
Abstract
Protective mechanisms of the brain may reduce the extent of injury after focal cerebral ischemia. Here, we explored in a mouse model of focal cerebral ischemia potential synergistic neuroprotective effects of two mediators of neuroprotection: (i) neuronal or glial precursor cells and (ii) the inhibitory neuromodulator adenosine. Embryonic stem (ES) cells, engineered to release adenosine by biallelic disruption of the adenosine kinase gene, and respective wild-type cells were induced to differentiate into either neural or glial precursor cells and were injected into the striatum of mice 1 week before middle cerebral artery occlusion. All stem cell-derived graft recipients were characterized by a significant reduction in infarct volume, an effect that was augmented by the release of adenosine. Neuroprotection was strongest in adenosine-releasing glial precursor cell recipients, which were characterized by an 85% reduction of the infarct area. Graft-mediated neuroprotection correlated with a significant improvement of general and focal neurologic scores. Histologic analysis before and after ischemia revealed clusters of implanted cells within the striatum of all treated mice. We conclude that ES cell derived adenosine-releasing brain implants provide neuroprotection by synergism of endogenous precursor cell-mediated effects and paracrine adenosine release.
Introduction
Ischemic stroke results from a transient or permanent reduction in cerebral blood flow that is restricted to the territory of a major brain artery. The reduction in flow is, in most cases, caused by the occlusion of a cerebral artery either by an embolus or by local thrombosis. With an incidence of approximately 250 to 400 per 100,000 per year and a mortality rate of around 30%, stroke remains the third leading cause of death in industrialized countries. In the USA alone, four million survivors are coping with its debilitating consequences. Extensive neuronal cell death may result not only as a consequence of ischemic injury in stroke but also after traumatic brain injury, or seizure activity in epilepsy (Dirnagl et al, 2003; Henshall and Simon, 2005).
Endogenous neuroprotective mechanisms are induced during the phenomenon of ischemic preconditioning or ischemic tolerance (IP/IT). In IP/IT, a subthreshold stimulus, for example, a transient ischemic attack, which does not cause structural damage, induces protection against a subsequent stroke (Dirnagl et al, 2003). Thus, understanding of neuroprotective strategies is a prerequisite for the development of novel therapies aimed at augmentation of the brain's endogenous neuroprotective capabilities. IP/IT in the brain consists of a rapid protective response within minutes after the stimulus and a delayed response, which develops within hours or days. While the acute response involves modulators, such as adenosine and the activation of ATP-sensitive K+ channels, the delayed response is thought to involve protein synthesis (Dirnagl et al, 2003). Gene expression profiling has recently revealed transcriptional changes during preconditioning, suggesting an involvement of the suppression of metabolic pathways and immune responses, the reduction of ion-channel activities, and a decrease in blood coagulation as integral components of IP/IT (Stenzel-Poore et al, 2003). It remains to be determined whether the tolerant state could be maintained and provide chronic protection. After a single temporary middle cerebral artery occlusion, IT persisted for approximately a week in rats (Barone et al, 1998) and epileptic tolerance in kindled rats was maintained for at least a month (Kelly and McIntyre, 1994). The mechanisms of ischemic and epileptic tolerance may be similar as both (ischemia and seizures) precondition, and protect the brain against injury by both (ischemia and seizures). This is referred to as cross-tolerance (Plamondon et al, 1999). Therefore, augmentation of endogenous tolerance mechanisms may provide prolonged protection to the brain.
Recently, neuroprotective functions have been attributed to stem cells, neural or glial precursor cells, and, in particular, to activated astrocytes (Kurozumi et al, 2005; Trendelenburg and Dirnagl, 2005). These types of cells are thought to provide neuroprotection by trophic support, scavenging of reactive oxygen species, and by the secretion of the neuroprotectant erythropoietin (Trendelenburg and Dirnagl, 2005). Conversely, ablation of astrocytes by a gliotoxin prevented astrocyte-mediated neuroprotection in a rat model of forebrain ischemia (Louw et al, 1998). Thus, it should be possible to augment these endogenous cell-based neuroprotective mechanisms by intracerebral transplantation of neural or glial precursor cells.
Apart from these cell-based neuroprotective effects, endogenous modulators play an important role in mediating neuroprotection. Adenosine is an endogenous neuromodulator and inhibits neuronal activity through activation of G protein-coupled adenosine receptors (A1, A2A, A2B, and A3) (Fredholm et al, 2005). The protective and regenerative functions of adenosine after acute injury are thus manifold (Cunha, 2005). As an acute response to a harmful stimulus, activation of A1 receptors by adenosine exerts a potent inhibitory presynaptic feedback mechanism to reduce the release of injurious excitatory neurotransmitters, in particular glutamate. Simultaneously, adenosine hyperpolarizes the postsynaptic membrane, limits the activation of N-methyl-
The involvement of the adenosine system in IP has been studied extensively in the heart. However, less is known about the involvement of the adenosine system for the induction and maintenance of IT in the brain. Recently, an involvement of the adenosine system in neuroprotection evoked by N-methyl-
Intracerebral implants of cells engineered to release adenosine have recently been used to provide seizure suppression in animal models of epilepsy. This was first accomplished by encapsulated fibroblasts engineered to lack ADK (Huber et al, 2001). In contrast to encapsulated adenosine-releasing cells, which exert their therapeutic effects exclusively by a paracrine mode of action, stem cell-derived brain implants may integrate directly into the brain and may therefore provide beneficial network-mediated effects. A protocol has been established, which allows the directed differentiation of embryonic stem (ES) cells into defined populations of either neural precursor (NP) cells (Okabe et al, 1996) or glial precursor cells (Brüstle et al, 1999).
