
Abstract
Xenon attenuates on-going neuronal injury in both in vitro and in vivo models of hypoxic-ischaemic injury when administered during and after the insult. In the present study, we sought to investigate whether the neuroprotective efficacy of xenon can be observed when administered before an insult, referred to as ‘preconditioning’. In a neuronal–glial cell coculture, preexposure to xenon for 2 h caused a concentration-dependent reduction of lactate dehydrogenase release from cells deprived of oxygen and glucose 24 h later; xenon's preconditioning effect was abolished by cycloheximide, a protein synthesis inhibitor. Preconditioning with xenon decreased propidium iodide staining in a hippocampal slice culture model subjected to oxygen and glucose deprivation. In an in vivo model of neonatal asphyxia involving hypoxic–ischaemic injury to 7-day-old rats, preconditioning with xenon reduced infarction size when assessed 7 days after injury. Furthermore, a sustained improvement in neurologic function was also evident 30 days after injury. Phosphorylated cAMP (cyclic adenosine 3′,5′-monophosphate)-response element binding protein (pCREB) was increased by xenon exposure. Also, the prosurvival proteins Bcl-2 and brain-derived neurotrophic factor were upregulated by xenon treatment. These studies provide evidence for xenon's preconditioning effect, which might be caused by a pCREB-regulated synthesis of proteins that promote survival against neuronal injury.
Introduction
Prolonged neonatal asphyxia is becoming a less frequent clinical event because of improvements in perinatal monitoring and early intervention by skilled practitioners. Yet, unexpected hypoxic–ischaemic injury still occurs, even in the developed world, with an overall incidence varying between 0.4 and 4.5 per 1000 live births (Finer et al, 1981; Adamson et al, 1995; Ekert et al, 1997; Smith et al, 2000; Wu et al, 2004). The mortality rate of this condition varies between 15% and 20%; of the survivors, 25% exhibit permanent, and often crippling, neurologic disability from cerebral palsy, learning disabilities, epilepsy, and mental retardation (Low, 1993; Perlman, 1999). Administration of a pharmacological agent to a mother in labour to protect her unborn child from perinatal neuronal injury may represent a method of reducing the devastating consequences of neonatal asphyxia.
The ‘preconditioning’ phenomenon has been best described in cardiac tissue, where a brief ischaemic insult before injury provides protection presumably by rendering vulnerable tissue more ‘tolerant’ to injury by a prior stress (Yellon et al, 1998). Similarly, a tolerant state can be induced by xenobiotics, referred to as pharmacological preconditioning (Mizumura et al, 1995; Cason et al, 1997); some general anaesthetics exhibit this feature in a neonatal asphyxia model (Zhao and Zuo, 2004).
The current investigation was motivated by the clinical goal of finding a pharmacological agent that can be administered to the parturient providing both labour analgesia and neuroprotection against an unexpected insult in the neonate. The decision to explore this with the noble anaesthetic gas xenon is predicated by its very rapid onset and recovery characteristics, its lack of side effects (Ma et al, 2002; Sanders et al, 2005) and the recent demonstration of its neuroprotective potential in both in vitro and in vivo models of acute neuronal injury (Ma et al, 2002, 2003a, b; Wilhelm et al, 2002; Petzelt et al, 2003; Homi et al, 2003). Thus, we have investigated whether xenon is capable of preconditioning using a series of models from an in vitro glial–neuronal cell co-culture system, through a hippocampal slice culture preparation, to an in vivo neonatal rat model.
Materials and methods
This study was approved by the Home Office (UK) and conforms to the United Kingdom Animals (Scientific Procedures) Act of 1986. All efforts were made to minimize animal suffering and the number of animals used. BALB/c mice, C57BL/6J mice and Sprague–Dawley rat pups were used for in vitro and in vivo experiments, respectively. Twelve rat pups of either sex were chosen randomly from different dams and then allocated to and housed with one dam in a 12-h-light/l2-h-dark schedule in temperature- and humidity-controlled conditions. The pups were housed with their dams until weaning on postnatal day 21 and then housed in groups of four per cage.
Cell Culture
The mixed cortical glial–neuronal cocultures derived from early postnatal and foetal BALB/c mice, respectively, have been described previously (Wilhelm et al, 2002). Briefly, whole cerebral neocortices were prepared from early postnatal (days 1 to 2) pups of BALB/c mice. After trypsination and resuspension, cells were plated at a density of 6.25 × 104 cells/cm2 on 24-multiwell plates and cultured in a medium consisting of Eagle's minimum essential medium augmented with 20 mmol/L glucose, 26 mmol/L NaHCO3, 10% foetal bovine serum, 10% heat-inactivated horse serum, antibiotic-antimycotic solution (Gibco, Paisley, UK), 2 mmol/L glutamine (Sigma, Poole, UK), and 10 ng/ml murine epidermal growth factor (Gibco, Paisley, UK). Glial cells reached confluence about 1 week after plating. Cortical neuronal cells were obtained from foetal BALB/c mice at 14 to 16 days of gestation and plated at a density of 1.25 × 105 cells/cm2 on the confluent monolayer of glial cells derived from the same genetic strain. The mixed glial–neuronal cells were used at 14 ± 1 days.
Cells were kept in purpose-built airtight, temperature-controlled, cell-culture chambers. Preconditioning conditions were achieved with the desired concentration of xenon (25% to 75% atm), 5% carbon dioxide, 20% oxygen (with the balance of one atmosphere of gas represented by nitrogen) for 120 mins. Cells were then returned to a culture incubator filled with air and 5% carbon dioxide.
Twenty-four hours later, cell injury was induced by oxygen and glucose deprivation (OGD) (Goldberg and Choi, 1993). Culture medium was replaced by deoxygenated balanced salt solution (116 mmol/L NaCl, 5.4 mmol/L KCl, 0.8 mmol/L MgSO4, 1.0 mmol/L NaH2PO4, 1.8 mmol/L CaCl2, 26 mmol/L NaHCO3, pH 7.4) in the absence of glucose and maintained in an anoxic chamber at 37°C for a period of 75 mins, which causes submaximal neuronal injury as reported previously (Ma et al, 2004). The injury was terminated by washing with Eagle's minimal essential medium enhanced with 25 mmol/L glucose and 38 mmol/L NaHCO3 and the cultures were returned to normoxic conditions. The medium was harvested 16 h later and the amount of lactate dehydrogenase (LDH) released into the medium was analysed using a standardized colorimetric enzyme kit (Sigma, Poole, UK). Neuronal protection provided by xenon preconditioning was expressed as the percentage of the maximal LDH released without intervention.
