
Abstract
Nix, a hypoxia-sensitive member of the Bcl-2 family, is upregulated at the mRNA level during hypoxia through induction of a hypoxia-inducible factor-1α (HIF-1α) response element in its promoter sequence. However, the mechanism(s) regulating Nix protein activation remain unclear. The present studies examine Nix protein expression and subcellular distribution in response to hypoxic stimuli in vivo and in culture and to two disparate apoptotic stimuli in vitro. Upregulation and translocation of Nix (by day 5) in hypoxic/serum-deprived CHO-K1 cells, was preceded by Bax activation (by day 4) and caspase-3 processing (by day 2), suggesting that initiation of cell death in vitro is a Nix-independent event. In contrast, an early Nix response (upregulation and translocation to the mitochondria) was observed after 6 h of middle cerebral artery occlusion in the rat. Nix translocation was observed in the ipsilateral cortex and striatum before other histological (infarct development, neuronal loss, apoptotic body formation) or biochemical (Bax activation or caspase-3 cleavage) markers of damage were detected. While fundamental differences between hypoxia/ischaemia in culture and in vivo likely explain the different temporal profiles of Nix, Bax, and caspase-3 activation observed, these studies show that like Bax, mitochondrial accumulation is a common event during Nix activation. These are the first studies to show upregulation and translocation of Nix in the ischaemic brain and suggest Nix to be a novel therapeutic target in ischaemic research. Moreover, Nix upregulation in staurosporine-treated SH-SY5Y cells and dexamethasone-treated A1.1 cells supports a more generalized role for Nix in apoptotic cell death.
Introduction
The ability to sense and respond to changes in oxygen levels is critical for many developmental (i.e. chondrocyte survival during bone development), physiologic (i.e. erythropoiesis) and pathologic processes (i.e. ischaemia, inflammation, and cancer) (Cramer et al, 2004; Melillo, 2004). Nix (Nip3-like protein X) and its homologue BNip3 (Bcl-2 and 19 kDa interacting protein-3) are considered to be mitochondrial-localized members of the Bcl-2 family which, courtesy of a functional hypoxia-inducible factor-1α (HIF-1α) responsive element in their promoter sequence, are activated at the level of transcription during hypoxia (Bruick, 2000). Upregulation of Nix and BNip3 mRNA have been shown in cultured Chinese hamster ovary (CHO-K1) cells in response to hypoxia (Bruick, 2000). While concomitant increases in BNip3 protein expression have been reported both in vitro (Bruick, 2000) and in vivo (Schmidt-Kastner et al, 2004) no such information is available on the Nix protein response.
Nix and BNip3 each possess a C-terminal transmembrane domain necessary for mitochondrial localization and death-promoting activity (Yasuda et al, 1998; Chen et al, 1999). While BNip3 is associated with a necrotic-like cell death (Vande et al, 2000), Nix appears to promote a more Bax-like cell death involving cytochrome c release and caspase activation (Imazu et al, 1999). Whether this mechanism reflects the endogenous Nix response to apoptotic stimuli remains unclear, as most studies have used over-expression as a tool to investigate Nix subcellular distribution and proapoptotic activity (Matsushima et al, 1998; Imazu et al, 1999; Yussman et al, 2002).
The biological roles of Nix are poorly defined. Elevated expression of Nix has, however, been reported in the hypertrophic myocardium where forced expression is associated with apoptotic cardiomyopathy and death (Yussman et al, 2002). Increases in Nix mRNA levels have also been described in the hypoxic (peri-necrotic) region of solid tumours (Sowter et al, 2001) while Nix protein expression appeared downregulated in malignant lung carcinoma tissue (Sun et al, 2004). In the present studies, we investigated the response of endogenous Nix protein to hypoxia/ischaemia in vitro and in vivo and to two disparate models of apoptotic cell death reported to involve p53 transcriptional activation and mitochondrial dysfunction (staurosporine-treated SH-SY5Y neuroblastoma cells and dexamethasone-treated A1.1 mouse T-cell lymphoma cells).
Materials and methods
Chemicals
All chemicals were obtained from Sigma Aldrich (Poole, UK) unless otherwise stated.
Middle Cerebral Artery Occlusion in the Rat
Procedures were performed under license by the UK authorities in accordance with The Animal (Scientific Procedures) Act of 1986. Middle cerebral artery (MCA) occlusion was performed according to Longa et al (1989) with modifications. The right MCA of male Sprague-Dawley rats (275 to 315 g; Charles River, Margate, UK) was occluded using a poly-
Histological Assessment of Ischaemic Damage
After transcardiac perfusion with 4% paraformaldehyde/phosphate-buffered saline (para/PBS) under deep anaesthesia, rat brains were removed and postfixed in 20% sucrose in para/PBS. Coronal brain sections (20 μm) were processed for histologic assessment. Infarct boundaries were identified by thionin staining and cell morphology was assessed by haematoxylin and eosin staining.
Cell Culture and Induction of Apoptosis
Chinese hamster ovary cells were cultured in HAM-F12 medium containing 10% foetal calf serum (FCS; Invitrogen Ltd, Paisley, UK), 2 mmol/L
Human SH-SY5Y neuroblastoma cells (passages 13 to 18) were grown to confluence in DMEM containing 10% FCS, 2 mmol/L L-glutamine, 100 U/mL penicillin and 100 mg/mL streptomycin. Apoptosis was induced by 500 nmol/L staurosporine (STS) for 5 h. Control cells were treated with DMSO vehicle (1:1,000).