We recently disrupted both alleles of ADK in mouse ES cells (Adk−/–) (Fedele et al, 2004) to induce therapeutic adenosine release. Intraventricular transplantation of encapsulated Adk−/– stem cell-derived embryoid bodies or glial precursor cells provided transient but complete seizure suppression in a rat model of epilepsy (Güttinger et al, 2005), thus demonstrating the therapeutic potential of stem cells engineered to lack ADK. In these previous studies of cell-based adenosine delivery, therapeutic effects were mediated by the paracrine release of adenosine. However, the potentially synergistic effect of cell-mediated adenosine release and of direct supportive effects from stem cell-derived precursors has not been addressed previously. As discussed above, stem cell-derived brain implants are likely candidates to provide neuroprotective agents and trophic factors. To address the question, whether Adk−/– ES cell-derived neural precursor cells (NPs) and glial precursor cells (GPs) synergistically combine adenosine-mediated and direct precursor cell-mediated neuroprotection, we compared neuroprotection in ischemic mice, which either received Adk−/– or wild-type ES cell-derived NPs or GPs, 1 week before middle cerebral artery occlusion.
Methods
Cells
Wild-type and genetically altered ES cells (Adk+/+ and Adk−/–, respectively) used in these experiments have been described previously (Fedele et al, 2004). Neural precursor cells were generated from Adk−/– and Adk+/+ ES cells using a well-established stepwise differentiation protocol (Okabe et al, 1996). Neural precursor cells were routinely cultured on poly-ornithine-coated dishes in N3 medium, which is based on a 1:1 mixture of Dolbecco's modified Eagle's medium with Ham's F12 supplemented with insulin (25 μg/mL), human apo-transferrin (100 μg/mL), progesterone (20 nmol/L), putrescine (100 μmol/L), sodium selenite (30 nmol/L), penicillin (100 U/mL), and streptomycin (100 μg/mL). In addition, laminin (1 μg/mL) was added when plating the cells. To keep the cells in a proliferative state, the growth factor FGF2 (10 ng/mL) was added daily. Glial precursor cells were derived from NPs by supplementing the NP culture medium with epidermal growth factor (20 ng/mL) as described previously (Fedele et al, 2004). Before transplantation, the cells were labeled with a fluorescent cell tracer (Vybrant CFDA SE Cell Tracer Kit, Molecular Probes, Inc., Eugene, OR, USA), which labeled up to 5% of the transplanted cells. For transplantation, the cells were harvested and resuspended at a concentration of 25,000 cells per μL of Ham's F12.
Adenosine Release
The release of adenosine from Adk−/– ES cell-derived brain implants has been described previously (Fedele et al, 2004; Güttinger et al, 2005). Briefly, for the analysis of the amount of adenosine released from ES cell-derived NPs or GPs, single-cell suspensions of precursors were plated at a density of 2.5 to 4 × 105 cells/cm2 onto poly-ornithinecoated six-well tissue culture dishes and cultured at 37°C under 5% CO2. Samples of medium were taken 2 h after a medium change from proliferating cells, grown in the presence of growth factors, or from fully differentiated cells, after withdrawal of growth factors for 7 days. After collecting samples, the cells were removed with trypsin and counted. This count was used for normalization of the amount of adenosine per number of cells. Adenosine was quantified using an enzyme-coupled bioluminescence assay of adenosine, which was performed as described previously (Fedele et al, 2004). Both wild-type and Adk−/– proliferating NPs did not release detectable amounts of adenosine. However, once allowed to differentiate, adenosine release from Adk−/– cells increased to 9.0 ± 4.6 ng adenosine per 105 cells in 2 h, while the amount of adenosine released from wild-type cells, 0.3 ± 0.2 ng adenosine per 105 cells per 2 h, was minimal. In comparison, proliferating and differentiated wild-type GPs released 0.8 ± 0.1 and 3.1 ± 0.6 ng adenosine per 105 cells per hour, respectively, while proliferating and differentiated Adk−/– GPs, released 11.7 ± 1.7 and 40.1 ± 6.0 ng adenosine per 105 cells per hour, respectively. Thus, disruption of the ADK gene in ES cells leads to the induction of adenosine release in differentiated progeny, with GPs releasing approximately eight times more adenosine than NPs after 7 days of growth factor withdrawal.
Animals
All animal procedures were conducted in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care in accordance with protocols approved by the Institutional Animal Care and Use Committee and the principles outlined in the NIH Guide for the Care and Use of Laboratory Animals. Male C57BL/6 mice (Charles River, Wilmington, MA, USA) were used at a body weight of 25 to 30 g. All mice were acclimatized for at least 1 week before being used in the experiments. The mice were housed under 12 h light/dark cycle (lights on from o800 hours) with food and water provided ad libitum.
