The fate of cells under anoxic or ischemic stress is determined by intracellular signaling pathways including the mitogen-activated protein kinases (MAPKs) and phosphatidylinositol 3-kinase (PI3K/Akt), which affect downstream members of the apoptotic cascade. The freshwater turtle Trachemys scripta is extremely tolerant of anoxia, surviving up to 48 h at room temperature and for weeks at 3°C in the complete absence of oxygen. We investigated the relationship between the neuroprotective purine adenosine, which increases greatly in the anoxic turtle brain, and MAPK and Akt activation during both short (1 h) and long-term (4 h) anoxia. ERK1/2 and Akt were significantly upregulated during the first hour of transition to full anoxia, but returned to baseline by 4 h anoxia. Conversely, p38MAPK levels were suppressed by a mean 71% at 1 h anoxia but also returned to baseline by 4 h anoxia. Systemic administration of the general adenosine receptor antagonist aminophylline abrogated the increases in both phosphorylated ERK1/2 and Akt, as well as the initial suppression of p38MAPK. The differential modulation of the MAPK/Akt pathways may be critical for neuronal protection during the initial transition to the hypometabolic state during anoxia, when physiologic stress is likely to be greatest.
Among mammals, oxygen deprivation is a pathologic event associated with occurrences like stroke and cardiovascular disorders. Under physiologic stress, cells activate multiple signaling cascades that determine their fate, and ischemic or anoxic survival may thereby depend on the balance between pro- and antiapoptotic molecular pathways (Martindale and Holbrook, 2002). Although the inhibition of death signaling pathways may enhance cell survival, promoting endogenous protective and repair mechanisms is likely to be equally important for neuroprotection (Downey et al, 2007). Among the signaling components affected by anoxia/reoxygenation and ischemia/reperfusion are the mitogen-activated protein kinase (MAPK) family and phosphatidylinositol 3-kinase (PI3K/Akt). The MAPKs are a superfamily of serine/threonine kinases involved in regulating a wide array of cellular processes, including cell proliferation, differentiation, stress adaptation, and apoptosis. They are divided into three multimember families, which include the extracellular signal-regulated kinases (ERK), c-Jun-N-terminal kinases (JNK), and the p38 kinases (for review see Martindale and Holbrook, 2002). These families are activated by independent, but sometimes overlapping, signaling cascades, so that activation through membrane or cytoplasmic receptors may lead to the translocation of a terminal kinase to the nucleus and ultimately result in alterations in gene expression and other cell functions. For example, ERK is activated in response to growth factors, cytokines, and ischemia (Wu et al, 2000; Sugino et al, 2000), and activation of the MAPK/ERK pathway leads to the phosphorylation of a number of cellular substrates that modulate cell growth, differentiation, and survival (Gu et al, 2001; Martindale and Holbrook, 2002).
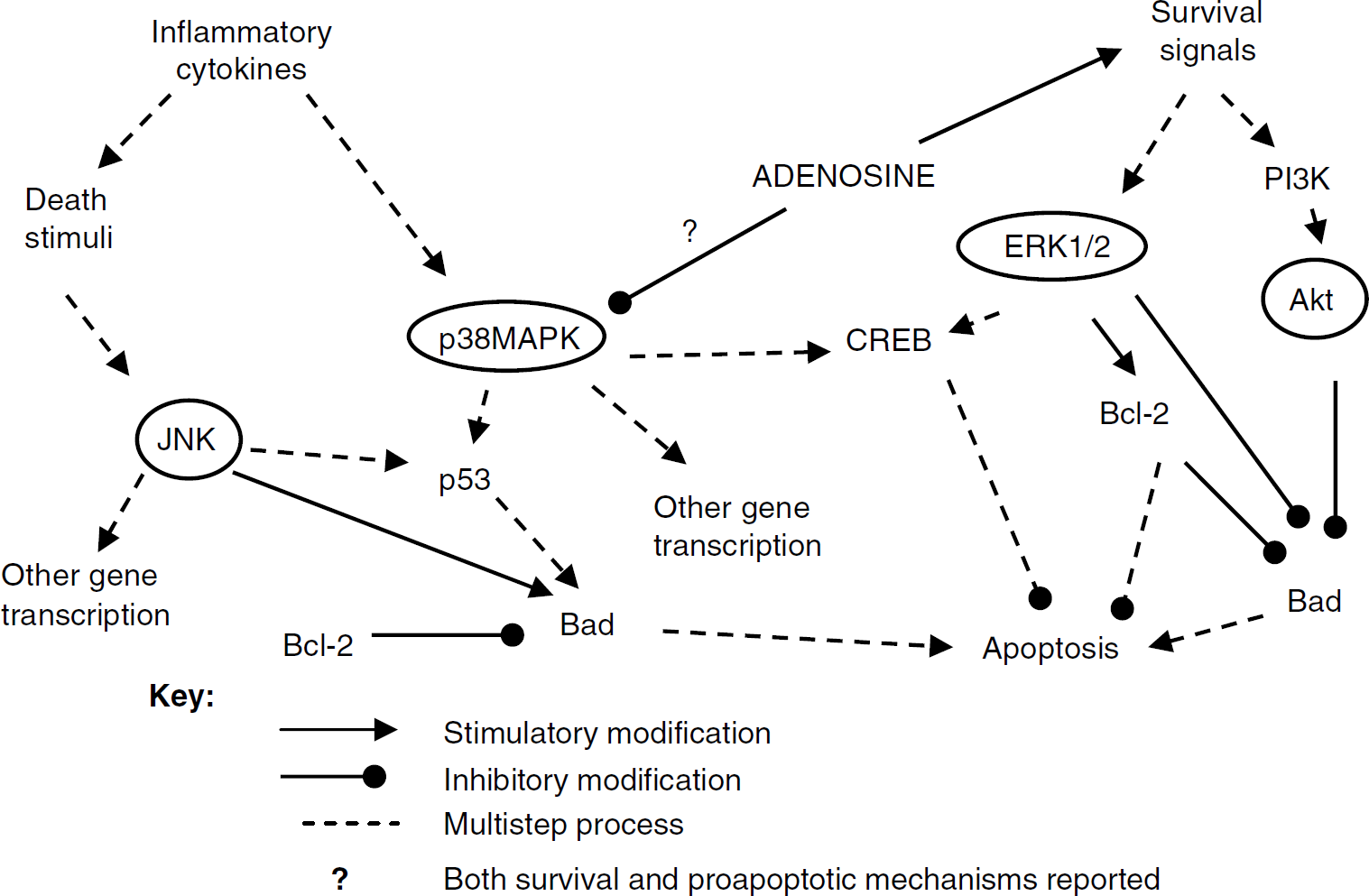
Despite a great deal of investigation, however, the exact role of individual MAPK pathways in hypoxic/ischemic cells is still uncertain, with studies indicating both prosurvival and proapoptotic roles, depending on the cell type and model system (Figure 1). Activation of the Akt pathway is generally accepted to be antiapoptotic (Kilic et al, 2006), whereas prolonged activation of p38-MAPK promotes neuronal apoptosis (Irving et al, 2000) and its inhibition increases neuronal survival in vitro (Horstmann et al, 1998) and in vivo after stroke (Barone et al, 1999). However, activation of p38-MAPK also prevents cell death during neuronal differentiation (Okamato et al, 2000). Activation of ERK has also been found to enhance cell survival under multiple conditions (Irving et al, 2000; Li et al, 2003), including oxidative stress (Guyton et al, 1996a, b), whereas other studies suggest that ERK activation can contribute to apoptosis in response to oxidant injury and after brain ischemia. Martindale and Holbrook (2002) commented that whether ERK is pro- or antiapoptotic may depend on the kinetics and duration of its activation, as ERK activity appears to be both more rapid and more transient in cases where survival is enhanced (Guyton et al, 1996a, b; Li et al, 2003), whereas delayed, sustained activation increases apoptosis (Martindale and Holbrook, 2002; Vartiainen et al, 2003).