Mouse A1.1 T-cell hybridoma cells were grown to confluence in RPMI 1640 medium containing 5% FCS, 2 mmol/L
Preparation of Rat Brain Cell Homogenates
Rats were killed by anaesthetic overdose and decapitation. Brains were removed and the cortex and striatum were dissected out. Samples (6 animals per treatment group) were homogenized in tissue extraction buffer (SCFT buffer: 10 mmol/L Tris-Cl pH 7.4, 0.32 mol/L sucrose, 1 mmol/L EGTA, 125 mmol/L NaCl, 0.1 mmol/L DTT, 20 μmol/L lactacystin, 1 mmol/L PMSF, and 10 μg/mL each aprotinin, pepstatin, and leupeptin) by performing 6 ‘loose’ followed by 10 ‘tight’ strokes using a glass Dounce homogenizer (Jencons, Sussex, UK). Samples were passed through 0.35 mm gauze to remove cell debris. Whole-cell homogenates were centrifuged at 1,000g for 3 mins and the cell debris pellet discarded. An aliquot of this fraction was removed, CHAPS (final concentration of 2%) was added and the sample centrifuged after 30 mins on ice (13,000 r.p.m. for 5 mins). The supernatant was retained as the whole-cell lysate. The remaining whole-cell homogenate was processed further to isolate cytosolic and mitochondrial fractions.
Isolation of Cytosolic and Mitochondrial Fractions from Rat Brain
All procedures were performed at 4°C. Whole-cell homogenates were centrifuged at 10,000g for 30 mins to obtain crude cytosolic (supernatant) and crude mitochondrial (pellet) fractions. The pellet was washed twice (10,000g for 30 mins) with SCFT buffer and the resultant mitochondrial-enriched fraction was resuspended in SCFT buffer containing 2% CHAPS. The crude cytosolic fraction was further centrifuged at 100,000g for 1 h and the supernatant retained as the S100 cytosolic fraction.
Preparation of Cell Lysates from Immortalized Cells
All procedures were performed at 4°C. Cells were collected by centrifugation (200g for 7 mins) and the resultant cell pellet was resuspended in cell extraction buffer (SCFC: 10 mmol/L Tris-Cl pH 6.7, 0.25 mol/L sucrose, 1 mmol/L EGTA, 10 mmol/L KCl, 0.15 mmol/L MgCl2, 0.1 mmol/L DTT, 1 mmol/L PMSF, 20 μmol/L lactacystin, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerolphosphate, 1 mmol/L sodium orthovanadate, and 10 μg/mL each aprotinin, pepstatin, and leupeptin) and homogenized using 15 ‘up/down’ strokes in a tight-fitting glass Dounce homogenizer (Jencons). Nuclei, insoluble material, and unbroken cells were removed by centrifugation at 600g for 5 mins. The resultant supernatant was retained as the whole-cell homogenate. CHAPS (final concentration of 2%) was added to in an aliquot of this homogenate, centrifuged after 30 mins on ice (13,000 r.p.m. for 5 mins) and the supernatant designated the whole-cell lysate. The remaining whole-cell homogenate was processed further to isolate cytosolic and mitochondrial fractions.
Isolation of Cytosolic and Mitochondrial Fractions from Immortalized Cells
All procedures were performed at 4°C. Whole-cell homogenates were centrifuged at 6,000g for 15 mins to separate the sample into crude cytosolic (supernatant) and crude mitochondrial (pellet) fractions. The pellet was washed twice (6,000g for 15 mins) with SCFC buffer and the resultant mitochondrial-enriched fraction was resuspended in SCFC buffer containing 2% CHAPS. The crude cytosolic fraction was further centrifuged at 100,000g for 1 h and the supernatant retained as the S100 soluble cytosolic fraction.
Assessment of Protein Concentration and Western Blotting
Protein content was measured using a standard colorimetric BCA assay (Pierce, Tattenhall, UK). Samples (30 μg protein) were separated using SDS polyacrylamide gels and transferred onto nitrocellulose membranes (Hybond C; Amersham Life Sciences, Little Chalfont, UK). Membranes were blocked in PBS containing 0.01% Tween 20, 5% Blotto, pH 7.4 (1 h room temperature) then incubated overnight at 4°C with one of the following antibodies: cleaved caspase-9 (1:1,000; Cell Signalling Technology, Hitchin, UK), cleaved caspase-3 (1:1,000, Cell Signalling Technology), BaxNT (1:1,000; Upstate Biotechnology, Milton Keynes, UK), Nix/BNip3L (1:500; ΨProSci. Inc., Moway, USA), β-tubulin (1:1,000; Sigma Aldrich), α-tubulin (1:5,000; Sigma Aldrich), or VDAC (1:500; CN Biosciences, Nottingham, UK). Bound complexes were visualized using the appropriate horseradish peroxidase-coupled anti-rabbit Ig, anti-mouse Ig (both 1:2,500; Amersham Life Sciences), or anti-goat Ig (1:20,000; Sigma Aldrich) secondary antibody and enhanced chemiluminescence (ECL; Amersham Life Sciences). Positive controls were run on all gels to ensure antibody specificity. For a negative control, the Nix antibody was preabsorbed with an equal volume of Nix blocking peptide (ΨProSci. Inc.) for 30 mins at 37°C.
Results
Nix is Upregulated in Hypoxia/Serum-Deprived CHO-K1 Cells
Upregulation of Nix and BNip3 mRNA and of BNip3 protein expression were previously reported in a CHO-K1 cell line exposed to hypoxia/serum deprivation (Bruick, 2000). However, Nix protein responses were not investigated. Preliminary studies showed that 7 days hypoxia without serum deprivation (0.5% O2/10% FCS) did not significantly affect CHO-K1 cell viability after 7 days, while 7 days normoxia with serum deprivation (20% O2/0.5% FCS) produced limited cell death (Figure 1A). In contrast, hypoxia with serum deprivation (0.5% O2/0.5% FCS) reduced cell viability by a maximum of ~60% over 7 days (Figure 1A). Accordingly, subsequent studies in CHO-K1 cells used hypoxia/serum deprivation as a model of hypoxic cell death and normoxia with serum deprivation as a negative control.