Cell Transplantation
Cells were transplanted 7 days before ischemia induction into the brain at the following coordinates relative to bregma: 2.0 mm lateral, 0.1 mm posterior, and 3.0 mm vertical. The cell injection was performed using a 5 μL Hamilton syringe, and 50,000 cells were injected in a volume of 2 μL using a drill hole above the right cortex. The animals were killed for histologic analysis, either 7 days after cell transplantation (nonischemia control group), or 8 days after transplantation (i.e. 24 h after middle cerebral artery occlusion (MCAO)). Four groups of mice were transplanted with either NP or GP cells, of either wild type (WT) or Adk−/– genotype (n = 8 for both NP and GP Adk−/–, n = 7 for NP WT, and n = 6 for GP WT). An additional group of control mice (n = 5) received vehicle injection only. All transplant recipients were subjected to 60 mins of MCAO, 1 week after cell transplantation. All animals were killed and analyzed after 24 h of reperfusion. Individuals masked to the treatment groups performed all manipulations and analyses.
Focal Ischemia
Transient focal ischemia was induced by suture occlusion of the middle cerebral artery in male mice anesthetized using 1.5% isofluorane, 70% N2O, and 28.5% O2 (Xiong et al, 2004). Ischemia was induced by introducing a coated filament (6.0; Doccol, Redlands, CA, USA) from the external carotid artery into the internal carotid artery and advancing it into the arterial circle of Willis, thereby occluding the middle cerebral artery (Longa et al, 1989). Rectal and temporalis muscle temperature was maintained at 37°C ± 0.5°C with a thermostatically controlled heating pad and lamp. Cerebral blood flow was monitored by transcranial laser doppler. All surgical procedures were performed under an operating stereomicroscope.
Evaluation of the Infarct Volume
Animals were killed with an overdose of isofluorane 24 h or 7 days after ischemia. Brains were quickly removed, sectioned coronally at 1 mm intervals, and stained by immersion in the vital dye (2%) 2,3,5-triphenyltetrazolium hydrochloride. The infarct volume was calculated by summing infarction areas of all sections and multiplying by slice thickness. The percentage of the infarct was calculated by dividing the infarct volume by the total ipsilateral hemispheric volume (Pignataro et al, 2004).
Evaluation of Neurologic Deficit Scores
In all animals, 24 h after ischemia, neurologic function was scored according to two scales: a general neurologic scale (general score) and a focal neurologic scale (focal score) (Clark et al, 1997). In the general score, the following six general deficits were measured: (a) hair conditions (0 to 2), (b) position of ears (0 to 2), (c) eye conditions (0 to 4), (d) posture (0 to 4), (e) spontaneous activity (0 to 4), and (f) epileptic behavior (0 to 12). For each of the six general deficits measured, animals received a score depending on the severity of the symptoms. The scores of investigated deficits were then summed to provide a total general score ranging from 0 to 28. For the determination of the focal score, the following seven parameters were assessed: (a) body symmetry, (b) gait, (c) climbing, (d) circling behavior, (e) front limb symmetry, (f) compulsory circling, and (g) whisker response. For each of these parameters, animals were rated between 0 and 4 depending on the severity. The seven parameters were then summed to give a total focal score ranging between 0 and 28.
Histologic Analysis
Mice with precursor cell implants were killed either before ischemia induction or 24 h after MCAO. Mice were intracardially perfused with saline followed with 4% paraformaldehyde. Brains were quickly removed and postfixed with 4% paraformaldehyde, cryoprotected with 20% sucrose for 24 h, and sectioned into 40-μm coronal slices with a vibratome (Leica, VT 1000s) and mounted onto gelatin-coated slides. The location of the graft was identified by Cresyl violet staining and microscopic inspection at low magnification. Graft-derived cells were identified at higher resolution by green fluorescence. Confocal images were acquired using a Leica TCS-SP2 confocal system with a Leica DM-R upright microscope fitted with a Plan APO 40.0 × 0.75 objective. The fluorescent cell tracer was excited at 488 nm with an Argon laser and emission filtered at 499 to 535 nm. To show graft survival after ischemia, brain slices from Adk−/– ES cell recipients, taken either before or 24 h after MCAO, were stained with 4′-6-diamidino-2-phenylindole to visualize nuclear morphology.
Statistical Analysis
Values are expressed as means ± s.e. Statistical analysis was performed with one-way ANOVA followed by Newmann—Keul's test. Statistical significance was accepted at the 95% confidence level (P < 0.05).
Results
Neuroprotection by Stem Cell-Derived Brain Implants
To investigate whether neuronal and glial precursor cells exert a protective role in the development of cerebral ischemia by an endogenous mechanism, 50,000 cells of each cell population (n = 7 and 6, respectively) were stereotactically transplanted into the cerebral cortex of adult male C57BL/6 mice. A group of control mice received cell culture medium (Ham's-F12) injections (n = 7). At 7 days after transplantation, the animals were subjected to an injurious ischemic insult, which consisted of 60 mins occlusion of the middle cerebral artery followed by 24 h of reperfusion. Achievement of ischemia was confirmed by monitoring regional cerebral blood flow in the area of the left middle cerebral artery. There were no differences in cerebral blood flow between treated and untreated animals. The percentage of infarct volume in the NP- and GP-treated mice was significantly reduced to 20.78 ± 0.72 and 14.0 ± 1.48 (P < 0.05, q = 18.09 and 21.00, respectively), respectively, compared with control animals, which had an infarct volume of 53.66 ± 2.61% after 60 mins of MCAO (Figure 1). We conclude that stem cell-derived neuronal and glial brain implants protect the brain from ischemic insults by an endogenous mechanism.