A diagrammatic overview of some of the effects and interactions of the MAPK and Akt pathways as they affect cell survival. It is important to note that, although not fully represented here, both pro-survival and proapoptotic functions have been suggested for all three MAPK pathways.
The role of the different isoforms of JNK in cell survival, too, is unclear. Many studies have shown that JNK activation is correlated with cell death or apoptosis, especially after brain ischemia (Sugino et al, 2000; Gu et al, 2001; Guan et al, 2006). But it is also possible that JNK is activated as part of a failed attempt at survival, and thus may be protective (Sommer et al, 1995). Part of the difficulty in determining whether a particular pathway is pro-survival or proapoptotic is that mammalian cells simultaneously exhibit both physiologic and pathologic responses to stressors such as oxidant injury, hypoxia, or ischemia—reperfusion (Bickler and Donohoe, 2002).
However, not all animals are equally vulnerable to oxygen deprivation. In particular, freshwater turtles, such as the red-eared pond slider (Trachemys scripta), are able to survive from up to 48 h at room temperature to weeks at 3°C in the total absence of oxygen (Lutz et al, 2003). The turtle is able to profoundly decrease its metabolic rate such that energy utilization is matched to anaerobic energy production. This is made possible by adaptations like decreasing membrane ion permeability (channel arrest), inhibiting the release of excitatory neurotransmitters, and increasing the release of inhibitory compounds, such as GABA and adenosine (for reviews see Lutz et al, 2003; Milton and Prentice, 2007).
It is thus of great interest to examine the MAPK and PI3K pathways and their interactions with adenosine in an animal model that can withstand long periods of anoxia without brain death, and thus one in which it is highly likely that the up- and downregulation of particular molecular pathways are adaptive rather than pathologic. Although a great deal is known regarding the physiology of vertebrate facultative anaerobes such as Trachemys and other anoxia-tolerant turtles, the molecular pathways of anoxia tolerance have hardly been investigated. A study by Greenway and Storey (2000) in anoxic submerged turtles reported increases in ERK and JNK in the spleen and liver, respectively, with activation of the immediate-early gene transcription factors c-fos and c-Myc in the brain after 20 h. About 2 to 3 weeks of hypoxic submergence increased JNK expression in the liver but not in the cortex of the similarly anoxia-tolerant Chrysemys picta (Haddad, 2007a), whereas ERK in the cortex increased by 5 h hypoxia (Haddad, 2007b). The heat shock proteins Hsp72 and Hsc73 were also altered in the anoxic turtle (Prentice et al, 2004), with high basal levels perhaps indicative of some degree of constitutive preconditioning (Prentice et al, 2004). The purpose of the current study was to examine the timing and kinetics of ERK, JNK, p38MAPK, and Akt activation in the brains of T. scripta subjected to both short-term (1 h) and longer (4 h) anoxia, and to determine if adenosine plays an additional role in survival by affecting MAPK activation.
Materials and methods
Tissue Preparation
All experiments were approved by the Florida Atlantic University Institutional Animal Care and Use Committee.
Freshwater turtles weighing 300 to 500 g were obtained from commercial suppliers (Clive Longdon, Tallahassee, FL, USA) and maintained in freshwater aquaria on a 12 h light-dark cycle. The turtles were fed to satiation 3 times weekly on commercial turtles' chow. The animals were divided into 7 experimental groups of N = 4 to 6 individuals and subjected to one of the following treatments: control (normoxic, no drug), 1 or 4 h anoxia (no drug), 1 or 4 h anoxia with systemic administration of the nonspecific adenosine receptor (ADR) blocker aminophylline (AP, 200 μmol/L by continuous intraperitoneal perfusion, 1 mL/kg/h), and 1 or 4 h normoxia plus AP. This concentration was chosen as similar doses of AP or theophylline have been shown to affect normoxic brain blood flow (Hylland et al, 1994), modulate anoxic dopamine release in vivo (Milton and Lutz, 2005), reduce channel arrest, and significantly alter the anoxic suppression of EEG patterns (PL Lutz, unpublished data). A dose-response curve showed that 200 μmol/L AP significantly reduced p-ERK activation (as mentioned in Results). For experiments, animals were placed individually in sealed 2-L plastic chambers at 24°C and subjected to anoxia (99.99% positive pressure (flow-through) nitrogen; Airgas, Miami, FL, USA) or normoxia (sealed chamber with flow-through air) with AP administered through an 18 g needle inserted intraperitoneally and externalized to a perfusion pump (Harvard Apparatus, Holliston, MA, USA). Control (normoxic/no drug treatment) animals were utilized directly from the aquaria. After treatment, animals were killed by decapitation and the brains removed into liquid nitrogen in less than 2 mins. For protein extraction, frozen samples were ground to powder in liquid nitrogen, resuspended in TRIzol reagent (Life Technologies, Rockville, MD, USA), homogenized in a handheld glass homogenizer, and extracted according to the manufacturer's instructions. Protein levels were determined by BCA analysis using the manufacturer's protocol (Pierce Biotechnology Inc., Rockville, IL, USA).
Immunodetection
Proteins were electrophoretically separated by SDS-poly-acrylamide gel electrophoresis (12%) at 100 V for 2 h and subsequently transferred to nitrocellulose membranes (Hybond ECL; Amersham Biosciences, Piscataway, NJ, USA). Actin was used to verify equal loading of proteins. Membranes were blocked overnight at 4°C in 5% nonfat dried milk in Tris-buffered saline (25 mmol/L Tris-Cl, pH 7.5, at 24°C, 150 mmol/L NaCl) and then incubated with the appropriate primary antibody: rabbit polyclonal antibodies to ERK1/2 (SC-93), JNKl (SC-571), and p38MAPK (SC-535), all from Santa Cruz Biotechnology (Santa Cruz, CA, USA); rabbit polyclonal antibody to Akt (no. 9272) and phospho-Akt (no. 9271), both from Cell Signaling Technology (Danvers, MA, USA); rabbit monoclonal antibodies to phospho-p38MAPK (no. 4631), phospho-JNK (no. 4671), phospho-ERK1/2 (no. 4376), also from Cell Signaling Technology. After washing thrice in Tris-buffered saline/Tween, membranes were incubated for 1 h with HRP-conjugated anti-rabbit secondary antibody (Southern Biotech no. 4050, Birmingham, AL, USA). The protein-antibody complex was visualized by standard chemiluminescence (Amersham Biosciences) and quantified by relative densitometry using NIH Image J image analysis software. Results were normalized to percentage of actin (utilized as a loading control, no change in anoxia (Prentice et al, 2004); described in the Results section), and the relative changes are expressed as percentage of control.
