
Abstract
This review will focus on inhalational anesthetic neuroprotection during cerebral ischemia and inhalational anesthetic preconditioning before ischemic brain injury. The limitations and challenges of past and current research in this area will be addressed before reviewing experimental and clinical studies evaluating the effects of inhalational anesthetics before and during cerebral ischemia. Mechanisms underlying volatile anesthetic neuroprotection and preconditioning will also be examined. Lastly, future directions for inhalational anesthetics and ischemic brain injury will be briefly discussed.
Introduction
Neurological sequelae from various neurosurgical, vascular, and cardiovascular procedures can include stroke, seizures, stupor, coma, neuropsychological deficits affecting personality, behavior, and cognitive function, and psychological problems such as depression, disorientation, and confusion (Arrowsmith et al, 2000). Perioperative stroke remains one of the more serious procedural complications (Kelley, 2001). Perioperative stroke incidence for coronary artery bypass grafting ranges from 1.6% to 5.2% (Arrowsmith et al, 2000; Dacey et al, 2005; Hogue et al, 1999) and from 0.25% to 7% for carotid endarterectomy (CEA) (Allain et al, 2005; Wilson and Ammar, 2005). In the North American Symptomatic Carotid Endarterectomy Trial, 35% of perioperative strokes occurred intraoperatively while 65% occurred after the patient left the operating room (delayed events) (Ferguson et al, 1999). A little more than half of these delayed events (56%) occurred within the first 24 h after surgery (Ferguson et al, 1999).
Clinically, there is much interest in determining the feasibility of pharmacologically protecting and preconditioning the human brain with anesthetics and its impact on perioperative stroke (Baughman, 2002; Hans and Bonhomme, 2004; Kawaguchi et al, 2005; Mortier et al, 2000; Patel, 2004; Traystman, 2004; Warltier et al, 2002; Warner, 2000, 2004a– c ) and other neurological and neuropsychological outcomes (Arrowsmith et al, 2000), since this has already been successfully performed with the human heart during cardiac surgery with observed cardioprotection (Belhomme et al, 1999; De Hert et al, 2002; Julier et al, 2003; Wang et al, 2004). Such anesthetic neuroprotection and preconditioning of the brain may therefore prevent or even delay neurological complications including perioperative stroke, thereby expanding the therapeutic window for other prospective neuroprotective agents.
Before reviewing the literature on the effects of inhalational anesthetics on perioperative ischemic brain injury, underlying mechanisms, and future research directions, a discussion of the research limitations in this area and how these limitations may affect conclusions regarding inhalational anesthesia and perioperative ischemic brain outcomes is warranted.
Research Limitations and Implications
Most of the experimental research has focused on volatile anesthetic administration during cerebral ischemia and short-term outcomes, with limited studies on evaluating long-term outcomes as well as pre- and postischemic inhalational anesthetic administration. Rodent studies suggest that the dose, timing, and duration of inhalational anesthetic administration in addition to the severity of ischemic brain injury is critical to outcome and may provide a possible explanation for the lack of benefit seen in clinical studies. More work is needed to clarify and optimize the role of these parameters in the anesthetic management of patients having surgical procedures in which the risk for perioperative stroke is high.
Most of the studies on inhalational anesthetics and perioperative ischemic brain protection in animal stroke models have compared outcomes of different anesthetics with other anesthetics, which were not always normalized for dose (i.e., comparable minimal alveolar concentration or MAC), making the assessment of any one anesthetic for clinical applications challenging. Few of these studies in ischemic brain have compared the effects of volatile anesthetics with the unanesthetized state, a more ʻideal' control. Of particular relevance is that hardly any of these studies have compared outcomes using different concentrations of a single inhalational anesthetic. A direct comparison among all of the clinically relevant inhalational anesthetics is also lacking (Baughman, 2002). Furthermore, the majority of these studies have evaluated use of one specific inhalational anesthetic agent peri-ischemia rather than anesthetic combinations. Only a few studies have evaluated combinations of various inhalational anesthetic agents with nitrous oxide (N2O), but even fewer animal studies have focused on sequential use of volatile anesthetics, combinations of volatile and injectible anesthetic agents as is commonly used in clinical practice, or even coadministration of inhalational anesthetic agents with other neuroprotectants.
Variability between studies may be due as well to the variety of outcomes used to assess the neuroprotective and preconditioning effects of inhalational anesthetics. Evaluation of histopathological damage and neurological function are the two most direct, sensitive, and commonly accepted end points utilized by researchers in animal and clinical studies. Many indirect and surrogate parameters have also been examined, such as electroencephalograms and various genetic, biochemical, and immunohistochemical markers. However, these types of assessments may not be as sensitive, predictive, or reproducible as histopathology and cognitive function tests, especially in the clinical setting. Another difficulty in comparing outcomes from experimental animal studies on inhalational anesthetics and perioperative ischemic brain protection is the diversity of global and focal cerebral ischemia models as well as animal species employed in these studies.