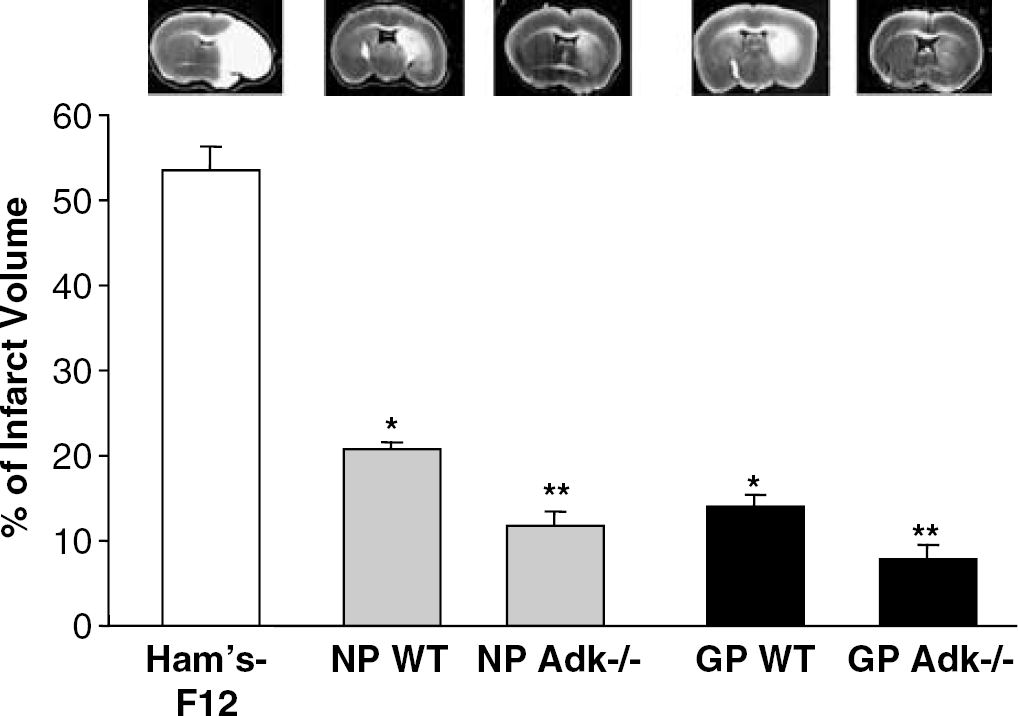
Effect of wild-type (WT) or Adk−/– neuronal progenitors (NP) or glial progenitors (GP) on cerebral ischemia induced in male C57BL/6 mice by 60 mins of MCAO. In all, 50,000 cells each (NP WT: n = 7; NP Adk−/–: n = 8; GP WT: n = 6; GP Adk−/–: n = 8) were transplanted into the striatum 7 days before MCAO. All animals were killed 24 h after MCAO and brains were analyzed by quantification of the infarct size after 2,3,5-triphenyltetrazolium hydrochloride staining. The percentage of infarct volume per ischemic brain hemisphere is given and representative 2,3,5-triphenyltetrazolium hydrochloridestained brain sections are shown. *P < 0.05 versus vehicle-treated animals. **P < 0.05 versus respective WT-treated animals and vehicle-treated animals.
Stem Cell-Mediated Adenosine Release Augments Neuroprotective Efficacy
To investigate whether stem cell-mediated adenosine release further improves neuroprotection to injurious ischemic brain injury by stem cell-derived brain implants, two additional groups of mice were transplanted with Adk−/– NPs and GPs (n = 8, each), releasing 2.3 ± 1.2 and 20.1 ± 3.0 ng adenosine per 50,000 cells per hour after 7 days of differentiation, respectively. At 7 days after transplantation, the animals were subjected to 60 mins of MCAO followed by 24 h of reperfusion. The percentage of infarct volume in these mice was further reduced to 11.8 ± 1.8 and 7.7 ± 1.7, in NP and GP recipients, respectively; a highly significant reduction (P < 0.01, q = 23.786 and 26.116, respectively) compared with control (Figure 1). Thus, NP- and GP-mediated adenosine release further augmented the reduction of the infarct volume in the graft recipients. We conclude that NP- and GP-mediated release of adenosine and endogenous neuroprotective effects of these cells act synergistically to provide nearly complete protection to ischemia.
Improvement of Neurologic Scores After Ischemia by Stem Cell-Derived Brain Implants
To evaluate whether WT and adenosine-releasing NP and GP implants provide neurologic benefit after 60 mins of MCAO, the behavior of the animals was assessed 24 h after reperfusion and compared with the vehicle-injected control group. The severity of the neurologic parameters analyzed was in line with the NP- or GP-mediated reduction of the infarct size. The general neurologic scores were 5.6 ± 0.2 in vehicle-treated animals, and reduced to 3.4 ± 0.2 and 2.3 ± 0.4 in NP- and GP-treated mice, respectively (P < 0.05). Likewise, the focal neurologic scores of 11.6 ± 0.5 in vehicle-treated animals were reduced to 8.0 ± 0.3 and 5.3 ± 0.2 in NP- and GP-treated mice, respectively (Figure 2). Again, implant-mediated adenosine release augmented the protective effect of the NP and GP implants with general scores of 2.4 ± 0.4 and 1.4 ± 0.2, and focal scores of 5.4 ± 0.7 and 3.8 ± 0.4 in Adk−/– NP- and GP-treated mice, respectively (Figure 2). We conclude that stem cell-derived brain implants provide neurologic benefit after ischemia.