Statistical Analysis
Results are expressed as mean ± s.e.m. Statistical significance was evaluated using analysis of variance (SAS-JMP). A value of P < 0.05 was considered statistically significant.
Results
Baseline Changes
The activation of presumptive pro-survival pathways (Akt and ERK) was evident in the anoxic turtle, with significant initial signals that disappeared as anoxia progressed. However, the JNK and p38MAPK pathways were suppressed or unchanged in anoxia. Alterations in MAPK levels were pathway-specific and not a generalized protein response to anoxia; levels of β-actin were unaltered by anoxia or by AP treatment (Figure 2).
Representative western blot of β-actin expression in the brain of T. scripta. Actin expression is unchanged by aminophylline (AP) treatment or anoxia. N = normoxic controls; 1NAP = 1 h normoxia, AP-treated; 4NAP = 4 h normoxia, AP-treated; 1A = 1 h anoxic control; 1AAP = 1 h anoxia, AP-treated; 4A = 4 h anoxic control; 4AAP = 4 h anoxia, AP-treated.
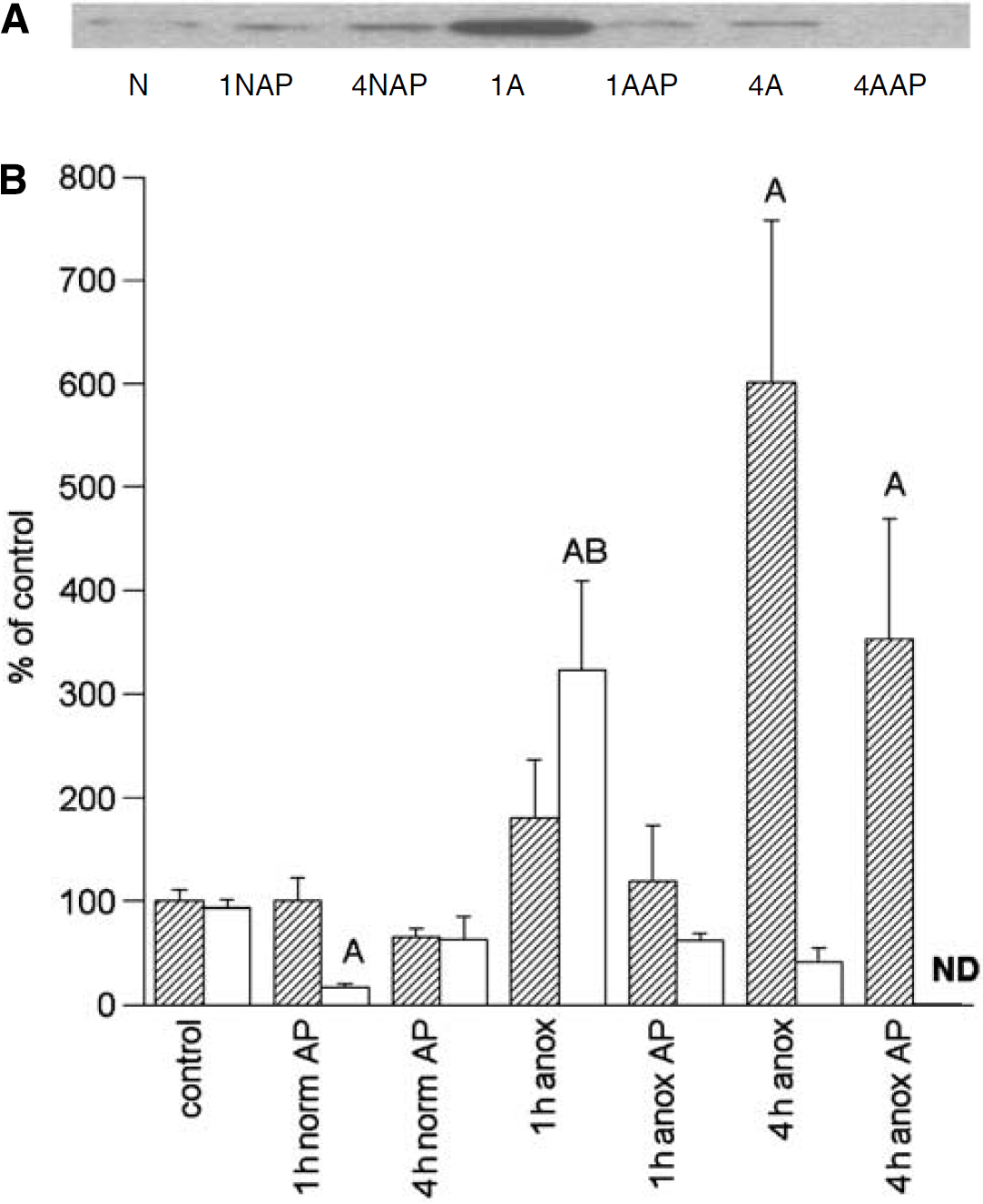
Akt/p-Akt: Phosphorylated (activated) Akt increased significantly but temporarily; by 1 h anoxic exposure, levels increased to 323% ± 86% of basal but had decreased by 4 h anoxia to less than half (mean 41%) of normoxic controls (Figure 3). Conversely, the nonphosphorylated form of Akt increased continuously from 180% ± 56% to 601% ± 157% of basal at 1 and 4 h anoxia, respectively, such that the sum of the phosphorylated and nonphosphorylated Akt levels at 1 and 4 h anoxia were similar to each other at these time points but elevated above normoxic controls.
Representative immunoblot of phospho-Akt (A) and densitometric analyses (B) of Akt and p-Akt levels in the normoxic and anoxic T. scripta brain in untreated controls and after 1 or 4 h intraperitoneal aminophylline (AP) perfusion. (A) N = normoxic controls; 1NAP = 1 h normoxia, AP-treated; 4NAP = 4 h normoxia, AP-treated; 1 A =1 h anoxic control; 1AAP = 1 h anoxia, AP-treated; 4A = 4 h anoxic control; 4AAP = 4 h anoxia, AP-treated. (B) Relative changes in Akt (hatched) and p-Akt (open), N = 4 to 8 individuals/time point. A = significantly different from normoxic controls. B = levels in aminophylline treatment (AP) animals s.d. from untreated animals at the same time point; P < 0.05.
