase induced by hypoxia (Figures 1 and 3). This may reflect the large unidirectional increase in shear stress following pMCAO versus accentuation of oscillatory shear stress during hypoxia.
Interestingly, in contrast to pial collaterals (Figures 1, 3, and 5) and peripheral arteries and arterioles, 61 , 62 hypoxia did not induce remodeling of the primary intracranial arteries, PCom collateral arteries or branches of the MCA tree (although a trend was evident), with the exception of the basilar artery (Figure 2). Boroujerdi and Milner 24 reported that 10% hypoxia caused a 9% increase in the diameter of intracerebral α-smooth muscle actin-positive vessels (presumably penetrating arterioles) in mice. The failure of cerebral and pial arteries to undergo remodeling may be a consequence of the increase in hematocrit being offset by the decrease in CBF seen during chronic hypoxia. 43 It is also possible that cerebral arterial vessels are less sensitive to hypoxic remodeling. This could be a specialization that reflects the requirement to maintain a near-constant volume within the calvarium. Remodeling of the basilar artery, on the other hand, may arise from its greater diameter thus volume flow, as well as to unique hemodynamic forces favored by its juxtaposition between the converging vertebral arteries and the variably present, small-diameter PComs present in C57BL/6 and certain other strains of mice (see Faber et al. 29 and references therein).
Twelve percent FIO2 had the same protective effect against infarct volume as 10% (Figure 3(d)). This suggests that hypoxia-induced collateral remodeling (Figure 3(b)) is not as important for protection as the increase in collateral number (Figure 3(a)), since 12% did not cause remodeling (presumably due to the lack of an increase in hematocrit thus shear stress – Figure 3(c)). Although flow is proportional to diameter to the 4th power, several factors likely reduced its impact and increased the effect of the increase in collateral number: Increased collateral tortuosity, which lessens flow due to increased path length and rheologic considerations, is well known to accompany post-stroke remodeling. 5 , 13 , 49 , 50 Second, if for example 10 collaterals cross-connect the crowns of the ACA and MCA trees, the aggregate collateral flow to the MCA territory post-occlusion encounters less aggregate resistance as it retrogradely perfuses the MCA tree over a wider area of inputs than would be encountered if it were mediated by a small number of collaterals/inputs. Third, since significant oxygen is well known to diffuse across small arteries and arterioles into the tissue parenchyma, oxygen delivery to the penetrating arterioles and capillaries supplying the MCA territory, and thus overall oxygen delivery, will be better achieved by a large number of smaller diameter collaterals than a small number of large collaterals.
Durability of hypoxic neo-collateral formation, collateral remodeling and infarct protection
The increase in collateral number and diameter induced by hypoxia was accompanied by a 50% decrease in infarct volume when pMCAO was performed five days after return to normoxia to allow time for re-acclimation plus procedural normalization of hematocrit to pre-pMCAO values. Importantly, the smaller infarct volume was also sustained following pMCAO done six weeks after return to normoxia when hematocrit had naturally normalized by neocytolysis. This is in accordance with the absence at six weeks of any pruning away of the neo-collaterals or reversal of collateral remodeling. Since neocytolysis normalizes hematocrit within one to two weeks after return to normoxia from levels and durations of hypoxia similar or in excess of the four weeks of 10% hypoxia used in this experiment, 30 , 31 the newly formed collaterals and remodeling were sustained well after polycythemia had resolved. This is an intriguing and unexpected finding. It suggests periods of sustained hypoxemia, such as sojourns to high altitude, may have trophic effects specifically on the collateral circulation that are retained well after returning to normoxia. Whether the neo-collaterals and remodeling of native collaterals persist for several months or indefinitely awaits additional study. Interestingly, age-adjusted mortality rates for males, but not females, with coronary artery disease decline from the lowest to the highest according to altitude of residence. 64
Hypoxia re-activates the embryonic collaterogenesis pathway or a similar pathway
As expected, hypoxia increased expression of Hif2α, Vegfa, Angpt2, Tie2 and Cxcr4, genes known to be induced by hypoxia in adult brain. 28 , 41 , 42 ,65–67 At baseline, VEGF-A is primarily expressed in neurons and glial end-feet, Hif2α in glia, Angpt2 and Tie2 in ECs, and Hif1α and Rabep2 are expressed ubiquitously. 10 , 42 , 68 Hypoxia also caused a sustained increase in expression of Rabep2, a novel gene that is critical in the VEGFA→Flk1→Rabep2→Notch→ADAM10/17→Clic4 pathway that directs collaterogenesis during development. 10 , 12 , 69 Our observation that hypoxic NCF was blocked in Rabep2−/− mice is consistent with evidence that reduced oxygen, which under baseline conditions in normoxia is lowest in the vessels within the watershed zone between adjacent arterial trees,70–75 induces the same or a similar pathway in the adult.