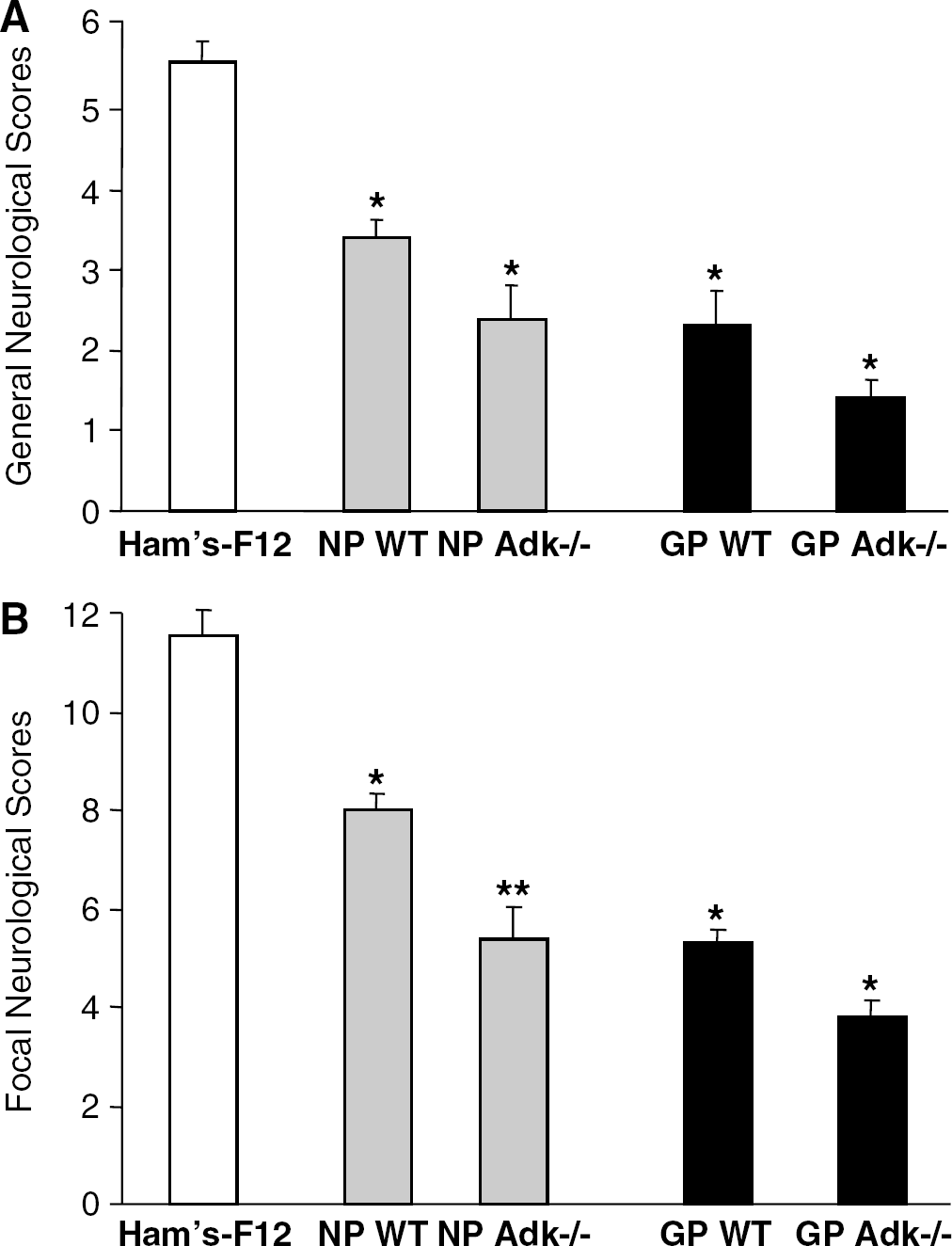
Effect of wild-type (WT) or Adk−/– neuronal progenitor (NP) or glial progenitor (GP) brain implants on neurologic deficits in male C57BL/6 mice subjected to 60 mins of MCAO. All animals were evaluated 24 h after MCAO. Each column represents the mean ± s.e. of the general (
Intracerebral Stem Cell-Derived Implants Survive After 60 mins of Ischemia
To evaluate whether 60 mins of ischemia had an impact on the grafted cells, an additional group of mice received intracerebral implants of the four cell types (NP-WT, GP-WT, NP-Adk−/–, GP-Adk−/–) (n = 4 per each group). The graft recipients were analyzed histologically either after 60 mins of MCAO on day 7 followed by 24 h of reperfusion (i.e. 8 days after cell transplantation), or 7 days after transplantation (i.e. at the time point of ischemia in the other experimental group). Coronal brain sections through the infarcted area were analyzed by Cresyl violet staining and by fluorescence microscopy to detect the fluorescence-labeled implanted cells (Figure 3). While Cresyl violet staining of control animals subjected to MCAO revealed the infarct area in the affected brain hemisphere (Figure 3A, light staining), the infarct was almost absent in mice treated with Adk−/– GP cells (Figure 3B1, showing the implant in the context of a protected brain). Thus, stem cell-derived brain implants (i) survive 60 mins of MCAO, and (ii) significantly reduce the infarct volume (Figure 3B1 and Figure 1). Cresyl violet staining demonstrated clusters of implanted cells consistently in the striatum (Figures 3B2 and 4A), being consistent with the stereotactic coordinates for the cell injections. The clusters of densely Cresyl violet-stained cells were graft derived as was verified by the expression of the green fluorescent label (Figure 3C), which was restricted to approximately 5% of the transplanted cells. Given the short-term nature of this cell transplantation experiment (8 days), we did not expect any significant cell migration or differentiation. Thus, based on the analysis of green fluorescence, we did not find any indication of cell migration into distal brain areas. We conclude that the observed therapeutic effect is exclusively because of trophic and adenosinergic effects from the clustered graft and therefore also effective in distal brain regions. In our analysis, we did not find any differences in graft location and gross assessment of cell number and morphology between NP or GP graft recipients. Likewise, the lack of ADK in Adk−/– cells (Figure 3C2) had no influence on graft survival, when compared with wild-type implants (Figure 3C1).