Hippocampal Slice Culture
The protocol for hippocampal slice culture was modified from a previous report (Lange-Asschenfeldt et al, 2004). Briefly, the slices were derived from the brain of 4- to 7-day-old postnatal C57/6J mouse pups. The harvested brain was sectioned sagitally and the hippocampal slices were placed on membranous inserts and cultured for 10 to 14 days with Eagle's minimum essential medium enhanced with heat-inactivated horse serum. Twenty-four hours before insult, the cultured slices were exposed to 75% xenon, 20% oxygen, and 5% carbon dioxide (or 75% nitrogen for the control group) for 45 mins at 37°C (n = 5 to 9/group). OGD was induced in a balanced salt solution for 2 h. Images were obtained before and 3 days after OGD to quantify dead cells with propidium iodide (PI) fluorescence; the intensity of PI fluorescence after OGD (reflective of the number of dead cells) (Sullivan et al, 2002), less the fluorescence at baseline, reflected the neuroprotective benefit derived from preconditioning. Propidium iodide intensity, corresponding to the number of dead neurons, was assessed in CA1 and dentate gyrus areas of the hippocampus by one author who was masked to the treatment groups.
In Vivo Hypoxia–Ischaemia
The model of neonatal asphyxia was adapted from a previous report (Rice et al, 1981). Briefly, 7-day-old postnatal rats (n = 7 to 8/group) were preconditioned by a 120 mins exposure to 70% xenon and 30% oxygen; control conditions consisted of 70% nitrogen and 30% oxygen. After a variable (2, 4, 8, and 24 h) delay, the animals underwent right common carotid artery ligation under surgical anaesthesia followed by hypoxia as described previously (Rice et al, 1981). Xenon concentration was measured continuously by an in-line gas analyser (Air Products Model No. 439Xe). For comparison, another cohort of pups (n = 5) were exposed to 70% nitrous oxide (N2O) balanced with oxygen for 120 mins. This gas has a similar anaesthetic potency in this species (Lynch et al, 2000; Russel and Graybeal, 1992). Four hours later, the pups underwent right common carotid artery ligation followed by hypoxia. To control the body temperature of the pups precisely, the exposure chambers were partially submerged in a water-bath and the desired (37°C) brain temperature, measured with a telemetry temperature monitoring system (VitalView, Mini-Mitter, OR, USA), was targeted using an appropriate water temperature in the bath. In each set of experiments, a temperature probe was implanted (−2 mm from bregma and 2 mm away from sagittal sinus; the tip of probe advanced to subcortex and fixed on the skull with glue) in a sentinel rat pup that was not further assessed for brain injury.
Four days after the injury, the pups were killed by an overdose of pentobarbitone (100 mg/kg, intraperitoneally) and perfused transcardially with heparinized 0.9% saline followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer (PB). The brains were removed and placed in 4% paraformaldehyde in 0.1 mol/L PB overnight. The blocks of brain were dehydrated, embedded in wax and sectioned into 5 μm slices. After staining with the cresyl violet (Nissl), six slices were selected from each pup to match predefined brain regions relative to the bregma (+2 mm, +1, 0, −2, −4, and −5) (Paxinos and Watson, 1998) (see Figure 3A). Once identified, each slice was photographed and the infarction size (mm2) was calculated using data analysis software (ImageJ v1.31, NIH image software, Web Public Software, USA) by one author who was masked to the treatment that the pups had received.
One cohort each of preconditioned and control animals (where the delay before carotid artery ligation was 4 h) were maintained for 30 days after injury at which time they underwent functional neuromotor testing with an established assay, which included performances of prehensile traction, strength, and balance beam graded on a zero to nine scale (best score = 9) as described previously (Ma et al, 2003b). Coordination was tested by placing rats on a rotarod (rotating at 30 r.p.m.) and the time spent on the rod was assessed (maximal latency was 300 secs). For each of the functional assays, rats were tested three times with a 10 mins interval between each assessment and the sum of the three assessments was used for data analysis.
Western Blot Analysis
Seven-day-old rat pups were killed under pentobarbitone anaesthesia at 0, 2, 4, 8, or 24 h after xenon exposure and both cortices were harvested, lysed in lysis buffer (pH 7.5, 20 mmol/L Tris-HCl, 150 mmol/L NaCl, 1 mmol/L Na2DTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, 2 mmol/L DL-dithiothreitol, 1 mmol/L phenylmethanesulphonyl and 1 μg/ml leupeptin), and centrifuged at 3,000g for 10 mins. Protein concentration in the supernatant was determined by Dc protein assay (Bio-Rad, Herts, UK). Protein extracts (30 μg/sample) and a biotinylated molecular weight marker (New England Biolab, Hitchin, UK) were denatured in Laemmli sample loading buffer (Bio-Rad, Herts, UK) at 100°C for 5 mins, separated by 10.5% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Amersham Biosciences, Little Chalfont, UK). The membrane was treated with blocking solution (5% nonfat dry milk in Tween-containing Tris-buffered saline (10 mmol/L Tris, 150 mmol/L NaCl, 0.1% Tween, pH 8.0)) and incubated with rabbit monoclonal antibody directed against phosphorylated cAMP (cyclic adenosine 3′,5′-monophosphate)-response element binding protein (pCREB), CREB, Bcl-2 (1:1000, New England Biolab, Hitchin, UK) brain-derived neurotrophic factor (BDNF) (1:250, Santa Cruz Biotech, Wembley, UK), or α-tubulin (1:2000, Sigma, Poole, UK) overnight at 4°C. The horseradish peroxidase-conjugated goat antibody to rabbit IgG (1:2000, New England Biolab) was used to detect the primary antibodies. The bands were visualized with the enhanced chemiluminescence system (ECL, Amersham Biosciences, Little Chalfont, UK).