Hypoxic NCF was abolished and reduced, respectively, by conditional knockdown of Vegfa and EC-specific Flk1, in agreement with the major role of VEGF-A→Flk1 signaling in collaterogenesis. 12 , 76 Neo-collateral formation was also abolished by knockdown of Cxcr4 which binds hypoxia- and Hif-induced release of SDF1 (CXCL12). While involvement of SDF1→CXCR4 in collaterogenesis in the brain has not been examined, SDF1 promotes vasculogenesis, ischemic neovascularization, and mobilization of hematopoietic stem/progenitor cells.77–79 Ischemia and inflammation stimulate microglia and astrocytes to release SDF1 post-stroke, which stimulates homing of CXCR4-expressing leukocytes and mesenchymal stem cells that secrete VEGF-A, angiopoietin-2 and other angiogenic factors.34,36,38,80–82 Mesenchymal stem cells can differentiate into myeloid, lymphoid and endothelial progenitor cells (EPCs) 80 and contribute to angiogenesis within the penumbra which is thought to play an important role in survival and regeneration of neurons post-stroke. It is possible that ligation of CXCR7 by SDF1 could also contribute to NCF. Stimulation of CXCR7 on EPCs promotes their recruitment, angiogenesis, smaller infarct volumes and improved behavioral scores after MCAO. 83 However, we found no residual NCF after knockdown of Cxcr4. SDF1 also stimulates proliferation, migration and differentiation of CXCR4-expressing neural stem/progenitor cells to the penumbra. 84 , 85 Interestingly, Das et al. 60 recently reported that coronary artery ligation in neonatal mice stimulated collateral formation by inducing CXCR4-expressing arterial endothelial cells to migrate onto SDF1-expressing capillaries. Our expression and knockdown findings suggest that hypoxia in the adult re-activates a signaling pathway similar to the pathway that drives collaterogenesis during gestation. Potential involvement of SDF1→CXCR4 in the latter awaits investigation.
Neo-collateral formation post-MCA occlusion is restricted to mice with poor-to-intermediate native collaterals
Permanent MCA occlusion stimulated NCF in C57BL/6 x BALB/cBy F2 mice with low but not high numbers of native collaterals. This extends our previous study 5 wherein pMCAO induced a 2-to-4 fold increase in MCA-ACA collaterals in BALB/cBy and 3 other strains with low collaterals at baseline, while no increase occurred in 11 strains with high collaterals. In that study, NCF reached a maximum between three and six days after pMCAO, which is faster than the seven days required for the onset of intracerebral angiogenesis induced by hypoxia. 42 , 44 The above findings were recently confirmed for the low-collateral BALB/c 86 and high-collateral CD1 52 strains. Neo-collateral formation in low- but not high-collateral mice could arise from a difference in their genetic backgrounds. However, the 162 F2 mice examined herein each have closely related genetic backgrounds made mosaic by random meiotic recombination. This suggests that NCF in low- but not high-collateral mice arises from a non-genetic mechanism, leading us to propose a more parsimonious hypothesis, namely that NCF requires reduced tissue oxygen as a stimulus and that it can only occur in the anatomic watershed zone where collaterals reside. Accordingly, oxygen levels following pMCAO in the watersheds of low-collateral strains 5 , 10 and F2 mice (Figure 6) would be reduced by their large nearby evolving infarct cores, but not reduced in the high-collateral strains whose small infarcts in the proximal MCA tree cause the core and penumbra to be well away from the watershed. In support, the fractional increase in collaterals in low-collateral strains was inversely related to their native number and distance between their infarct core and watershed. 5
Proposed mechanism for neo-collateral formation
Our findings that: (1) NCF is induced by both systemic hypoxia and pMCAO—the latter in low- but not high-collateral mice,( 2) that both scenarios require Rabep2, (3) that hypoxic NCF is abolished or diminished when Vegfa, Flk1 and Cxcr4 are reduced, (4) that hypoxemia increases expression of these and other genes in the collaterogenesis pathway, and (5) that little proliferation is involved suggesting that NCF results from muralization-then-enlargement of pial watershed capillaries that is stimulated by reduced oxygen and increased fluid shear stress, support the following model/hypothesis: The capillaries in the watersheds of the pia (and other tissues) are arranged in a plexus. 