Supporting a role for Nix in this model of cell death, upregulation of Nix protein expression was observed after 4 to 7 days hypoxia/serum deprivation (0.5% O2/0.5% FCS; Figure 1A), but not in control (serum deprived) CHO-K1 cells. The Nix antibody also recognized a 48 kDa migrating band in a K562 cell lysate (positive control supplied with the antibody) and in an apoptotic mouse A1.1 cell line (caspase-3 positive control). The hypoxia-responsive protein, Glut-1 produced a similar temporal profile as Nix (Figure 1B). In contrast, onset of caspase-3 cleavage was observed after 2 days of hypoxia/serum deprivation with levels steadily increasing over 7 days (Figure 1B). Antibody specificity was confirmed by preincubating the Nix antibody with its corresponding competing peptide, which resulted in selective loss of the ~48 kDa immunoreactive band (Figure 1C). A cross-reacting 30/60 kDa immunoreactive band corresponding to the Nix homologue, BNip3 was not observed. A 96 kDa band was, however, detected in CHO-K1 cells (Figure 1B and Figure 1C), but preincubation with the competing peptide had no effect on detection of this band.
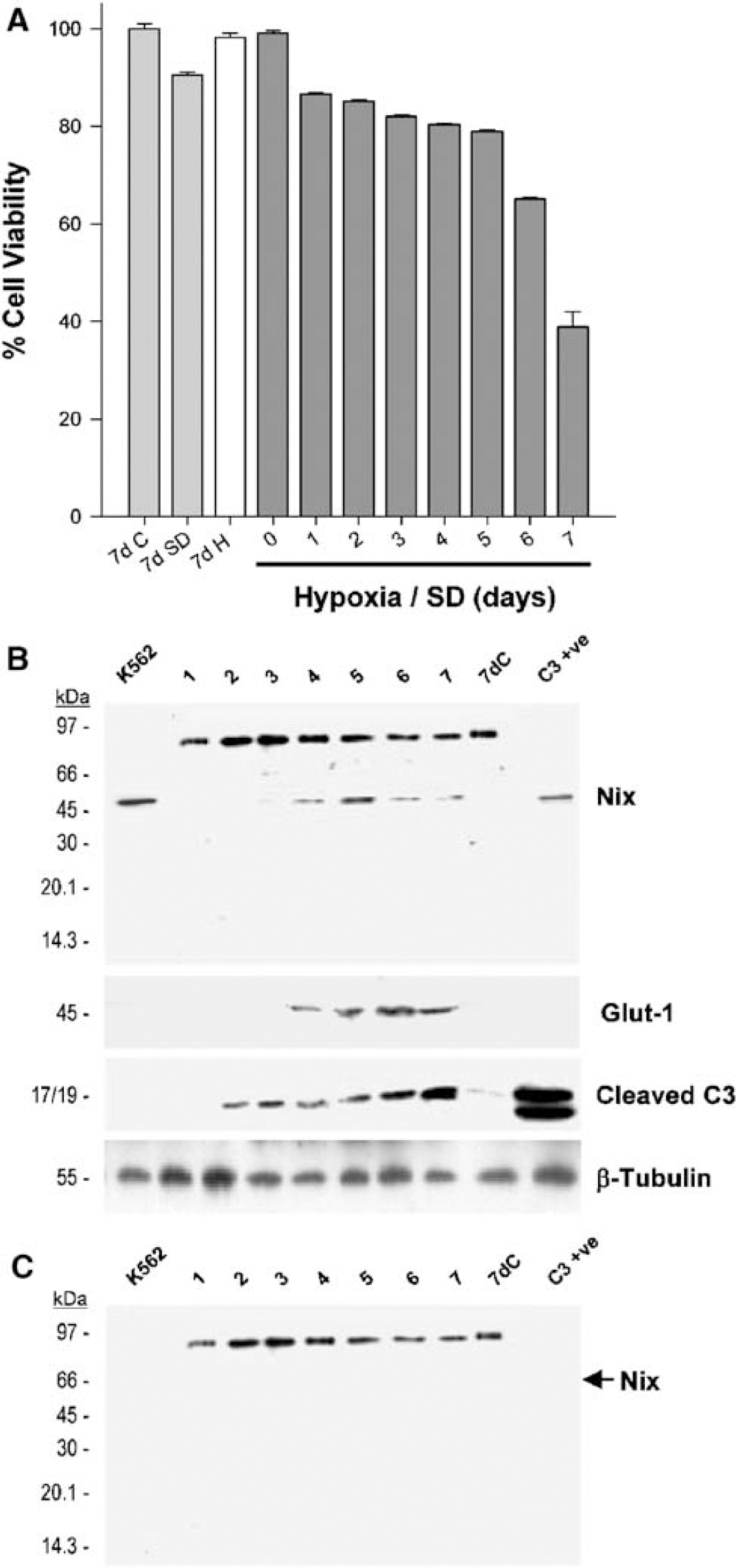
Nix protein expression in hypoxic CHO-K1 cells. CHO-K1 cells were exposed to hypoxia with serum withdrawal for 1 to 7 days (0.5% O2/0.5% FCS) or hypoxia alone (7dH; 0.5% O2/10% FCS) for 7 days. Control cells were maintained under normal culture conditions (7dC; 20% O2/10% FCS) or normoxia with serum deprivation (7dSD; 20% O2/0.5% FCS) for the duration of the experiment (
Nix and Bax Subcellular Distribution After Hypoxia—Serum Deprivation in CHO-K1 Cells
The effects of hypoxia/serum deprivation on the subcellular distribution of Nix were investigated in CHO-K1 cells. Whole-cell homogenates from control and apoptotic samples were separated into cytosolic and mitochondrial-enriched fractions. After 4 days of hypoxia, upregulation of Nix was observed in the cytosol without obvious translocation to the mitochondria; however, mitochondrial translocation was evident by day 5 (Figure 2). In contrast, proapoptotic Bax was detected in both cytosol and mitochondria fractions of untreated CHO-K1 cells. Bax translocation to the mitochondria preceded Nix translocation and was evident after 4 days of hypoxia (Figure 2).