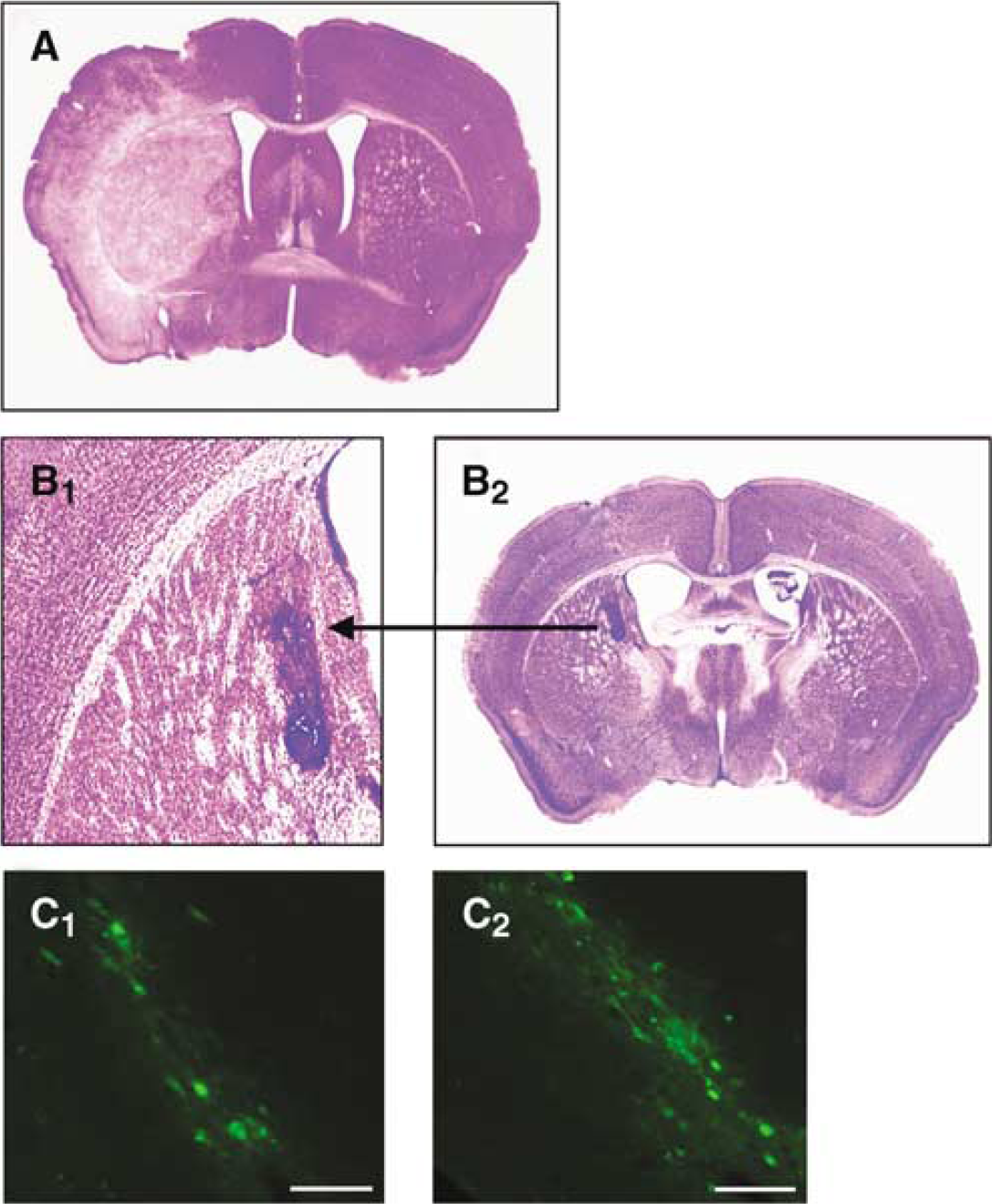
(
To further validate graft survival after MCAO, using the same stereotactic coordinates as defined above, we transplanted 50,000 Adk−/– ES cells into each mice, which were either not subjected to MCAO, or which were subjected to 60 mins of MCAO 7 days after transplantation (n = 6 mice each). Animals were killed on day 8 after transplantation, that is 24 h after ischemia in the MCAO group. Brain sections of these animals were stained with 4′-6-diamidino-2-phenylindole to visualize nuclear morphology as indicator for cell survival. Both groups of mice displayed grafts of similar location and size (Figure 4A, arrows). Higher magnification revealed intact cell nuclei in both groups, thus demonstrating survival of the transplanted cells in the MCAO-treated mice (Figures 4B and 4C).