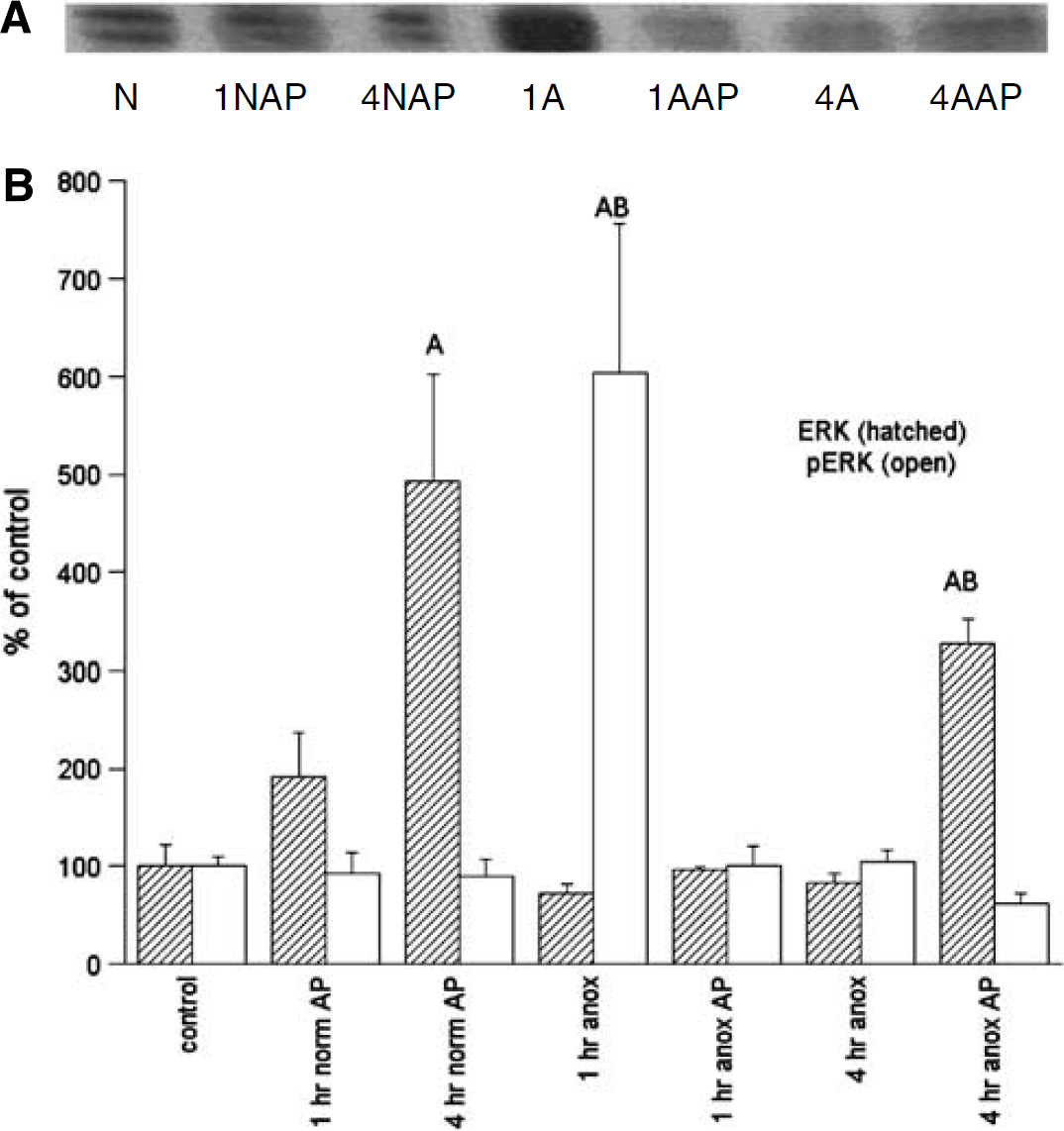
ERK/p-ERK: Changes in activated ERK were the most dramatic of the protein alterations measured, with a sixfold increase in p-ERK by 1 h anoxia (Figure 4). Like Akt activation, however, the increase was temporary, with levels returning to basal (104% ± 12%) by 4 h anoxia. The ERK levels did not change significantly from normoxic controls over 1 or 4 h anoxia.
Representative immunoblot of phospho-ERK (A) and densitometric analyses (B) of ERK1/2 and p-ERK1/2 levels in the normoxic and anoxic T. scripta brain in untreated controls and after 1 or 4 h intraperitoneal aminophylline (AP) perfusion. (A) N = normoxic controls; 1NAP = 1 h normoxia, AP-treated; 4NAP = 4 h normoxia, AP-treated; 1A = 1 h anoxic control; 1AAP = 1 h anoxia, AP-treated; 4A = 4 h anoxic control; 4AAP = 4 h anoxia, AP-treated. (B) N = 4 to 8 individuals/time point. A = significantly different from normoxic controls. B = levels in aminophylline treatment (AP) animals s.d. from untreated animals at the same time point; P < 0.05.
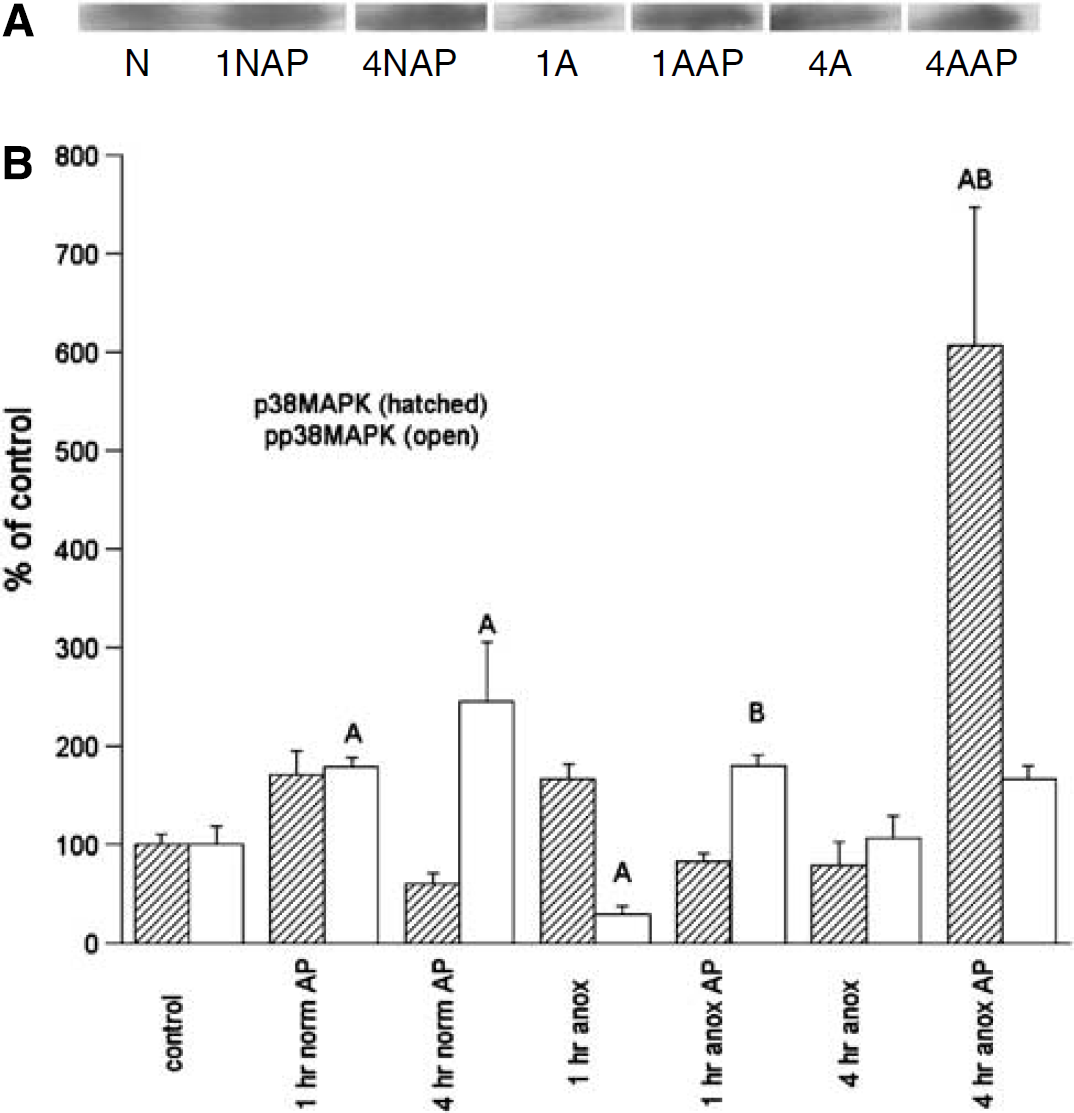
p38MAPK/p-p38MAPK: Unlike Akt and ERK, p38MAPK activation was clearly suppressed in early anoxia, with p-p38 levels decreasing significantly to only 29% of normoxic controls by 1 h anoxia, and returning only to basal by 4 h anoxia (Figure 5). The increase in nonphosphorylated p38MAPK from normoxic controls was not statistically significant, with an increase only to 166% of basal at 1 h anoxia, which returned to baseline by 4 h anoxia. Total p38MAPK remained unchanged from control levels over the anoxic exposure.