Data Analysis
The statistical analysis was performed by analysis of variance, unpaired t-test, or Mann–Whitney test where appropriate. A P < 0.05 was considered to be statistically significant.
Results
Time- and Concentration-Dependent Effect of Xenon
In the in vitro coculture system, LDH release induced by OGD was significantly inhibited by preconditioning with xenon for 120 mins; no additional preconditioning was achieved with exposure for 180 mins (Figure 1A). Xenon preconditioning was concentration-dependent resulting in reduction of LDH release of 55% ± 7% and 38% ± 6% of control at 50% and 75% xenon, respectively (P < 0.01) (Figure 1B).
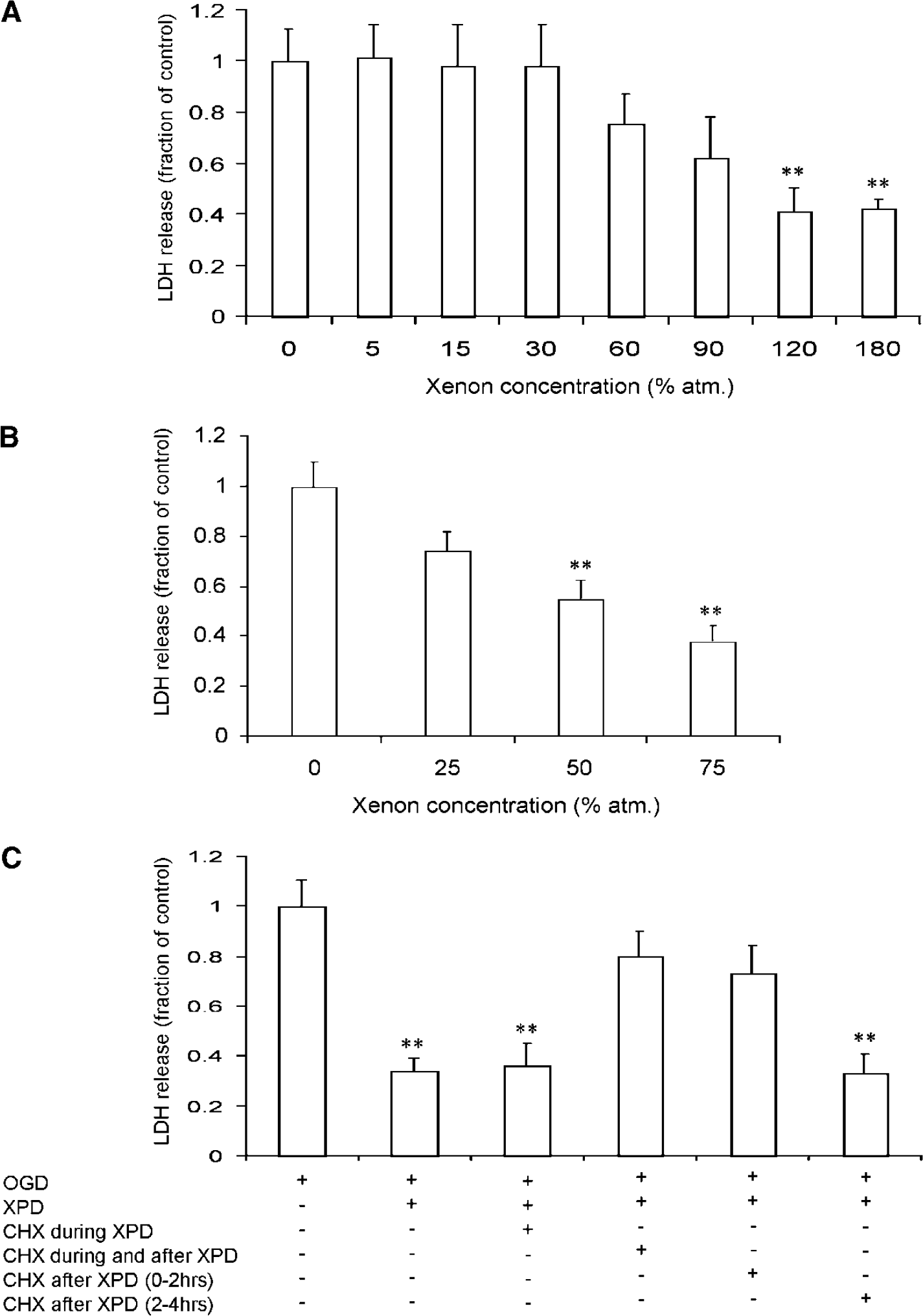
Xenon preconditioning (XPD) reduces lactate dehydrogenase (LDH) release in cell culture. (
Requirement of New Protein Synthesis for Xenon Preconditioning
To study whether protein synthesis was involved in the neuroprotective preconditioning afforded by xenon, the cells were treated with a protein synthesis inhibitor, cycloheximide (CHX, 30 μmol/L) either during preconditioning, and/or for 120 mins immediately after 120 mins xenon exposure, or from 120 to 240 mins after xenon withdrawal (Figure 1C). The protective effect of xenon preconditioning was abolished by CHX when administered for 120 mins after a 120 mins xenon exposure. These data indicate that preconditioning requires new protein synthesis during the first 2 h after xenon exposure.
Preconditioning with Xenon in Hippocampal Slice Cultures
In both CA1 and dentate gyrus areas of hippocampus, OGD-induced PI intensity was significantly reduced by preconditioning with xenon; in the CA1 region of the hippocampus, PI intensity after OGD was 22% ± 6% after xenon preconditioning versus 75% ± 6.5% under control conditions after OGD (P < 0.01). A smaller but still significant protective effect was found in the dentate gyrus, namely, 51% ± 4% in the xenon preconditioned group against 66% ± 5% in the controls (P < 0.05) (Figure 2).