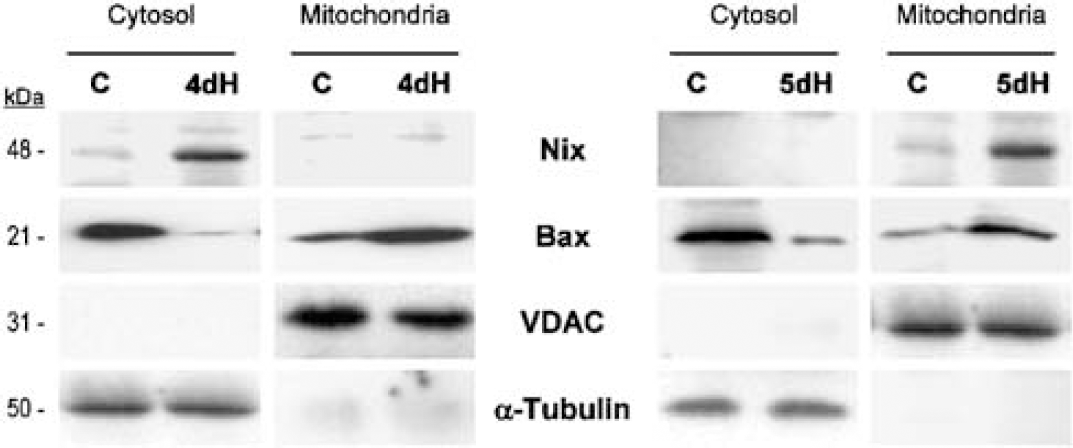
Nix and Bax translocation in hypoxic CHO-K1 cells. CHO-K1 cells were exposed to hypoxia with serum withdrawal (0.5% O2/0.5% FCS) for 4 (4dH) or 5 (5dH) days. Control (
Nix and Bax Responses to Non-Hypoxic Insults In vitro
Because both Nix- and Bax-activation have been suggested to involve similar mitochondria-dependent cell death pathways (Imazu et al, 1999), we investigated whether Nix could be activated by nonhypoxic stimuli reported to involve the Bax pathway. Apoptotic cell death was induced in CHO-K1 cells by hypoxia/serum deprivation (0.5% O2/0.5% FCS), in SH-SY5Y cells by staurosporine treatment (STS, 500 nmol/L per 5 h) and in A1.1 cells by dexamethasone treatment (DEX, 200 nmol/L per 18 h). Induction of apoptosis in each cell type was confirmed by the detection of cleaved caspase-3 immunoreactivity (Figure 3). Apoptosis in each model was associated with upregulation of Nix and Bax protein expression (Figure 3).
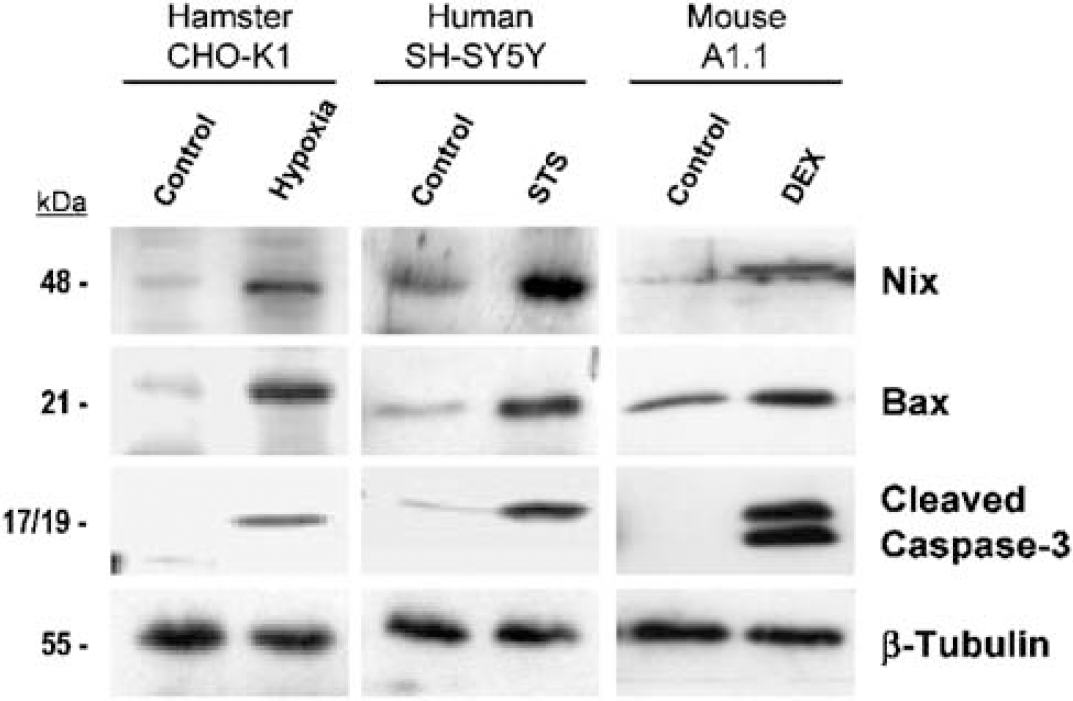
Nix and Bax responses to hypoxic and nonhypoxic stimuli. Apoptosis was induced in several in vitro models of apoptosis: CHO-K1 cells exposed to hypoxia/serum withdrawal (0.5% O2/0.5% FCS for 4 days); staurosporine-treated SH-SY5Y cells (STS–500 nmol/L for 5 h) or dexamethasone-treated A1.1 cells (DEX–200 nmol/L for 18 h). CHO-K1 control cells were maintained under normoxia with serum deprivation (20% O2/0.5% FCS). Control SH-SY5Y and A1.1 cells were treated with DMSO vehicle (1:1,000) for the duration of the experiment. Cell lysates were prepared and analysed for changes in Nix (48 kDa), Bax (21 kDa), and cleaved caspase-3 (17/19 kDa) expression. Equal loading was confirmed by stripping the membranes and blotting for β-tubulin (55 kDa). The results are representative of at least three independent experiments.