Representative immunoblot of phospho-p38MAPK (A) and densitometric analyses (B) of p38MAPK and p-p38MAPK levels in the normoxic and anoxic T. scripta brain in untreated controls and after 1 or 4 h intraperitoneal aminophylline (AP) perfusion. (A) N = normoxic controls; 1NAP = 1 h normoxia, AP-treated; 4NAP = 4 h normoxia, AP-treated; 1A = 1 h anoxic control; 1AAP = 1 h anoxia, AP-treated; 4A = 4 h anoxic control; 4AAP = 4 h anoxia, AP-treated. (B) N = 4 to 8 individuals/time point. A = significantly different from normoxic controls. B = levels in aminophylline treatment (AP) animals s.d. from untreated animals at the same time point; P < 0.05.
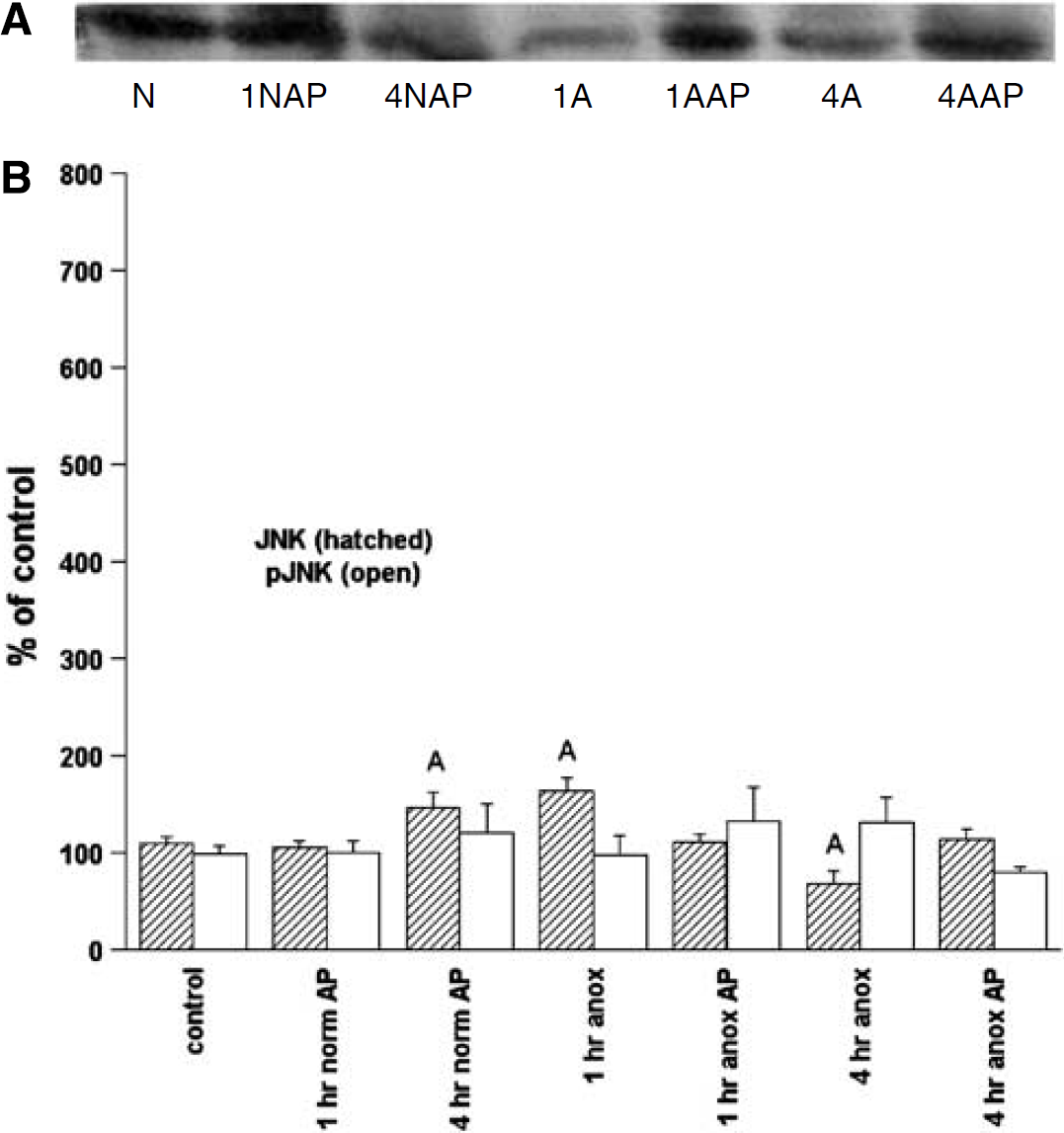
JNK/p-JNK: Changes in JNK and p-JNK were small, with a modest, but statistically significant, increase in JNK at 1 h anoxia to 63% above basal, which decreased to a mean 33% below basal by 4 h anoxia (Figure 6). There were no significant changes in p-JNK levels in anoxia compared with controls.
Representative immunoblot of phospho-JNK (A) and densitometric analyses (B) of JNK and p-JNK levels in the normoxic and anoxic T. scripta brain in controls and after 1 or 4 h intraperitoneal aminophylline (AP) perfusion. (A) N = normoxic controls; 1NAP = 1 h normoxia, AP-treated; 4NAP = 4 h normoxia, AP-treated; 1 A = 1 h anoxic control; 1AAP = 1 h anoxia, AP-treated; 4A = 4 h anoxic control; 4AAP = 4 h anoxia, AP-treated. (B) N = 4 to 8 individuals/time point. A = significantly different from normoxic controls. B = levels in aminophylline treatment (AP) animals s.d. from untreated animals at same time point; P < 0.05.