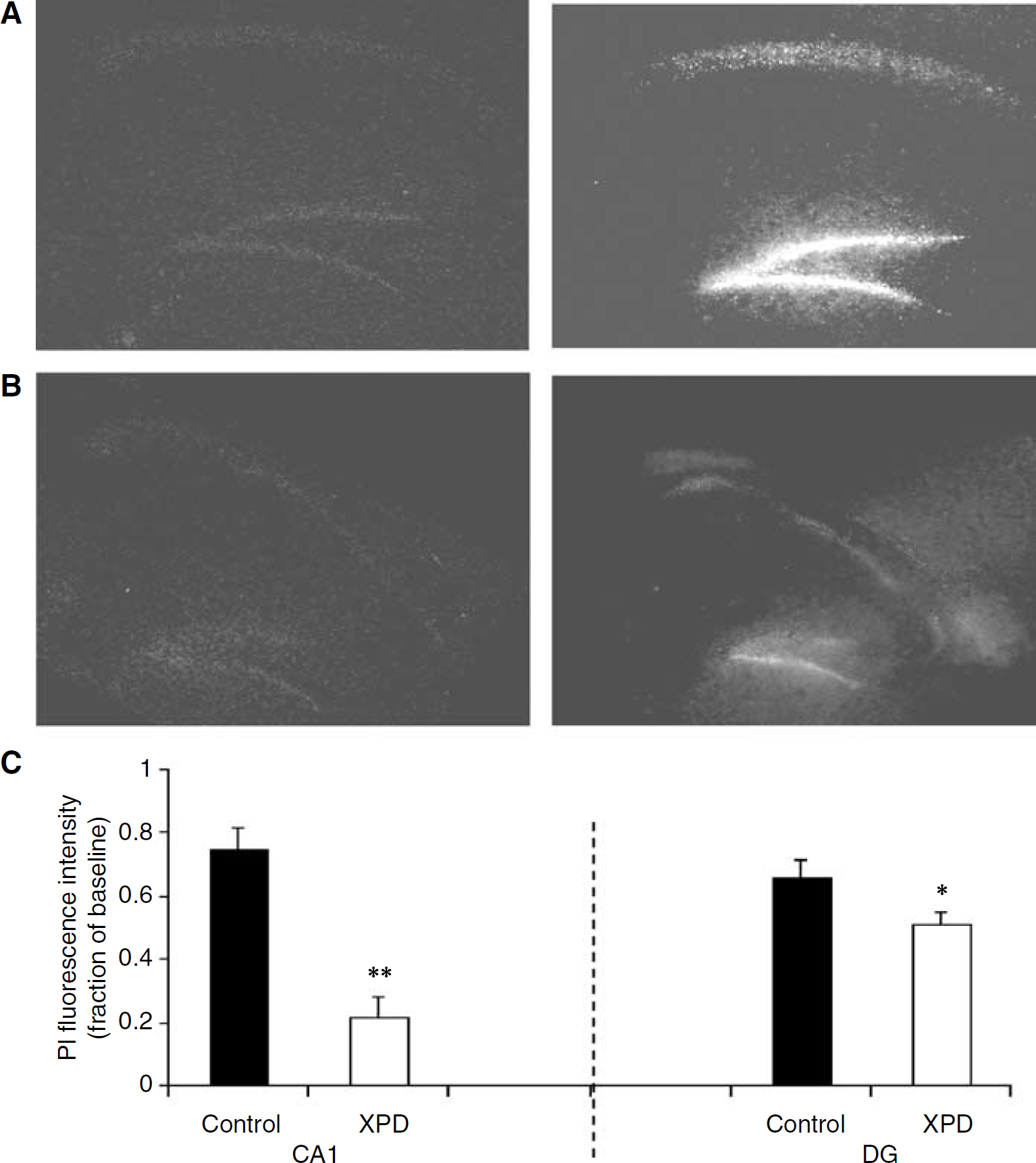
Xenon preconditioning (XPD) against oxygen–glucose deprivation (OGD)-induced neuronal death in the hippocampal slice culture. Hippocampal slices were cultured for 3 weeks and then exposed to 75% xenon for 2 h. Twenty-four hours after exposure, slices were treated with OGD for 2 h. Cell death was assessed with propidium iodide (PI) staining and measured by fluorescence intensity in CA1 and dentate gyrus (DG) of hippocampus 3 days after OGD. (
Comparison Between Xenon and N2O Preconditioning In Vivo
We sought to determine whether N2O, a gaseous anaesthetic that is also able to antagonize the N-methyl-D-aspartate (NMDA) subtype of glutamate receptors (Jevtovic-Todorovic et al, 1998), also exerts preconditioning. While rat pups exposed to xenon for 120 mins were protected from a subsequent hypoxic–ischaemic insult 4 h later, N2O was ineffective (Figures 3B and 3C).