Focal Cerebral Ischaemia Involves the Apoptotic Cell Death Pathway
Focal cerebral ischaemia (0 h/sham, 6 h or 24 h) was induced in rats using the intraluminal filament model of MCA occlusion. Ischaemic brain damage was readily discernable in thionin-stained coronal sections by light microscopy where lesion boundaries were identified by loss of thionin staining (Figure 4A). Occlusion of the MCA for 6 h resulted in an area of pallor that was restricted to the striatum. No damage was observed in the surrounding cortex. In contrast, a lesion encompassing the cortex and striatum (MCA vascular territory) was apparent by 24 h that affected the lateral parts of the frontal cortex extending through the parietal, insular, and occipital cortices of the ipsilateral hemisphere. ‘Ghost-like’ cells resembling the end stages of infarction were detected within the cortex and striatum after 24 h ischaemia. Apoptotic bodies appeared most prevalent in the haematoxylin and eosin-stained core striatal lesion at 6 and 24 h (arrows; Figure 4B). While relatively few apoptotic-like structures could be detected in the cortex after 6 h ischaemia, closer examination revealed numerous apoptotic bodies within the cortex at 24 h (Figure 4C). No damage was detected in the contralateral hemisphere (data not shown) or in the brain of sham-operated animals.
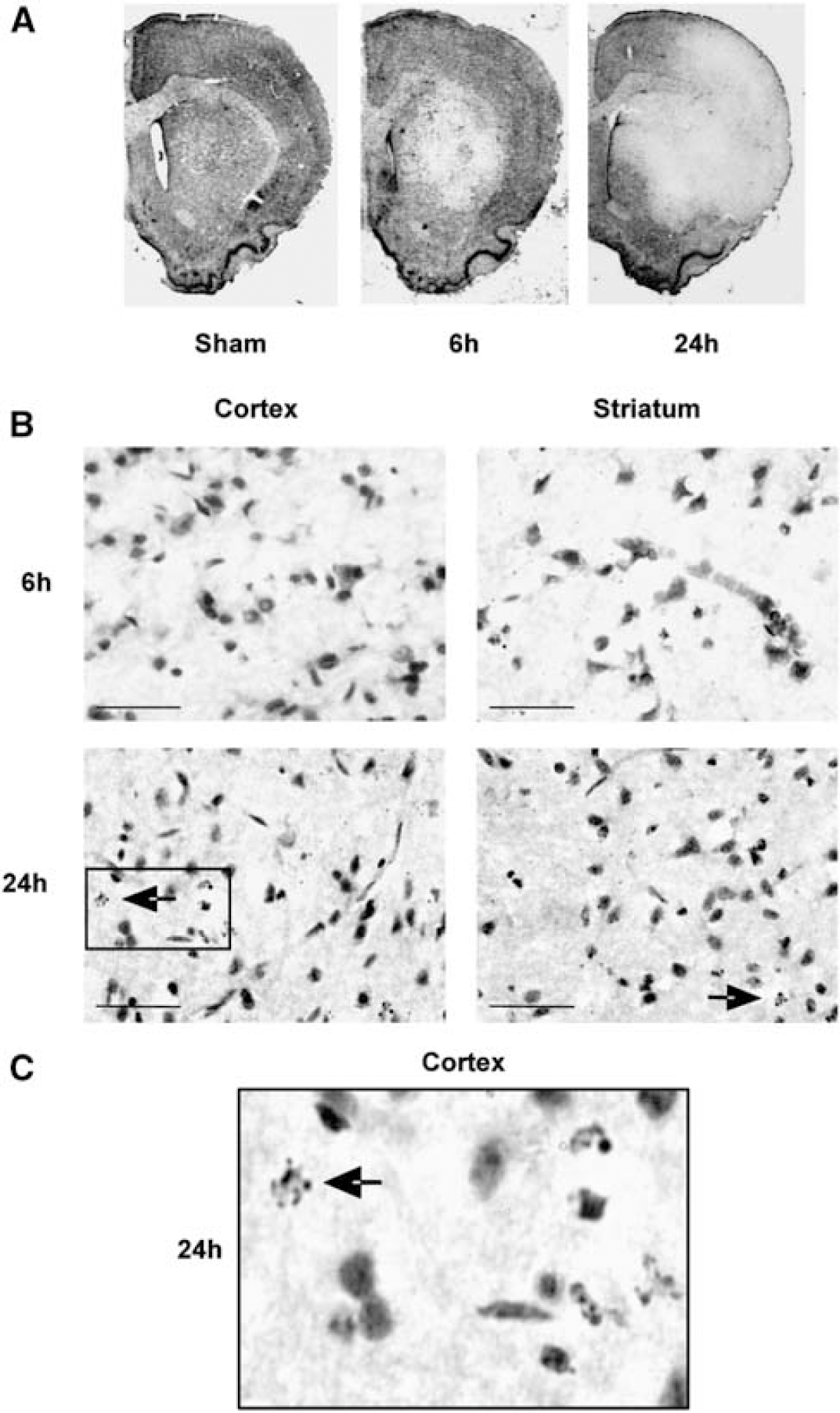
Histologic assessment of apoptotic cell death in the ischaemic rat brain. Middle cerebral aretery (MCA) occlusion was induced for 0 h (sham), 6 or 24 h in male Sprague—Dawley rats and the brains were assessed by histology. The pattern of ischaemic damage was analysed by thionin staining of coronal brain sections (
Proapoptotic Nix and Bax Responses to Middle Cerebral Artery Occlusion
Whole-cell lysates prepared from sham and MCA occluded (6 or 24 h) rat striatum and cortex were analysed for the appearance of cleaved caspase-3, as a biochemical marker of apoptosis. Cleaved caspase-3 was not detected in the sham brain. In keeping with published data, cleaved caspase-3 detected at 6 h in the core striatal lesion and in both the cortex and striatum after 24 h ischaemia (Figure 5A). Nix immunoreactivity was detected in the cortex and striatum of sham animals. Increased Nix expression was observed in the striatum after 6 and 24 h ischaemia, expanding into the overlying cortex by 24 h (Figure 5A). In contrast, a comparatively delayed upregulation of Bax was only observed in the core striatal lesion after 24 h ischaemia.