11 , 12 Perfusion of the plexus by the adjacent arterial trees normally proceeds down the trees’ pressure gradients and venous outflow pathways.87–89 At baseline, oxygen content within watershed capillaries is low because they are the “furthest away from the aorta,” which is consistent with the watershed stroke phenomenon70–75 ; the above-mentioned negligible flow in collaterals at baseline would also favor their PO2 values being closer to tissue PO2. In the case of systemic hypoxemia, the PO2 in watershed capillaries declines further and activates the collaterogenesis signaling pathway. Accompanying this hypoxic stimulus, the increase in CBF early during hypoxemia 42 , 43 increases the magnitude of oscillatory flow within the plexus, which can also convert to unidirectional flow during regional changes in neuronal activity induced by awake behaviors. These changes plus the slowly increasing hematocrit/viscosity result is an increase in shear stress in the plexus capillaries, particularly in the small fraction of large-diameter capillaries extant in capillary beds. 90
Continuing with this hypothesis, in the case of pMCAO, occlusion favors a shift in the point of convergence of MCA with ACA and PCA flows, which at baseline is in the center of the watershed plexus and its collaterals, toward the crown of the MCA tree. When collaterals are in abundance, this result in the outer MCA tree being retrogradely perfused with oxygenated blood from the ACA and PCA trees, displacing the “functional watershed” towards the proximal MCA region surrounding the ischemic core (dashed line in Figure 6(a)). Hence, capillaries within the “anatomic” watershed experience no hypoxia and thus no NCF occurs. However, in individuals with intermediate or poor collaterals, PO2 is reduced in the plexus due to its proximity to the ischemic MCA tree. And as with hypoxemia, the largest-diameter capillaries will evidence the largest increase in flow/shear stress following pMCAO. In both scenarios, the increased shear stress induces their ECs to migrate and close off their venous side-connections, 40 , 60 resulting in them becoming “collateralized,” followed by muralization by migrating or pericyte-transformed smooth muscle cells, then lumen enlargement and formation of a neo-collateral. In support of this “capillary collateralization” hypothesis, within 36 h after arterial occlusion in the chick yolk sac and zebrafish embryo, a small fraction of the capillaries within the plexus between the opposing trees begin to undergo collateralization. 39 , 40 , 91 Furthermore, something similar to the above occurs in skeletal muscle of BALB/c mice after arterial occlusion. 18 , 19
The above model/hypothesis proposes that a combination of reduced oxygen and increased shear stress drive NCF. Furthermore, collaterals, by definition, only exist in the watershed zone, thus neo-collaterals can only form there because the substrate for their formation—an inter-tree capillary plexus—is only found there. In agreement, hypoxemia did not increase the number of branches or branchlets within the MCA tree, nor the number of collateral-like intra-tree anastomoses between MCA branches (Figure 1). These vessel locations are proximal to the watershed region and thus have high (i.e. arterial) oxygen levels and continuous orthograde flow at baseline as well as after pMCAO. 11
The findings in our study examining hypoxemia suggest a corollary hypothesis to the above model, i.e. that collaterals, besides providing protection against ischemia in occlusive disease, also serve a physiological function in healthy tissues by interconnecting adjacent arterial trees and thus optimizing oxygen delivery to meet oxygen demand when oxygen availability is limited. And that hypoxic NCF is therefore an adaptive response. Support for this hypothesis derives from the fact that remodeling of the primary intracranial arteries, PComs and branches of the MCA induced by hypoxemia, which was absent in wildtype mice, became robust in Rabep2−/− mice. We propose this arises because of the reduced density and diameter of their collaterals at baseline and loss of NCF during hypoxia. These deficiencies would favor additional cerebral vasodilation and increased flow/shear stress within the primary intracranial arteries and pial artery trees, resulting in their remodeling which would help maintain oxygen delivery. Evidence from several studies support the concept that hypoxic NCF is an adaptive response: Patients with chronic obstructive pulmonary artery disease, cyanotic heart disease and sleep apnea have unusually abundant coronary collaterals,92–99 as do piglets kept at low FIO2. 100 Chronic anemia in dogs is accompanied by increased coronary collateral conductance,101–103 and angiography in humans with coronary artery disease finds the same. 104 , 105 In addition, moyamoya syndrome occurs in sickle cell anemia. 54 , 55