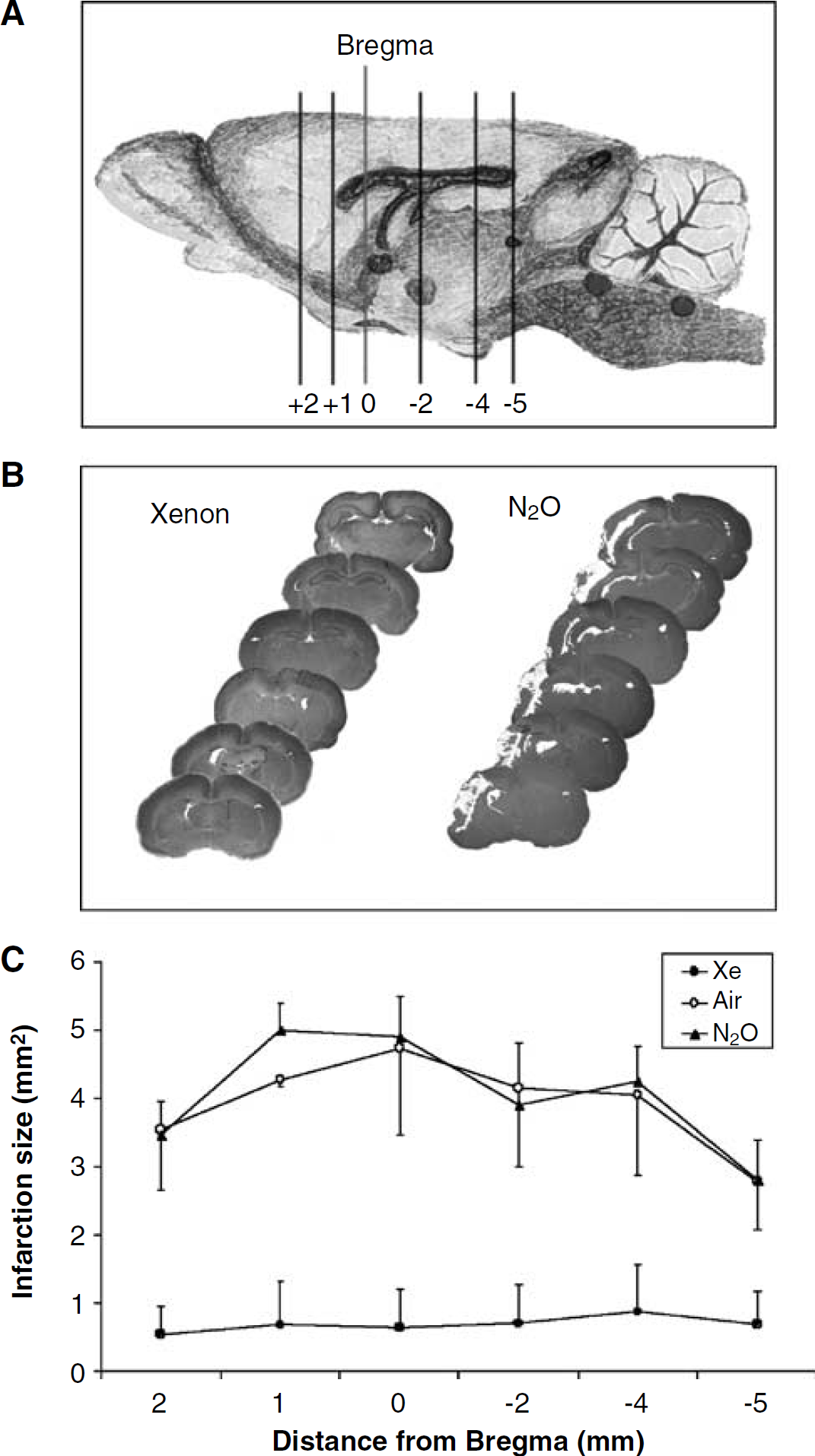
Effect of preconditioning on focal infarction size. After exposure to xenon (70%), N2O (70%), or air, 7-day-old rat pups underwent hypoxic–ischaemic injury 4 h later. Infarct size was assessed 4 days later. (
Time Course Between Xenon Preconditioning and Hypoxia–Ischaemia In Vivo
To determine the time required for the neuroprotective effect afforded by xenon preconditioning, pups were treated with xenon, and 2, 4, 8, or 24 h later, they underwent hypoxic–ischaemic injury for 90 mins.
When the preinjury interval after xenon exposure was only 2 h, there was no statistically significant decrease in the area under curve (AUC) of infarction between the treatment and control groups (16.9 ± 5.5 versus 15.4 ± 5.9 arbitrary unit, P > 0.05) (Figure 4A). However, when administered 4, 8, or 24 h before hypoxia–ischaemia, xenon preconditioning significantly reduced infarction size when compared with corresponding controls (Figures 4B, 4C, and 4D).
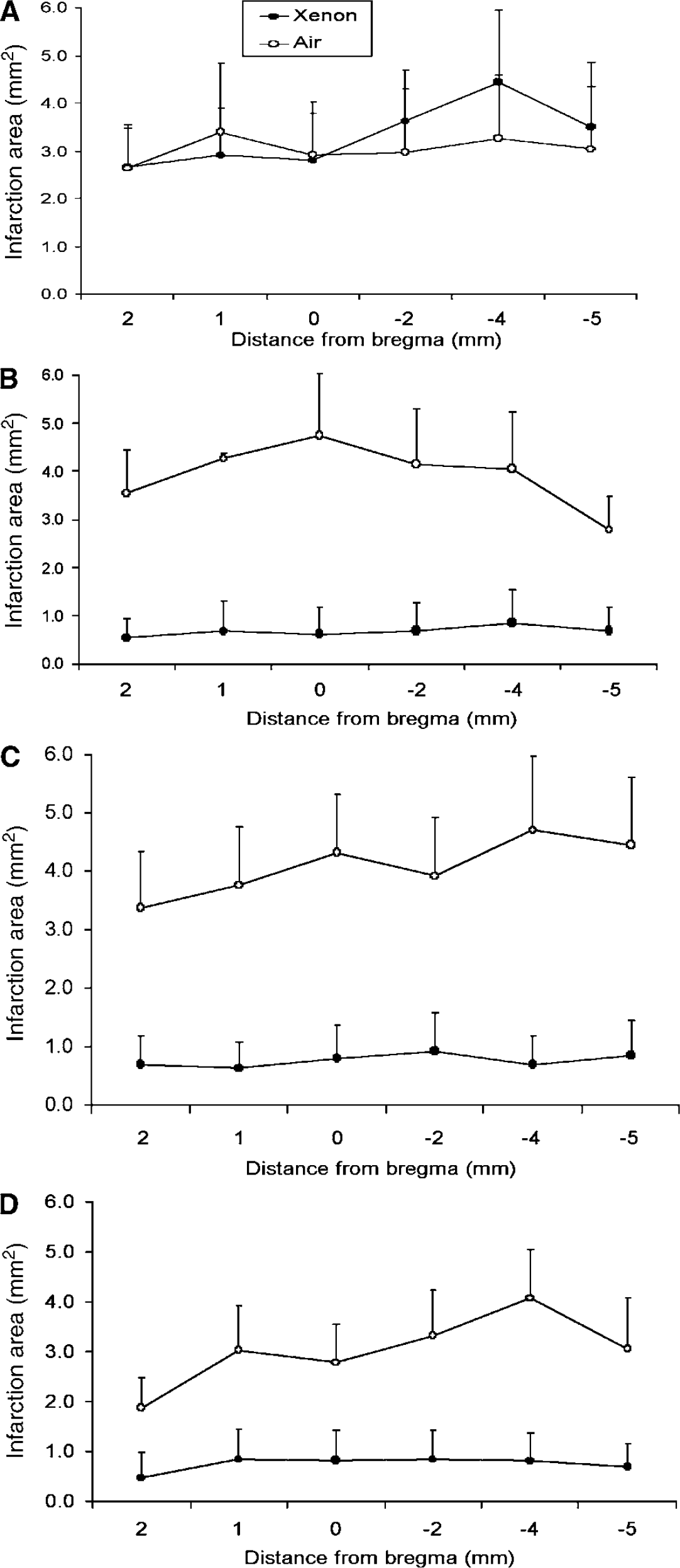
Time course of xenon preconditioning (XPD) effect in neonatal rats. After 2 h xenon (70%), rat pups underwent 90 mins of hypoxic–ischaemic injury, a variable time after xenon exposure. Cerebral infraction was measured 4 days later. (
Effect on Long-Term Neurologic Function
We chose one time point (xenon preconditioning administered 4 h before hypoxia–ischaemia) to investigate whether the morphologic improvement by xenon preconditioning extended to a long-term improvement in neurologic function. Thirty days after the injury, the impairments in neurologic motor function (Figure 5A) and coordination (Figure 5B) were both significantly improved by xenon preconditioning.