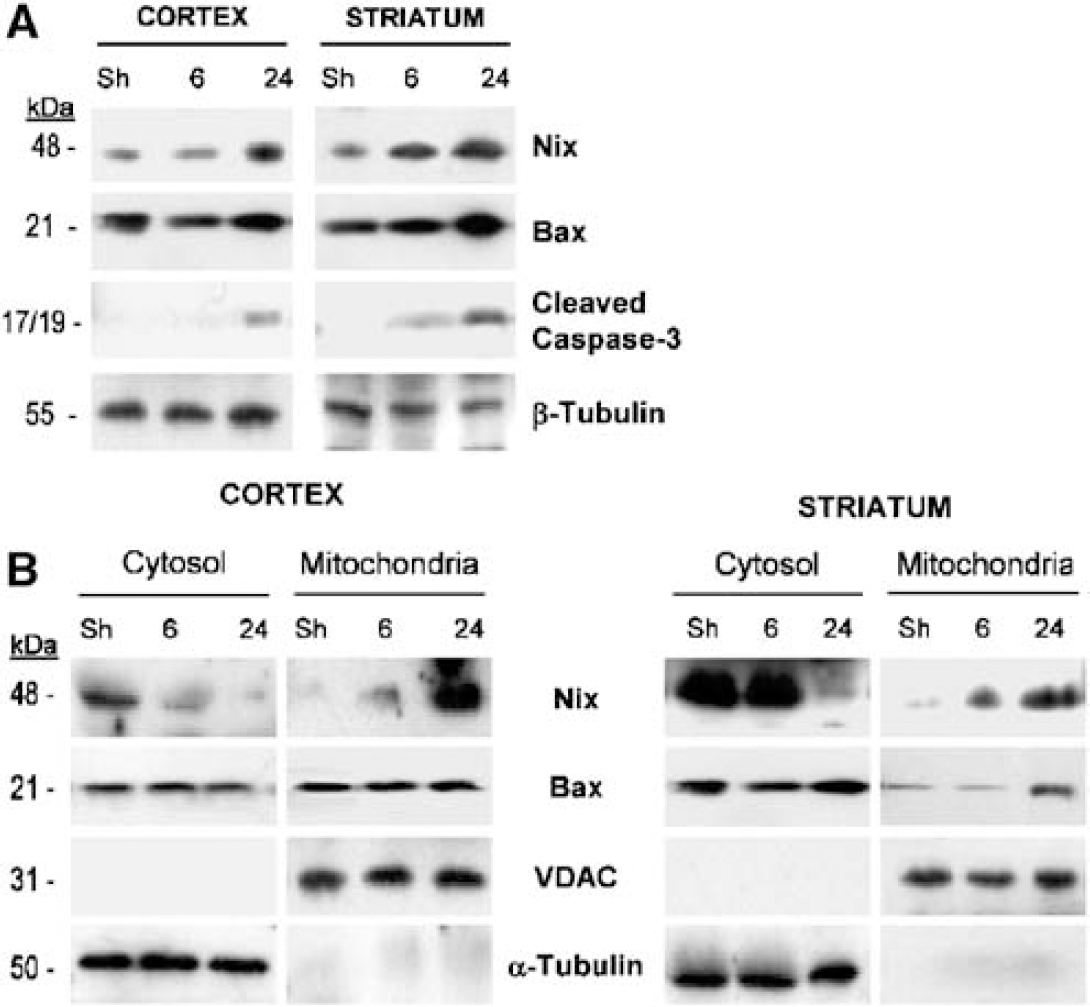
Ischaemia-induced changes in Bax and Nix protein expression. Brains were removed from sham (Sh) and MCA occluded (6 h or 24 h) rats. Whole-cell lysates were prepared from the ipsilateral striatum and cortex (MCA vascular territory) and analysed for changes in Nix (48 kDa), Bax (21 kDa), and cleaved caspase-3 (19 kDa) immunoreactivity by SDS-PAGE and Western blotting (
Whole-cell homogenates were subsequently separated using subcellular fraction methods into cytosolic and mitochondrial-enriched fractions to investigate the effects of ischaemia on the subcellular distribution of endogenous Nix and Bax. VDAC and α-tubulin were used as mitochondrial and cytosolic marker proteins, respectively. Fraction purity was confirmed by the absence of each protein in the other fraction. Nix was detected in the cytosolic, and less so mitochondrial, fraction of the sham rat brain (Figure 5B). A progressive translocation of Nix to the mitochondria was observed in both the cortex and striatum after 6 and 24 h ischaemia (Figure 5B). In contrast, Bax translocation was only evident after 24 h ischaemia in the striatum.
Discussion
Several studies have implicated Nix in hypoxia-induced cell death (Chen et al, 1999; Bruick, 2000; Yussman et al, 2002; Aerbajinai et al, 2003). Increases in Nix mRNA expression were first reported by Bruick who exposed CHO-K1 cells to hypoxia/serum deprivation (Bruick, 2000), although in that study the effect on Nix protein was not investigated. The present studies confirm that hypoxia/serum deprivation in CHO-K1 cells does increase levels of Nix protein expression. These findings are comparable to the hypoxia-induced BNip3 protein response (upregulation after 4 days hypoxia) reported by Bruick (2000). The present studies militate against a constitutive mitochondrial localization for both Nix and BNip3 (Chen et al, 1999), but instead suggest that, in CHO-K1 cells, Nix is predominately a cytosolic protein that migrates to the mitochondria after a hypoxic challenge. Because preincubation of the Nix antibody with its corresponding competing peptide resulted in selective loss of the ~48 kDa immunoreactive band, it is likely that we were observing the response of Nix to hypoxia/serum withdrawal and not its homologue BNip3. The cytosolic localization of Nix (observed in the present study by subcellular fractionation and Western blotting analysis) is in keeping with recent immunohistochemical data where Nix (Yussman et al, 2002; Aerbajinai et al, 2003) and BNip3 (Schmidt-Kastner et al, 2004) were observed to be cytosolic/mitochondrial and primarily cytosolic proteins, respectively. These observations contrast with reports where overexpressed Nix and BNip3 appear to colocalize with the mitochondrial heat shock protein, HSP60 (Chen et al, 1999; Yussman et al, 2002). Discrepancies in the subcellular localization of native and overexpressed Bcl-2 family proteins have been documented: for example, endogenous BNip3 has been reported to be associated with the outer mitochondrial membrane in human and murine skeletal muscle, but inserted into the membrane when overexpressed (Vande et al, 2000). Bcl-Xs, another Bcl-2 member, was described as an endogenous cytosolic protein, but mitochondrial-localized when overexpressed (Lindenboim et al, 2000).