Aminophylline Exposure
Blocking ADRs through the systemic administration of the general adenosine antagonist AP significantly decreased the activation of both presumed survival pathways, p-ERK and p-Akt, but resulted in a concomitant increase in p-p38MAPK activation. Systemic administration of a range of AP concentrations from 20 to 400 μmol/L showed that responses in the turtle are dose-dependent, with mean p-ERK levels (N = 4/treatment) of 402% (1 h anoxic control animals), 395% (20 μmol/L AP), 169% (100 μmol/L AP), 105% (200 μmol/L AP), and 57% (400 μmol/L AP) of normoxic basal levels.
Akt/p-Akt: Aminophylline treatment during normoxia did not significantly alter Akt or p-Akt levels; interestingly, the initial activation of p-Akt at 1 h anoxia was abolished in AP-treated turtles (Figure 3), with a further suppression by 4 h anoxia to below detectable levels, although 4 h anoxic p-Akt in untreated animals was also reduced below normoxic controls. Levels of the nonphosphorylated Akt in AP-treated anoxic animals also appeared to decrease compared with anoxic, untreated animals, although the differences were not statistically significant.
ERK/p-ERK: Similar to Akt, AP treatment abolished the activation of p-ERK at 1 h anoxia; however, AP treatment had no effect on p-ERK activation at 4 h anoxia; p-ERK had already returned to baseline in untreated animals and there was no further significant depression of p-ERK with AP (Figure 4). Four hours of AP treatment in both normoxic and anoxic animals, however, did result in significant increases in the nonphosphorylated form of ERK.
P38MAPK/p-p38MAPK: Coincident with the abolition of ERK and Akt activation by AP, there was an increase in p-p38 MAPK. In contrast to the suppression evident in untreated, anoxic animals, p-p38 MAPK expression levels were expressed at 180% ± 11% of basal in AP-treated animals, a sixfold increase over 1 h anoxic controls (Figure 5). At 4 h anoxia, p-p38 MAPK in AP-treated animals was only 167% of normoxic controls, which was not significantly different from normoxic or anoxic, untreated animals; by contrast, the nonphosphorylated form of p38 had increased more than sixfold over 4 h anoxia. Aminophylline treatment also significantly increased p-p38 MAPK in normoxic animals to 178% ± 8% and 245% ± 60% of untreated controls, suggesting a basal tone of p38 suppression that is maintained by adenosine even in normoxia.
JNK/p-JNK: Generally, AP treatment did not alter JNK or p-JNK levels compared with normoxic or anoxic controls, with the exception of a small (less than 50%) but statistically significant increase in nonphosphorylated JNK after 4 h normoxic AP treatment (Figure 6).
Discussion
Although the physiologic mechanisms underlying the remarkable survival exhibited by anoxia-tolerant turtles have been investigated for more than 30 years, there has been relatively little work on the molecular adaptations that permit neuronal survival at the cellular level, particularly during the initial transition phase to the hypometabolic state. In this study, we show the significant but temporary activation of both ERK1/2 and PI3K/Akt pathways in the T. scripta brain in response to anoxia. Coupled to the increase in these putative pro-survival factors is the again temporary, but significant, suppression of p38 MAPK by more than two-thirds during the initial hour of anoxia, without any significant changes in p-JNK. Conversely, in mammalian hypoxia/ischemia models, increases in brain p-Akt and p-ERK (Wang et al, 2003; Li et al, 2003) are accompanied by increases in p-JNK (Ferrer et al, 2003; Guan et al, 2006) and p-p38 MAPK (Irving et al, 2000; Sugino et al, 2000; Ferrer et al, 2003), as oxygen deprivation triggers both pro-survival and pathologic responses. Blocking p-JNK activation or p38 MAPK activation has been shown to be neuroprotective in a number of ischemia/reperfusion and toxin studies (Sugino et al, 2000; Ma et al, 2005). Conversely, the activation of ERK1/2 and Akt is a hallmark of the preconditioning response in the brain and heart, and is thought to regulate components of the apoptotic pathway. If one assumes that strongly upregulated pathways in the anoxic turtle are indeed protective, then it is likely that an initial upregulation of ERK and Akt increases cell survival during an oxygen deficit, and also that the suppression of p38MAPK activation is protective. The turtle, then, represents a model of anoxic survival physiology without pathology. Changes in p-JNK were minimal and were not affected by ADR blockade, and for this reason, the results of this study cannot support either a pro-survival or a proapoptotic role for JNK activation.
During the initial hour of anoxia in a turtle respiring nitrogen, blood oxygen levels decline to zero (making this a period of progressively increasing hypoxia), and as the animal has not yet entered a fully hypometabolic state, energy demand temporarily outstrips energy supply, leading to a limited but significant temporary decline in ATP and ADP (Lutz et al, 2003). The decrease in high-energy phosphates, in turn, opens ATP-dependent potassium channels (KATP) and increases AD release, with extracellular AD increasing more than 12-fold to peak at about 100 mins anoxia before declining again to basal levels (Nilsson and Lutz, 1991). By 4 h anoxia, the turtle has entered a state of greatly reduced metabolism, where energy demand can be fully met by anaerobic glycolysis (Lutz et al, 2003): AD levels decline, KATP channels close as ATP levels return to basal, and neurotransmitters are again released and recycled, albeit at reduced rates (Milton and Prentice, 2007). Thus, the initial 1 to 2 h anoxia, the transition phase, is likely to be a time of physiologic stress and rapid metabolic change even in the anoxia-tolerant turtle, and it is during this early transition phase that we see significant changes in MAPK pathways and PI3/Akt activation. The downregulation of glutamate and dopamine release because of ADR stimulation and its effects on K+ channel conductance also occur during the first 1 to 2 h anoxia in the turtle (Milton and Prentice, 2007). By 4 h anoxia, other mechanisms not related to AD maintain the hypometabolic state (Milton and Lutz, 2005). It is noteworthy that research in other model systems has suggested that a temporary upregulation of ERK1/2 provides cellular protection, but that sustained elevated levels increase cell death (Vartiainen et al, 2003; Wang et al, 2003). Neuronal protection by an early, transient increase in Akt activation has also been reported in rats subjected to arterial occlusion (Shibata et al, 2002).
In this model, both the upregulation of ERK1/2 and Akt and the suppression of p38MAPK appear to be linked functionally to adenosine signaling, although it cannot be determined from this in vivo study whether there are direct links between the ADRs and each of these pathways or the regulation is because molecular crosstalk. The pro-survival effects of adenosine are so numerous that it has been referred to as a ‘retaliatory metabolite’, balancing energy supply and demand in the energy-deficient brain (Lutz et al, 2003). In the turtle, adenosine has been shown to impact NMDA receptor and potassium channel arrest, increase brain blood flow, and decrease the release of excitatory amino acids (for reviews see Lutz et al, 2003; Lutz and Milton, 2004).