Lastly, many of the animal studies discussed in this review often had no or poor control of physiological parameters that could affect ischemic outcomes like infarct size and cerebral blood flow (CBF). Monitoring of pericranial and core body temperatures and physiological variables such as blood gases, blood pH, blood pressure, and blood glucose is essential when utilizing experimental animal ischemic models as these parameters can be a source of infarct size variability if poorly controlled within and between treatment groups (Graham et al, 2004). Much of the experimental literature on anesthetic neuroprotection and preconditioning did not report or did not attempt to control these parameters via ventilation of the animals, administration of appropriate fluids, or use of warming blankets.
While the focus of this review is on the applicability of pharmacologically protecting and preconditioning the brain with inhalational anesthetics and the impact on ischemic injury, the effects of anesthetics on ischemic brain could have broader implications for stroke and perhaps other forms of brain injury research. Many experimental animal stroke models require some form of anesthesia, even briefly, to induce, maintain, or terminate cerebral ischemia. Consequently, animal studies evaluating genetic interventions or pharmacological agents other than anesthetics in cerebral ischemia may be confounded by anesthetic use in surgical models. As discussed below, inhalational anesthetics can modulate a number of cell death and survival signaling pathways and may accordingly impact on the pathophysiology of brain ischemia. Therefore, selection and use of anesthetic agents need to be considered carefully in all experimental cerebral ischemia studies regardless of the overall experimental aims.
Literature Search Strategy
In performing the literature search for this review, the following inhalational anesthetics were examined: isoflurane, halothane, sevoflurane, desflurane, enflurane, N2O, methoxyflurane, xenon, ether, and cyclopropane. The following search strategies were used for each inhalational anesthetic agent: inhalational agent AND brain AND preconditioning; inhalational agent AND brain AND injury; inhalational agent AND brain AND anesthetic preconditioning; inhalational agent AND brain AND ischemia; and inhalational agent AND brain AND tolerance. Two reviewers independently searched PUBMED (1965 to March 2006). The reviewers also searched the reference lists of articles identified through PUBMED searches.
Selection Criteria, Data Collection, and Analysis
Studies were included in which the inhalational agent was administered before, during, or after experimental cerebral ischemia and in which the effect or mechanism of an inhalational anesthetic in ischemic brain was being evaluated. Studies were excluded if the inhalational agent was administered as part of the experimental model methodology but was not the focus of the study's aims, or if the study was specifically evaluating the effect or mechanisms of inhalational anesthetics in nonischemic or uninjured brain. One reviewer selected studies for inclusion and another independently checked the decisions. One reviewer independently extracted the data and assessed study quality and design.
Neuroprotection During Ischemic Brain Injury
Studies that evaluate volatile anesthetics in experimental ischemic brain injury may provide a better understanding of the pathophysiology of intraoperative stroke and identify new neuroprotective strategies with clinical utility. This section will review the effects of inhalational anesthetic agents during experimental cerebral ischemia and in clinical scenarios.
Isoflurane
Although much work has been performed examining the potential cerebroprotective effects of isoflurane and its underlying neuroprotective mechanisms (Warner, 2000), isoflurane's ability to acutely protect the brain during an ischemic event remains unclear. Data from animal stroke models suggest that isoflurane either delays or ameliorates injury up to 7 days postinjury (Table 1). In these studies, histological injury and functional outcomes were compared with ʻawake' ischemic controls or other inhalational or injectible anesthetic agents. However, an equivalent number of experiments suggest that isoflurane exposure during experimental focal or global stroke offers little or no protection and may even worsen histological and functional outcomes evaluated from 2 h to 3 months postinjury (Table 1). In aggregate, available data suggest that isoflurane dose and the severity and duration of the cerebral ischemia are key factors in determining outcomes for intraoperative stroke, with neuroprotective effects more readily shown in mild to moderate ischemic models, at lower isoflurane concentrations, and after shorter postinjury intervals.