We observed a 96 kDa Nix-interacting immunoreactive band, which decreased in expression with increasing hypoxia duration. Preincubation with a Nix-competing peptide had no effect on detection of this 96 kDa band and was therefore not considered to represent a Nix-specific protein/protein complex. Interestingly, a similar expression profile was observed by Bruick (2000) when investigating BNip3 responses to hypoxia/serum deprivation in CHO-K1 cells. Therefore, while it seems unlikely that the 96 kDa band represented an oligomer of either Nix or BNip3, the reduction in immunoreactivity observed with increasing hypoxia duration possibly reflects degradation of a Nix-containing SDS-resistant complex, which might act to hold Nix and/or BNip3 in an inactive complex in healthy cells. Such inactive protein complexes have been reported for other proapoptotic Bcl-2 family members, for example Bad with 14-3-3 (Zha et al, 1996), Bim (Puthalakath et al, 1999; Chen and Zhou, 2004) and Bmf with the cytoskeleton (Puthalakath et al, 2001), and recently Bax with the scaffolding protein 14-3-3 (Nomura et al, 2003; Tsuruta et al, 2004). Studies to identify this 96 kDa protein complex are ongoing.
In CHO-K1 cells, hypoxia with serum deprivation was associated with activation of the apoptotic cell death pathway, as confirmed by detection of caspase-3 cleavage products. Interestingly, caspase-3 cleavage (by day 2) appeared to precede any changes in Nix expression (by day 4). These observations suggest that hypoxic-induced cell death in CHO-K1 cells is initiated by Nix-independent mechanisms. One possible mechanism might involve activation of Bax (Bonavita et al, 2003) or other p53-regulated Bcl-2 family proteins such as Noxa (Kim et al, 2004) and Puma (Yu et al, 2003). Consistent with our findings, Bax upregulation has been observed in response to hypoxia/ischaemia both in vitro (Bonavita et al, 2003; Brunelle et al, 2004) and in vivo (Isenmann et al, 1998; Cao et al, 2001). However, other groups have reported either no change or a reduction in Bax expression during hypoxia in vitro (Greijer and van der, 2004; Fei et al, 2004). For example, Fei and colleagues showed an early p53-regulated Nix protein response to hypoxia (between 5 h and 24 h) in PA1 and USOS colonic cell lines, but found Bax protein expression to decrease (Fei et al, 2004). Similarly, Erler et al (2004) failed to detect any changes in Bax expression levels in hypoxic CHO-K1 cells or in solid tumours. While these results appear at variance with the present findings, one explanation may be that Bax, like other proapoptotic Bcl-2-like proteins, is regulated primarily by posttranslational modifications, rather than transcriptional activation, where translocation to the mitochondria is a key event (Gross et al, 1999). An alternative explanation may be that the aforementioned studies used hypoxia alone, without serum deprivation or low glucose. Many cell types, including neurons, are reported to undergo excitotoxic/necrotic-like cell death rather than apoptosis under conditions of hypoxia alone (Kitano et al, 1996; Webster et al, 2005). Unfortunately, apoptotic cell death was not measured in the studies by Fei et al (2004) and Erler et al (2004). Furthermore, the early Nix response (by 6 h) reported by Fei and colleagues might be an HIF-1α-dependent response to hypoxia (Bruick, 2000) independent of any Bax-regulated apoptotic event. In the present studies, hypoxia/serum deprivation resulted in an upregulation of Nix after 4 days. These findings are consistent with upregulation of the Nix homologue, BNip3 reported by Bruick (2000) using the same in vitro cell model. In the present studies, hypoxia alone (0.5% O2/10% FCS) did not significantly reduce CHO-K1 cell viability, while serum deprivation alone produced only limited cell death. In contrast, hypoxia with serum deprivation (0.5% O2/0.5% FCS) resulted in approximately 60% cell death by day 7. In this in vitro hypoxia/ischaemia model, caspase-3 cleavage was detected within 2 days hypoxia/serum deprivation, thus suggesting hypoxia/serum deprivation to be an appropriate system to model aspects of ischaemic cell death in vivo. The present findings support a model where, unlike BNip3-mediated necrotic-like cell death (Vande et al, 2000), Nix promotes a more Bax-like cell death, involving translocation to the mitochondria and subsequent caspase-3 activation (Rosse et al, 1998). Interestingly, Bax translocation and caspase-3 cleavage preceded mitochondrial accumulation of Nix suggesting that Nix is unlikely to play a key role in the initiation of the apoptotic signalling cascade after hypoxia/serum deprivation in vitro.
Apoptosis has been implicated in the brain's cellular response to transient global ischaemia, although the importance of such an energy-dependent mechanism in focal cerebral ischaemia remains controversial (Li et al, 1995; Chopp and Li, 1996; Lipton, 1999). The rat MCA occlusion model of focal cerebral ischaemia model produces well-demarcated infarcts of reproducible size and distribution restricted to the MCA territory, encompassing a core striatal lesion and a surrounding zone of varying damage largely in neocortical tissue (Longa et al, 1989; Nagasawa and Kogure, 1989). Damage to the penumbra, but less commonly in the core lesion, can be ameliorated by a range of pharmacological or physiologic interventions when initiated around the time of vessel occlusion (Dirnagl et al, 1990; Gill et al, 1992), suggesting differences in the underlying mechanisms of cell death in each region. It has been proposed that cells within the core lesion die primarily via necrosis, whereas those in the penumbra, which are electrically silent neurons but with their ionic gradients largely intact, go on to die by either apoptosis or necrosis, depending on available energy resources (Benchoua et al, 2001). However, a growing body of evidence suggests that many proapoptotic molecules (including the Bcl-2 and caspase families) exist in an inactive ‘proform’ in healthy cells and require posttranslational modifications rather than ATP-dependent de novo synthesis for activation (Cory et al, 2003). We therefore examined the proapoptotic response to permanent focal cerebral ischaemia in the core striatal lesion and in the overlying cortical tissue, specifically assessing the role of Nix in each of these two ischaemic regions.