Administration of the general ADR blocker, AP, to anoxic Trachemys does not alter cardiac output, heart rate, or arterial blood pressure (Stecyk et al, 2007), so the observed effects are likely because of direct effects of ADR blockade on neuronal tissues, rather than systemic effects such as decreased blood flow. This is supported by recent data from our lab indicating that the direct application of the specific AD1R blocker DPCPX in neuronal cultures blocks the upregulation of p-ERK and p-Akt but increases p-p38 MAPK, whereas stimulation of AD1R has the opposite effect (Nayak et al, in preparation). It has already been demonstrated that treatment of turtle neuronal cultures with a specific AD1R agonist increases anoxic and anoxia/reoxygenation cell survival and decreases ROS (reactive oxygen species) production, whereas an AD1R antagonist increases ROS release and cell death (Milton et al, 2007).
In addition to its action as an ADR blocker, AP is utilized as a nonselective inhibitor of cyclic nucleotide phosphodiesterases (PDEs). The many isoforms of PDEs are enzymes that hydrolyze cAMP and/or cGMP (Soderling and Beavo, 2000), and their inhibition can thus alter intracellular cAMP levels. In cultured spinal motor neurons, AP has been found to be protective against both acute ROS-induced and chronic glutamate-induced neurotoxicity, possibly through PDE5 inhibition, as specific inhibitors of PDE 1 to 4 offered no protection (Nakamizo et al, 2003). Phosphodiesterase activity has not been measured in the turtle brain and the overall trend in our current and previous studies is for blockade of ADRs to be injurious to the anoxic turtle brain. These data suggest that potential protective effects via PDE blocking (perhaps against ROS) are likely to be relatively minor compared with the clear toxicity induced by anoxic ADR blockade.
Other factors may also affect cell survival through ERK or Akt activation. It has been hypothesized that the cellular protection provided by the rapid activation of ERK1/2 and the suppression of proapoptotic factors is due in part to the opening of KATP in mammals (Zhang et al, 2007). Although beyond the scope of this study, it is of interest to note that the pattern of ERK and Akt activation does mirror known changes in KATP channel activity in the turtle brain (Milton and Prentice, 2007), or KATP may interact with ADRs (Rogel et al, 2005). Similarly, the stimulation of delta-opioid receptors increases ERK and Bcl2 activity and counters the hypoxic activation of p38MAPK and cytochrome c release in mammals (Ma et al, 2005). And as the turtle brain has both a much higher density and lower dissociation constant of delta-opioid receptors compared with the rat (Xia and Haddad, 2001), it has been suggested that this factor may also increase turtle anoxic survival.
Although numerous cardiac studies in mammals have shown AD effects on ERK and Akt activation, a lesser number have examined any link between AD and these neuroprotective pathways in the brain. A1 and A2 ADRs are linked to ERK activation in neuronal differentiation (Canals et al, 2005), whereas AD induces both ERK and Akt phosphorylation in a human glioma cell line (Jacques-Silva et al, 2004). In astrocytes, AD agonists reduce apoptosis and induce a rapid activation of the PI3K and ERK1/2 pathways, concomitantly reducing both JNK and p38MAPK activation; levels of the proapoptotic protein Bad and antiapoptotic Bcl-X(L) are also altered to promote cell survival (Ciccarelli et al, 2007; D'Alimonte et al, 2007). Similarly, AD agonists administered during myocardial infarction increased Akt activation and Bcl-2 expression in the rat amygdala, thereby reducing apoptosis (Boucher et al, 2006).
Other researchers have also reported changes in these pathways in hypoxic and anoxic submerged turtles, although under somewhat different conditions. It was recently reported that weeks of hypoxic submergence did not alter JNK expression in the cortex of the similarly anoxia-tolerant turtle C. picta, although ERK activation was significantly increased after 5 h hypoxia (Haddad, 2007a, b). Anoxia versus submergence can result in different responses, as has been previously noted, for example, in the expression and upregulation of heat shock proteins (Prentice et al, 2004). Other studies also differ in being cold-temperature submergence studies, where an attempt was made to replicate the turtle's natural overwintering strategy of underwater hibernation, which results in hypoxic or anoxic conditions. Thus, for example, Greenway and Storey (1999) found no increase in ERK activity in anoxic turtle hatchlings held at 5°C, although brain JNK activity increased; there were significant ERK increases with freezing. Likewise, anoxic submergence at 7°C did not alter brain ERK activity in adult turtles (Greenway and Storey, 2000). It may be that at low temperatures, short-term anoxia is an insufficient stress to trigger ERK activation without the additional stress of freezing. At room temperature, however, 1 h anoxic exposure results in a number of physiologic changes, including a decline in ATP, the opening of KATP channels, and AD release. Our results indicate that AD, in turn, results in changes at the molecular level that are likely to increase cell survival, including ERK and Akt activation and the suppression of p38MAPK.
Footnotes
Acknowledgements
We thank J Boatright, S Kesaraju, and G Nayak for their technical assistance.
References
1.
BaroneFCFeuersteinGZWhiteRFIrvingEAParsonsAAHadinghamSJRobertsJHunterAJArcherGEKumarSLeeJCSmithBR (1999) Inhibition of p38 mitogen-activated protein kinase reduces brain injury. Neurological deficits and inflammatory cytokines in rat focal stroke. J Cereb Blood Flow Metab19(Suppl 1):613.
2.
BicklerPEDonohoePH (2002) Adaptive responses of vertebrate neurons to hypoxia. J Exp Biol205:3579–86.
3.
BoucherMWannBPKaloustianSCardinalRGodboutRRousseauG (2006) Reduction of apoptosis in the amygdala by an A2A adenosine receptor agonist following myocardial infarction. Apoptosis11:1067–74.
4.
CanalsMAnguloECasadóVCanelaEIMallolJViñalsFStainesWTinnerBHillionJAgnatiLFuxeKFerréSLluisCFrancoR (2005) Molecular mechanisms involved in the adenosine A1 and A2A receptor-induced neuronal differentiation in neuroblastoma cells and striatal primary cultures. J Neurochem92:337–48.
5.
CiccarelliRD'AlimonteIBalleriniPD'AuroMNargiEBuccellaSDi IorioPBrunoVNicolettiFCaciagliF (2007) Molecular signaling mediating the protective effect of A1 adenosine and mGlu3 metabotropic glutamate receptor activation against apoptosis by oxygen/glucose deprivation in cultured astrocytes. Mol Pharmacol71:1369–80.
6.
D'AlimonteIBalleriniPNargiEBuccellaSGiulianiPDi IorioPCaciagliFCiccarelliR (2007) Staurosporine-induced apoptosis in astrocytes is prevented by A1 adenosine receptor activation. Neurosci Left418:66–71.
7.
DowneyJMDavisAMCohenMV (2007) Signaling pathways in ischemic preconditioning. Heart Fail Rev12:181–8.
8.
FerrerIFrigutsBDalfoEPlanasAM (2003) Early modifications in the expression of mitogen-activated protein kinase (MAPK/ERK), stress-activated kinases SAPK/JNK and p38, and their phosphorylated substrates following focal cerebral ischemia. Acta Neuropathol105:425–37.