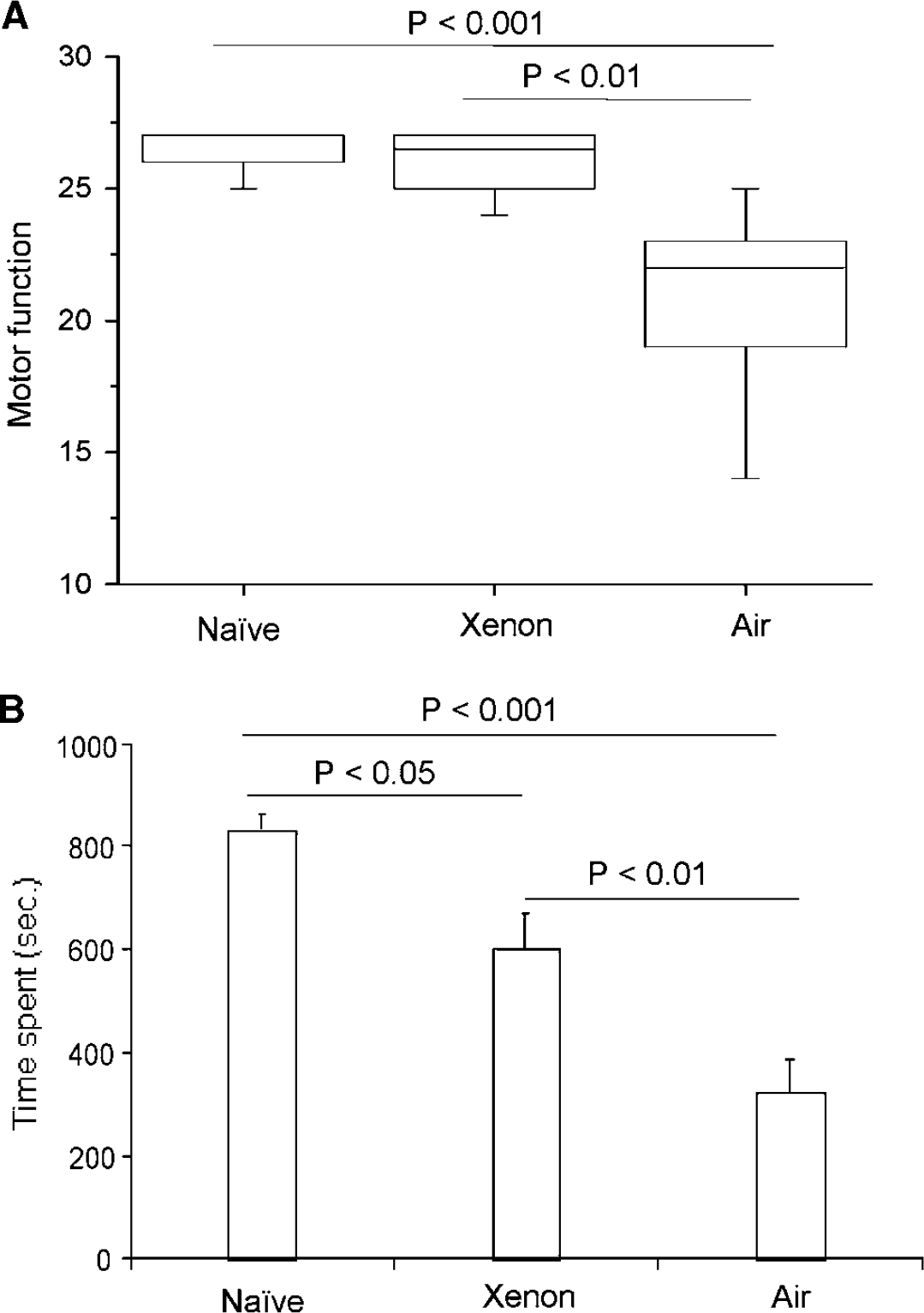
Long-term functional neurologic outcome after xenon preconditioning (XPD). Seven-day-old Sprague–Dawley pups were exposed for 2 h to 70% xenon or air; 4 h later, pups underwent hypoxic/ischaemic injury for 90 mins. Neuromotor function (
pCREB, Bcl-2, and BDNF
It has been postulated that preconditioning stimuli exert their effects by triggering signalling cascades that ultimately converge onto nuclear transcription factors. There are a large number of transcription factors (CREB being the prototype) that are pivotally involved in growth, survival, and synaptic connectivity of developing neurons (Lonze and Ginty, 2002). Therefore, we examined whether xenon exposure to neonatal mice triggers CREB phosphorylation (pCREB). A significant increase in pCREB was noted 4 h after xenon exposure (Figure 6A); conversely, N2O exposure, which does not precondition, did not induce pCREB (Figure 6B). Phosphorylation of CREB at serine 133 allows CREB to recruit coactivators such as CREB-binding protein (CBP) and RNA polymerase II to activate transcription of CREB-dependent genes including the prosurvival protein, Bcl-2 (Wilson et al, 1996), and BDNF (Tao et al, 1998). Bcl-2 and BDNF were significantly upregulated 4 h after xenon treatment (Figure 6C).