The spatio-temporal patterns of damage observed after 24 h ischaemia in the present studies were consistent with literature (Memezawa et al, 1992; Siesjo, 1992; Gillardon et al, 1996). Analysis of DNA fragmentation coupled with morphologic data suggests that most biochemical changes occur within the first 24 h of ischaemia (Tominaga et al, 1993; Charriaut-Marlangue et al, 1996; Gillardon et al, 1996; van Lookeren and Gill, 1996). We therefore examined the responses of endogenous Nix and other markers of apoptosis to 6 and 24 h focal cerebral ischaemia. Detection of apoptotic bodies and cleaved caspsase-3 in the core striatal lesion at 24 h supported an apoptotic component to this model of cerebral ischaemia (van Lookeren and Gill, 1996). Interestingly, increased Nix immunoreactivity was detected in the core striatal lesion after 6 h (the earliest time point investigated), expanding into the striatum and overlying cortex by 24 h ischaemia.
Subcellular fractionation studies revealed that Nix was present in both cytosolic and mitochondrial fractions of sham-operated rat brain tissue. In keeping with the concept of a proapoptotic Nix response, Nix translocation to the mitochondria was evident at 6 h and at 24 h in both brain regions investigated. In contrast, Bax upregulation and translocation was observed only in the striatum and only after 24 h of ischaemia. Evidence of Bax upregulation and translocation to the mitochondria has been described as early as 6 h after transient (with reperfusion) MCA occlusion (Cao et al, 2001), although the response to 6 h sustained (without reperfusion) ischaemia, as used in the present studies, has not been examined. Our observation that mitochondrial accumulation of Nix appeared to precede, or indeed occur in the absence of Bax translocation, suggests that Nix translocation may represent an early proapoptotic response in the ischaemic rat brain.
The rapidity of the upregulation and translocation of Nix in the rat stroke model contrasts with the comparatively delayed response of Nix (4 days) to hypoxia observed in vitro. Discrepancies in the temporal profiles of Nix and Bax activation suggest fundamental differences between cellular responses observed in vitro and in vivo. Such inconsistencies are however, not uncommon: for example, although a necrotic-like cell death has been reported for BNip3 in vitro (Vande et al, 2000; Schmidt-Kastner et al, 2004), upregulation of this protein has been reported during global cerebral ischaemia (Schmidt-Kastner et al, 2004); a model commonly associated with apoptosis rather than necrosis (Back et al, 2004). Furthermore, the mitochondrial localization of BNip3 observed in vitro contrasts with the nuclear accumulation of this protein observed in vivo (Schmidt-Kastner et al, 2004). The nuclear accumulation of BNip3 during global ischaemia contrasts with the mitochondrial targeting of its homologue, Nix during focal cerebral ischaemia described in the present study. While it is tempting to speculate that BNip3 and Nix target different organelles after an ischaemic insult, it could equally be argued that this simply reflects differences in the severity of the insult and rapidity of the apoptotic response in each model. For example, while the BNip3 response to global ischaemia (reported at 24 h) appeared to be comparatively delayed to the Nix response to focal ischaemia (observed at 6 h) in the present study, upregulation of BNip3 protein has been reported within 6 h in a rat model of hypobaric hypoxia (Schmidt-Kastner et al, 2004). Although not examined in the present study, it is plausible that Nix also translocated to the nucleus during focal cerebral ischaemia. These data highlight the need to take cognisance of cell type, stimulus intensity and duration of insult when comparing data obtained from different experimental models. Moreover, while in vitro systems are pivotal tools for the characterization of protein function and protein-protein interactions, the present studies highlight the need for in vivo animal models when attempting to characterize molecular mechanisms underlying disease.
Recent observations support a p53-regulated Nix response (Fei et al, 2004). In support of such a mechanism, transcriptional activation (phosphorylation and/or increased expression) of p53 has been shown in culture after 6 to 12 h hypoxia (Fei et al, 2004; Li et al, 2004) as well as in the ischaemic rat brain after 24 h MCA occlusion, the earliest time point investigated (Chopp et al, 1992; Park et al, 2000). We therefore questioned whether Nix could also be activated cytotoxic agents reported to involve p53 signalling. We next characterized the Nix protein response to nonhypoxic cytotoxic agents, staurosporine and dexamethasone, both of which have been reported to induce translocation within the nucleus and transcriptional activation of p53 (Crochemore et al, 2002; Charlot et al, 2004) as well as Bax activation and mitochondrial dysfunction (McGinnis et al, 1999; Yoshino et al, 2001; De Sarno et al, 2003; Su et al, 2004). Using models of staurosporine-treated human SH-SY5Y neuroblastoma cells and dexamethasone-treated mouse A1.1 T-cell hybridoma cells, we confirmed that apoptosis in each of the aforementioned models was associated with upregulation of Bax and cleaved caspase-3 immunoreactivity. Nix protein expression was also upregulated in response to both stimuli. Our data, together with two recent reports describing Nix upregulation during erythroid differentiation (Aerbajinai et al, 2003) and cardiomyopathy (Yussman et al, 2002), support a more generalized proapoptotic role for Nix protein within the cell.
Several novel observations emanate from these studies: (a) Nix was upregulated and translocated to the mitochondria in response to hypoxic stimuli in vitro and in vivo; (b) The rapidity of the Nix response to focal cerebral ischaemia, which preceded other markers of ischaemic damage (i.e. Bax activation, caspase-3 cleavage, and infarction), suggests that Nix might be a viable therapeutic target in ischaemic research; and finally (c) Nix upregulation in response to staurosporine and dexamethasone (two dissimilar apoptotic stimuli involving p53 transcriptional activation and mitochondrial cell death) in neuronal and nonneuronal cell types supports a more expansive role for Nix in both hypoxic and nonhypoxic disorders, possibly acting to regulate more generalized cell stress signals. While the present studies support a model of p53-regulated Nix activation, it remains unclear whether Nix can be activated by p53-independent mechanisms. Such studies are in progress.