9.
GreenwaySCStoreyKB (1999) Discordant responses of mitogen-activated protein kinases to anoxia and freezing exposures in hatchling turtles. J Comp Physiol B169:521–7.
10.
GreenwaySCStoreyKB (2000) Mitogen-activated protein kinases and anoxia tolerance in turtles. J Exp Zool287:477–84.
11.
GuZLJiangQZhangGY (2001) Extracellular signal-regulated kinase 1/2 activation in hippocampus after cerebral ischemia may not interfere with post-ischemic cell death. Brain Res901:79–84.
12.
GuanQHPeiDSLiuXMWangXTXuTLZhangGY (2006) Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res1092:36–46.
13.
GuytonKZLiuYGorospeMXuQHolbrookNJ (1996a) Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem271:4138–42.
14.
GuytonKZGorospeMKenslerTWHolbrookNJ (1996b) Mitogen-activated protein kinase (MAPK) activation by butylated hydroxytoluene hydroperoxide: implications for cellular survival and tumor promotion. Cancer Res56:3480–5.
15.
HaddadJJ (2007a) Discordant tissue-specific expression of SAPK/MAPK(JNK)-related cofactors in hypoxia and hypoxia/reoxygenation in a model of anoxia-tolerance. Protein Pept Lett14:373–80.
16.
HaddadJJ (2007b) The role of Bax/Bcl-2 and pro-caspase peptides in hypoxia/reperfusion-dependent regulation of MAPK(ERK): discordant proteomic effect of MAPK(p38). Protein Pept Lett14:361–71.
17.
HorstmannSKahlePJBorasioGD (1998) Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res52:483–90.
18.
HyllandPNilssonGELutzPL (1994) Time course of anoxia-induced increase in cerebral blood flow rate in turtles: evidence for a role of adenosine. J Cereb Blood Flow Metab14:877–81.
19.
IrvingEABaronebFCReithaADHadinghamaSJParsonsaAA (2000) Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Mol Brain Res77:65–75.
20.
Jacques-SilvaMCBernardiARodnightRLenzG (2004) ERK, PKC and PI3K/Akt pathways mediate extracellular ATP and adenosine-induced proliferation of U138-MG human glioma cell line. Oncology67:450–9.
21.
KilicEKilicUWangYBassettiCLMartiHHHermannDM (2006) The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF's neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J20:1185–7.
22.
LiFOmoriNJinGWangSJSatoKNaganoIShojiMAbeK (2003) Cooperative expression of survival p-ERK and p-Akt signals in rat brain neurons after transient MCAO. Brain Res962:21–6.
23.
LutzPLMiltonSL (2004) Negotiating brain anoxia survival in the turtle. J Exp Biol207:3141–7.
24.
LutzPLNilssonGEPrenticeH (2003) The Brain without Oxygen: Causes of Failure Molecular and Physiological Mechanisms for Survival. 3rd ed.Dordrecht: Kluwers Press.
25.
MaMCQianHGhassemiFZhaoPXiaY (2005) Oxygen-sensitive {delta}-opioid receptor-regulated survival and death signals: novel insights into neuronal preconditioning and protection. J Biol Chem280:16208–18.
26.
MartindaleJLHolbrookNJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol192:1–15.
27.
MiltonSLLutzPL (2005) Adenosine and ATP sensitive potassium channels modulate dopamine release in the anoxic turtle (Trachemys scripta) striatum. Am J Physiol289:R77–83.
28.
MiltonSLNayakGKesarajuSKaraLPrenticeHM (2007) Suppression of reactive oxygen species production enhances neuronal survival in vitro and in vivo in the anoxia-tolerant turtleTrachemys scripta. J Neurochem101:993–1001.
29.
MiltonSLPrenticeHM (2007) Beyond anoxia: the physiology of metabolic downregulation and recovery in the anoxia-tolerant turtle. Comp Biochem Physiol A Mol Integr Physiol147:277–90.
30.
NakamizoTKawamataJYoshidaKKawaiYKankiRSawadaHKiharaTYamashitaHShibasakiHAkaikeAShimohamaS (2003) Phosphodiesterase inihibitors are neuroprotective to cultured spinal motor neurons. J Neurosci Res71:485–95.
31.
NilssonGELutzPL (1991) Release of inhibitory neurotransmitters in response to anoxia in turtle brain. Am J Physiol261:R32–7.
32.
OkamatoSKraincDShermanKLiptonSA (2000) Anti-apoptotic role of p38 mitogen-activated protein kinase-monocyte enhancer factor 2 transcription factor pathway during neuronal differentiation. Proc Natl Acad Sci USA97:7561–6.
33.
PrenticeHMMiltonSLScheurleDLutzPL (2004) The upregulation of cognate and inducible heat shock proteins in the anoxic turtle brain. J Cereb Blood Flow Metab24:826–8.
34.
RogelABrombergYSperlingOZoref-ShaniE (2005) Phospholipase C is involved in the adenosine-activated signal transduction pathway conferring protection against iodoacetic acid-induced injury in primary rat neuronal cultures. Neurosci Lett373:218–21.
35.
ShibataMYamawakiTSasakiTHattoriHHamadaJFukuuchiYOkanoHMiuraM (2002) Upregulation of Akt phosphorylation at the early stage of middle cerebral artery occlusion in mice. Brain Res942:1–10.
36.
SoderlingSHBeavoAJ (2000) Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr Opin Cell Biol12:174–9.
37.
SommerCGassPKiesslingM (1995) Selective c-Jun expression in CA1 neurons of the hippocampus during and after acquisition of an ischemic tolerant state. Brain Res5:135–44.
38.
StecykJAWStensløkkenK-ONilssonGEFarrellAP (2007) Adenosine does not save the heart of anoxia-tolerant vertebrates during prolonged oxygen deprivation. Comp Biochem Physiol A Mol Integr Physiol147:961–73.
39.
SuginoTNozakiKTakagiYHattoriIHashimotoNMoriguchiTNishidaE (2000) Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus, J Neurosci20:4506–14.
40.
VartiainenNGoldsteinsGKeksa-GoldsteineVChanPHKoistinahoJ (2003) Aspirin inhibits p44/42 mitogen-activated protein kinase and is protective against hypoxia/reoxygenation neuronal damage. Stroke34:752–7.
41.
WangXZhuCQiuLHagbergHSandbergMBlomgrenK (2003) Activation of ERK 1/2 after neonatal rat cerebral hypoxia-ischaemia. J Neurochem86:351–62.
42.
WuDCYeWCheXMYangGY (2000) Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain. J Cereb Blood Flow Metab20:1320–30.
43.
XiaYHaddadGG (2001) Major difference in the expression of delta- and mu-opioid receptors between turtle and rat brain. J Comp Neurol436:202.
44.
ZhangSZhouFDingJ-HZhouXQSunX-LHuG (2007) ATP-sensitive potassium channel opener iptakalim protects against MPP(+)-induced astrocytic apoptosis via mitochondria and mitogen-activated protein kinase signal pathways. J Neurochem103:569–79.