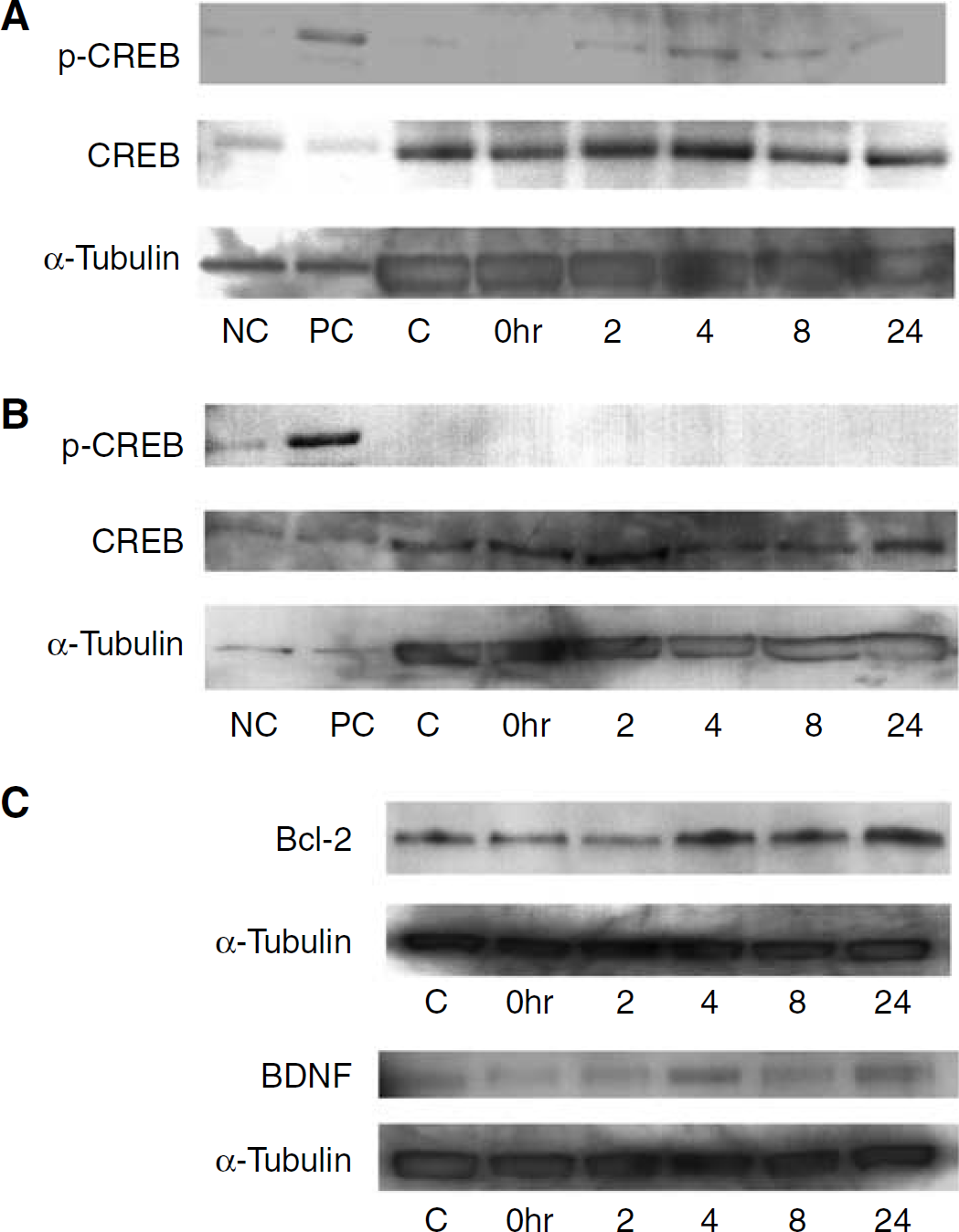
Phosphorylated cAMP (cyclic adenosine 3′,5′-monophosphate)-response element binding protein (pCREB) immunoreactivity in pups' cerebral cortex after exposure to 70% xenon (
Discussion
The noble anaesthetic gas xenon provides long-lasting protection against the consequences of acute neurologic injury when delivered up to 24 h before the insult. This robust preconditioning action of xenon can be demonstrated in several paradigms of acute hypoxic–ischaemic injury including in vitro cell and organotypic cultures as well as an in vivo model of neonatal asphyxia. Initial studies exploring the mechanisms point towards the upregulation of prosurvival proteins.
Preconditioning was first described in a dog model of myocardial injury (Murray et al, 1986) in which sublethal ischaemia enabled cells to better tolerate subsequent usually lethal ischaemia. Elucidation of the molecular pathways mediating protection by ischaemic preconditioning provided rational targets for pharmacological intervention. Several pharmacological agents are able to mimic ischaemic preconditioning in the myocardium, including volatile anaesthetics (Yellon et al, 1998). Other interventions, including temperature elevation, precondition by the phenomenon of crosstolerance in which a sublethal stress confers tolerance to a subsequent stress that is qualitatively different from the first one (Kirino, 2002).
Both ischaemic and pharmacological preconditioning induce intracellular signalling via protein kinase C (PKC) and CREB (Uecker et al, 2003; Hara et al, 2003; Lee et al, 2004; Eliseev et al, 2004; Weber et al, 2005). Inhibition of PKC activation via the PKC inhibitor staurosporine can prevent induction of ischaemic preconditioning and activators of PKC can mimic ischaemic preconditioning (Yellon et al, 1998). Activation of the NMDA subtype of the glutamate receptor inhibits PKC; conversely, NMDA antagonists, such as MK-801, increase PKC activity (Tremblay et al, 2000) and exert a preconditioning effect. Xenon, which is also an NMDA antagonist (Franks et al, 1998), also activates PKC, which is thought to be the mechanism for the myocardial preconditioning produced by xenon (Weber et al, 2005). Surprisingly, N2O does not precondition, although it too exerts an NMDA antagonistic action at the concentration used (Jevtovic-Todorovic et al, 1998). Consistent with its lack of efficacy as a preconditioner, N2O did not increase pCREB, an effect that was noted after exposure to xenon (Figures 6A and 6B). Upstream of pCREB, N2O and xenon exhibit divergent effects on PKC signalling; preconditioning with xenon upregulates PKC activity (Weber et al, 2005), while N2O has been shown to inhibit PKC activity (Fukura et al, 2000).