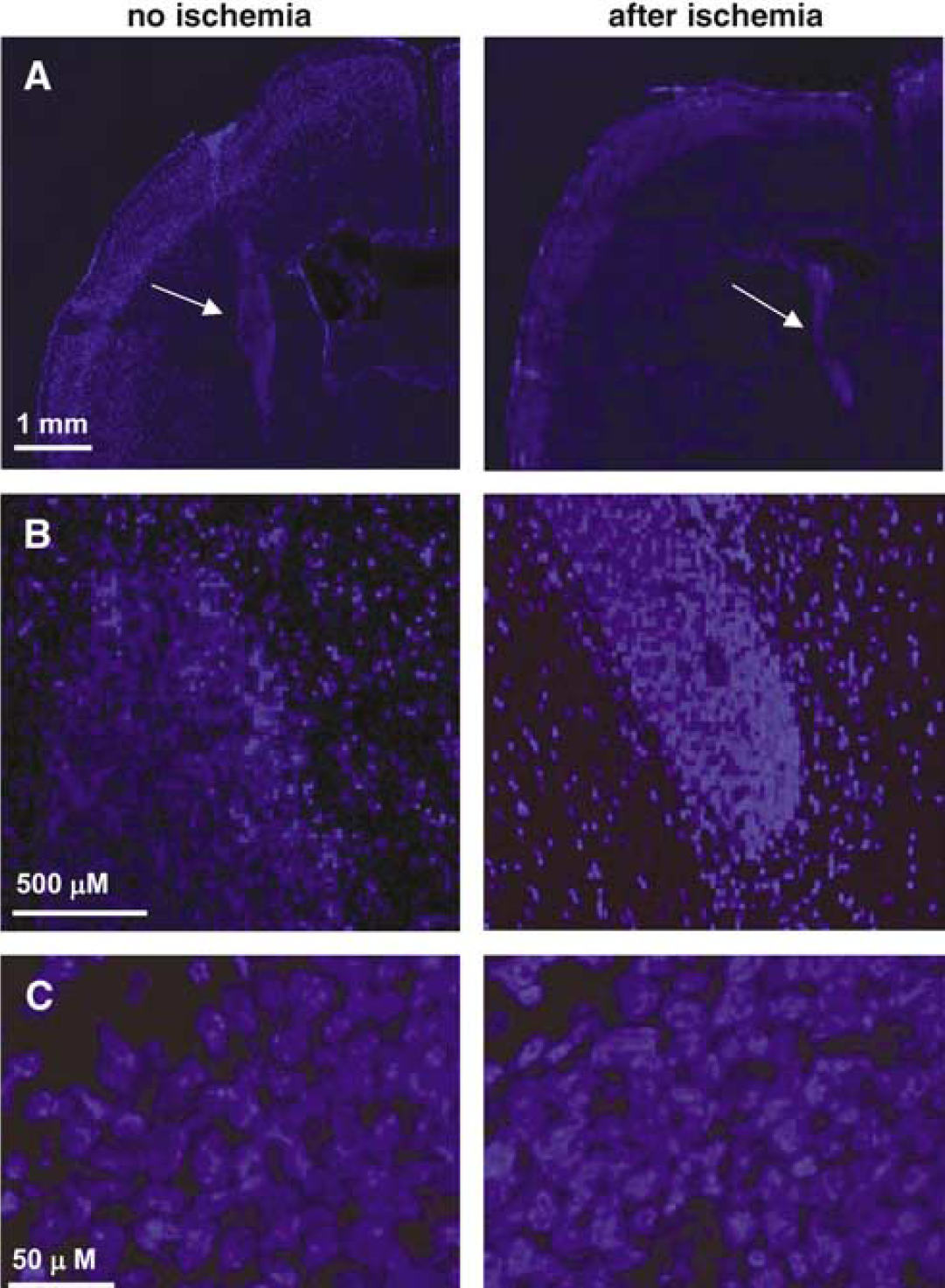
Representative 4′-6-diamidino-2-phenylindolestained coronal brain sections from mice 8 days after intrastriatal transplantation of 50,000 Adk−/– ES cells. Animals from sections shown on left panel were not subjected to MCAO, while animals from sections shown on right panel were subjected to 60 mins of MCAO 24 h before killing. (
We conclude that the neuroprotective effect exerted by the grafted cells not only prevents an extensive ischemic lesion but also protects the implanted cells from ischemic cell death.
Discussion
Extensive neuronal cell death may result as a consequence of ischemic injury in stroke, traumatic brain injury, or seizure activity in epilepsy (Dirnagl et al, 2003; Henshall and Simon, 2005). Previous strategies to prevent neuronal cell loss have mostly focused on the NMDA receptor, which has long been considered to be the main target responsible for excitotoxic Ca2+ overload in the ischemic brain (Meldrum, 1995; Simon et al, 1984). Recent experimental strategies to prevent cell death after stroke include caspase inhibitors to prevent apoptosis (Liou et al, 2003) or blockers of calcium-permeable acid-sensing ion channels to prevent glutamate receptor-independent Ca2+ toxicity (Xiong et al, 2004). Although many neuroprotective agents have proven efficacious in animal models, so far no human study has shown a statistically significant benefit in patients with acute ischemic stroke on primary end point measures (Wahlgren and Ahmed, 2004). The effects of neuroprotective agents on infarct size are time dependent, and clinical treatment has often been initiated much later than in successful experimental stroke models. Thus, a strategy that would augment neuroprotection during the acute phase after an ischemic insult would be an important therapeutic advance. In the present contribution, we therefore studied the role of two neuroprotective mechanisms, which are based on neural or glial precursor cells and adenosine.