Study limitations
This study has several limitations. An increase in capillary density induced by hypoxia 28 , 42 , 44 could contribute to the 50% reduction in infarct volume seen in the 10% hypoxia group five days after return to normoxia (Figure 3(d), hematocrit corrected group). However, infarct volume remained reduced by the same amount six weeks after return to normoxia in both the 10 and 12% hypoxia groups, well after the two to three weeks required for capillary density to return to normal. 28 , 42 , 44 We did not assess the contribution of NCF versus remodeling of native collaterals to the decrease in infarct volume in the 7, 8.5 and 10% hypoxia groups. However, in the 12% group where remodeling was absent, infarct volume was reduced the same amount as in the 10% group. In support of our finding that the hypoxia-induced 40% increase in collateral number in C57BL/6 mice is primarily responsible for the 50% decrease in their infarct volume, we previously found that the related C57BLKS strain, which has 12% more native collaterals of slightly smaller diameter than C57BL/6 mice (whose MCA tree territory is slightly smaller and hematocrit higher), sustains 78% smaller infarctions. 5 Unlike knockdown of Flk1 which was targeted to endothelial cells, knockdown of Vegfa and Cxcr4 was ubiquitous. Additional studies will be required to identify the cell type(s) that release VEGF-A and SDF1, respond to SDF1, and whether SDF1 induces VEGF-A release from peripherally recruited leukocytes or a cerebral cell type(s). This will require differentiating neo-collaterals from native collaterals, since peripheral and/or resident macrophages and microglia may be recruited to the perivascular site of both newly forming as well as to remodeling native collaterals. Systemic hypoxia was undoubtedly accompanied in our animals by pulmonary hypertension and changes in metabolism, gene expression, and humoral and other factors. However our aim was not to study hypoxia-induced NCF toward a potential “hypoxia therapy” for stroke or obstructive disease. Rather, we sought to determine if hypoxia causes NCF and then characterize it as a model to dissect mechanisms of NCF for future study of NCF induced by MCAO or other occlusive cerebrovascular diseases. Nevertheless, it is intriguing to contemplate a possible “collateral benefit” of residence at high altitude. 64 Whether NCF also occurs in models of anemia, sleep apnea and intracranial atherosclerosis awaits future studies, as does the question of whether occlusion-induced NCF is fast enough to affect evolution of the penumbra.
In conclusion, systemic hypoxemia caused new collaterals to form, remodeling of the native collaterals, and a decrease in infarct volume—effects that persisted for at least six weeks after return to normoxia. Permanent MCA occlusion also caused NCF. Hypoxic NCF provides a model to study NCF that avoids the complex milieu of injury and cell death induced by MCA occlusion and other models of stroke. Importantly, hypoxic NCF can be studied in the high-collateral C57BL6 strain, which does not exhibit occlusion-induced NCF, thus allowing use of the many genetically modified mutant lines available in this strain. Understanding the process of NCF could lead to strategies aimed at augmenting it in models of stroke and steno-occlusive disease.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20924107 - Supplemental material for Hypoxia induces de novo formation of cerebral collaterals and lessens the severity of ischemic stroke
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20924107 for Hypoxia induces de novo formation of cerebral collaterals and lessens the severity of ischemic stroke by Hua Zhang, Wojciech Rzechorzek, Amir Aghajanian and James E Faber in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health, National Institute of Neurological Diseases and Stroke grant NS083633.
Acknowledgements
The authors thank Brian Buckley for assistance with animal husbandry and hypoxia chamber management.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
HZ assisted in animal husbandry and hypoxia chamber management, performed angiography, morphometry, pMCAO and statistical analysis; WJ conducted morphometry for the data in Figures 1(g) and 2; AA contributed valuable discussion; JF designed the study and wrote the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.