PKC is able to phosphorylate CREB (Roberson et al, 1999); together with its transcriptional coactivator, CBP, pCREB upregulates the expression of a number of genes, including Bcl-2 and BDNF (Wilson et al, 1996; Tao et al, 1998) and their corresponding proteins. In our study, we did show upregulation of pCREB and Bcl-2 as well as BDNF and this pathway may contribute to the mechanism for the preconditioning effect exerted by xenon (Figure 7).
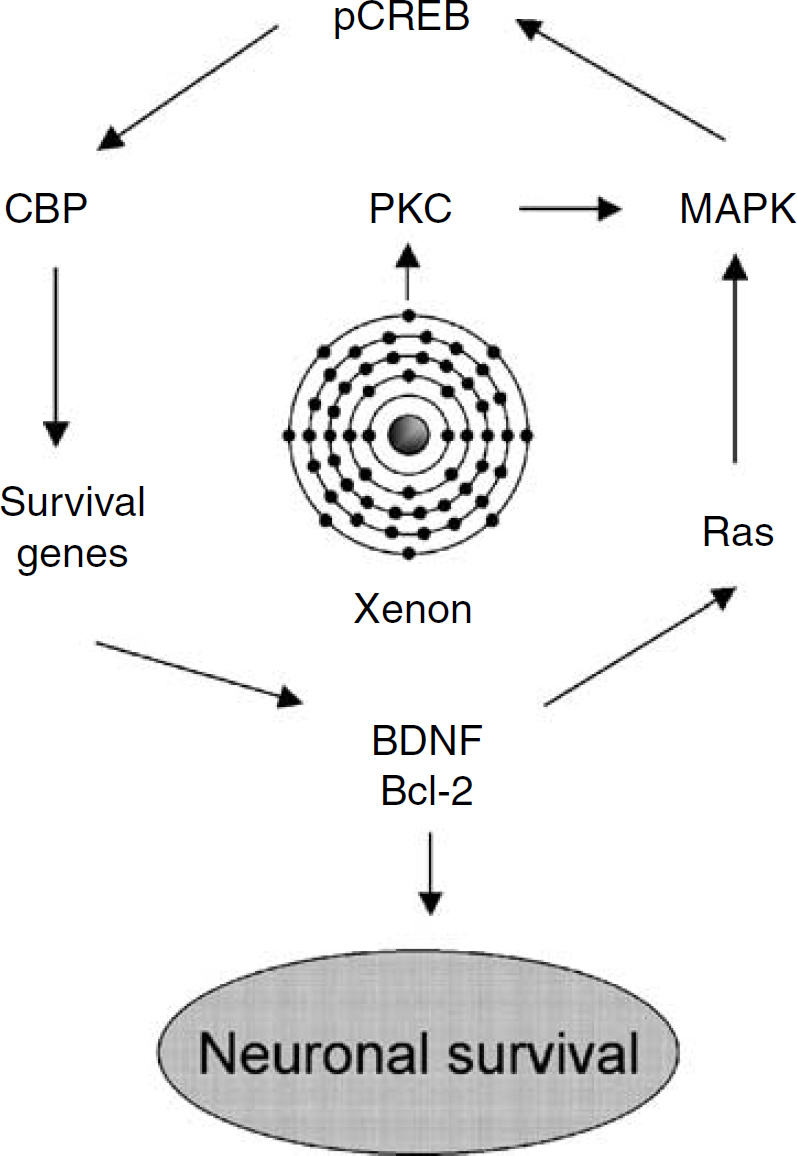
Putative modulation of xenon on CREB transcriptional pathway associated with survival signals. Xenon activates protein kinase C (PKC) which triggers phosphorylation of mitogen-activated protein kinases (MAPK) including p38 MAPK and extracellular signal-regulated kinase (ERK) (Weber et al, 2005). Consequently, cAMP-response element-binding protein (CREB) is transformed into the phospho-CREB (pCREB) form (Roberson et al, 1999). The CREB and CREB-binding protein (CBP) interaction can then induce transcription of a battery of CREB-dependent survival genes. The transcribed cAMP (cyclic adenosine 3′,5′-monophosphate)-response element binding (CREB)-dependent proteins, including brain-derived neurotrophic factor (BDNF) and prosurvival protein Bcl-2, can be enhanced, which promotes cells to tolerate hypoxia–ischaemic insult. The activation of Ras (a small guanine nucleotide-binding protein that is essential for the proliferation, differentiation, and survival of cells) by neurotrophic factors and prosurvival proteins can in turn initiate more MAPK phosphorylation and by so doing it will enhance the survival signals.
Pathogenic pathways that contribute to the development of hypoxic–ischaemic neonatal brain injury include oxygen-free radical formation, release of excitatory neurotransmitters, and consequent elevation of intracellular calcium; these result in energy failure within 20 h after insult to produce irreversible brain damage (Vannucci et al, 2004). Therefore, for an intervention to be maximally effective, it may need to be used before, during, or immediately after birth. Xenon's preconditioning properties coupled to its powerful neuroprotective capabilities and absence of fetotoxicity (Lane et al, 1980; Fukura et al, 2000) might have clinical implications in terms of both the prevention as well as the treatment of neonatal asphyxia.
In summary, we have used in vitro and in vivo models to show that xenon is capable of preconditioning against neuronal injury induced by OGD or hypoxia–ischaemia. Its effect is long lasting as evidenced by the improvement of both morphology and neurologic function outcome assessed 4 and 30 days after the injury, respectively. Our data suggest that the mechanism of xenon preconditioning may be due to the upregulation of genes and synthesis of the CREB-dependent survival proteins, Bcl-2 and BDNF. Future elaboration of the mechanisms involved can exploit the fact that xenon's preconditioning effect is also present in an organotypic slice preparation. Thus, we can explore the involvement of putative signalling pathways through both pharmacologic (antagonists and toxins) and genetic (blockers of transcription) manipulations and assess the effect not only morphologically but also functionally using long-term potentiation.