Neuroprotection by Stem Cell-Derived Brain Implants
Glial cells contribute to important regulatory aspects in the physiology and pathophysiology of brain function and brain injury. Thus, specific neuroprotective pathways have been identified recently, by which glial cells can protect or even help to regenerate brain tissue after an ischemic insult (Trendelenburg and Dirnagl, 2005). Similarly, stem cells have inherent neuroprotective capabilities in stroke models. Thus, intravenous infusion of human mesenchymal stem cells reduced infarction size and ameliorated functional deficits in rat cerebral ischemia models, a benefit, which could be augmented when the cells were engineered to release brain-derived neurotrophic factor (Nomura et al, 2005). Endogenous progenitor cells may also play a role in IP. Thus, it was recently demonstrated that IP in adult rat brain led to an upregulation of progenitor cell proliferation in the adult rat hippocampus (Naylor et al, 2005). Conversely, the ablation of endogenous neuroprotective cells (e.g. astrocytes) abolished cell-mediated neuroprotection (Louw et al, 1998). We, therefore, explored the possibility, whether intracerebral implants of either neural or glial precursor cells would protect the brain from a subsequent ischemic event. We chose to use ES cells, which can be differentiated into pure populations of either neural (Okabe et al, 1996) or glial (Brüstle et al, 1999) precursor cells. Here, we clearly show a strong neuroprotective effect from ES cell-derived intrastriatal implants on the development of subsequent ischemic brain damage. In line with the documented neuroprotective role of astrocytes (Trendelenburg and Dirnagl, 2005) in our hands, the strongest neuroprotection was achieved with ES cell-derived glial precursor cells (Figure 1), thus further demonstrating the pivotal role exerted by glial cells in neuroprotective mechanisms.
Neuroprotection by Cell-Based Adenosine Release
Cerebral ischemia and transient periods of hypoxia are known to induce an increase in extracellular adenosine, which is thought to provide neuroprotection during cerebral ischemia. Thus, adenosine has widely been characterized as an endogenous neuroprotectant of the brain (Cunha, 2005; Fredholm, 1997; Ribeiro, 2005). In fact, excessive adenosine release appears to be one of the mechanisms by which the brain attempts to protect itself from cell injury. Sustained elevation of extracellular adenosine was recently demonstrated as a consequence of oxygen glucose deprivation in a rat brain slice model of ischemia and post-ischemic inhibition of neuronal function was because of activation of A1 receptors by increased adenosine (Pearson et al, 2006). The potentially beneficial effects of adenosine may be limited by a rapid turnover of this purine ribonucleoside by the astrocytic enzyme adenosine kinase (ADK), which removes (protective) adenosine via phosphorylation to AMP. However, ADK is rapidly downregulated after ischemia in vitro (Lynch et al, 1998) and after status epilepticus in vivo (Gouder et al, 2004), thus being causal for the rapid elevation of protective adenosine levels after brain injury. Conversely, overexpression of ADK and thus a reduction of ambient levels of adenosine can be a direct cause for seizures (Fedele et al, 2005). Likewise, we have recently demonstrated that overexpression of ADK dramatically increased the susceptibility to cerebral damage induced by focal ischemia (Pignataro et al, 2006). In ADK-overexpressing mice, a short period of ischemia (15 mins of MCAO in mice) led to a fourfold increase in the infarcted area compared with wild-type control mice, while 60 mins of MCAO was lethal in the mutant animals (Pignataro et al, 2006). This study clearly demonstrated that the brain normally is under neuroprotective control by endogenous adenosine. Thus, further increasing the levels of ambient adenosine by intracerebral transplantation of adenosine-releasing cells should further augment the neuroprotective potential of adenosine. Thus, an additional group of mice received intrastriatal implants of ES cell-derived neuronal and glial precursor cells with a biallelic genetic disruption of the Adk gene (Fedele et al, 2004). We have previously demonstrated that ES cell-derived brain implants prevent seizures in epilepsy by the paracrine release of adenosine (Güttinger et al, 2005). Therefore, Adk−/– ES cell-derived precursor cells constitute an ideal tool to study the effect of cell-mediated adenosine release on cerebral ischemia. By combining the neuroprotective potential of ES cell-derived precursor cells with cell-based paracrine adenosine delivery, we were able to address potential synergistic effects on cerebral ischemia mediated by two neuroprotective mechanisms (Figure 1). Our results clearly indicate that progenitor cells and cell-based adenosine release act synergistically to result in a profound protection from an ischemic infarct. Both, Adk−/– NPs and Adk−/– GPs, reduced the infarcted area by 50% in comparison with the respective non-adenosine-releasing NP and GP implants, an effect, which was statistically significant (P < 0.05).
Conclusion
Our results show a possible synergistic neuroprotective effect mediated by a combination of progenitor cell transplantation with paracrine adenosine release. In particular, the combination of intrastriatal glial precursor cell implants with cell-mediated adenosine release (20 ng adenosine/h per 50,000 cells) reduced the infarct area by approximately 85%, which is a significant finding. Thus, therapeutic approaches, which augment precursor cell-mediated neuroprotection and/or augment the endogenous neuromodulatory adenosine system, may provide neuroprotection in patients at risk for cerebral infarction.