Effect of isoflurane exposure during experimental cerebral ischemia on histological and functional outcomes
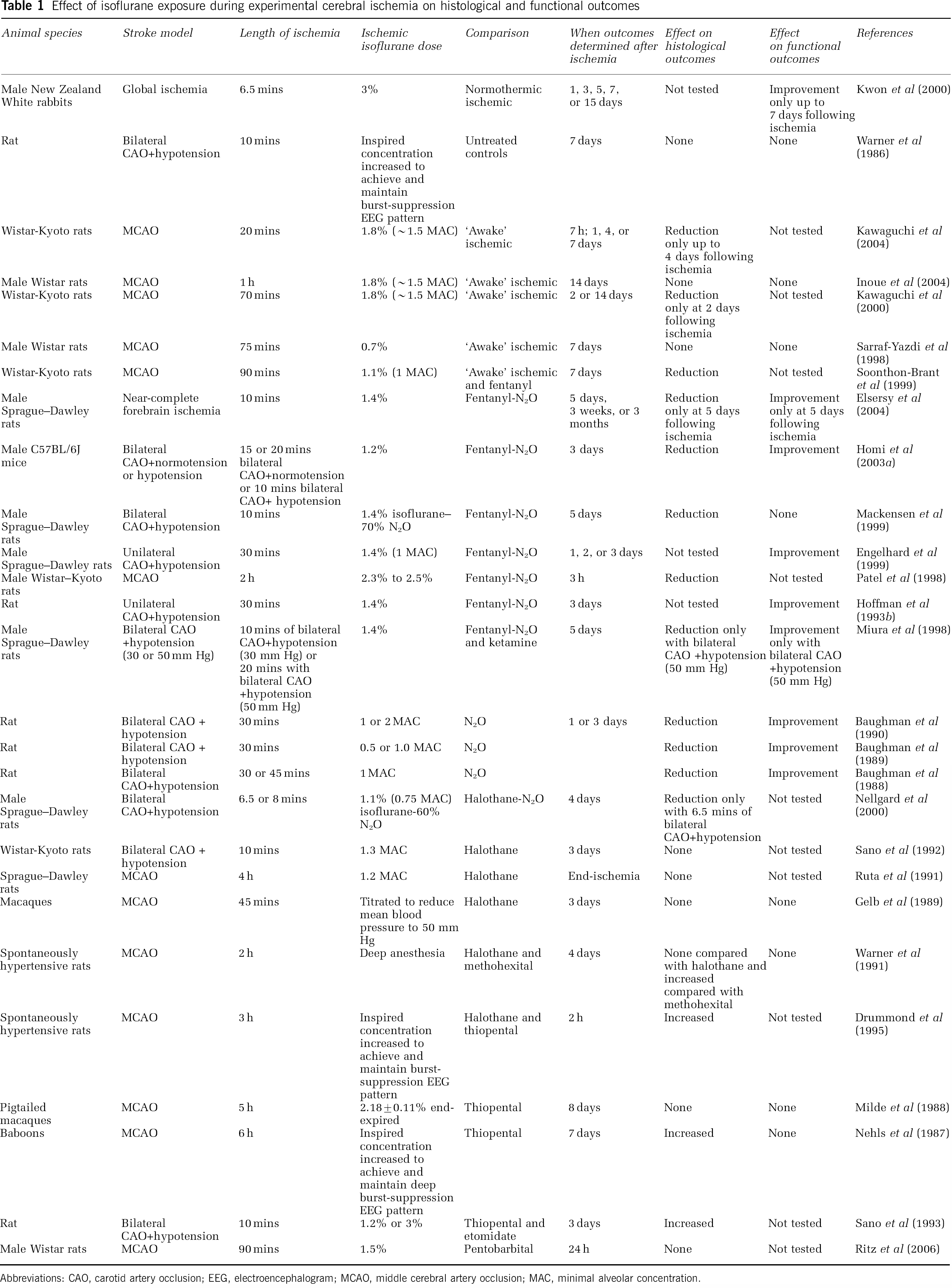
Abbreviations: CAO, carotid artery occlusion; EEG, electroencephalogram; MCAO, middle cerebral artery occlusion; MAC, minimal alveolar concentration.
Although all in vitro studies with isoflurane have been limited to rat brain cultures and slices, these in vitro models suggest a neuroprotective role for isoflurane during ischemia or other forms of neuronal injury. For example, in rat cerebellar and hippocampal slices, isoflurane administered concurrently with oxygen—glucose deprivation (OGD), hypoxia, or α-amino-3-hydroxyl-5-methyl-4-isoxazol propionic acid (AMPA) reduced or prevented neuronal cell death and degeneration as measured from 5 h to 14 days postinsult (Gray et al, 2005; Li et al, 2002; Liniger et al, 2001; Popovic et al, 2000; Sullivan et al, 2002). In rat primary mixed neuronal—glial cultures, isoflurane during N-methyl-
Halothane
As with isoflurane, halothane's ability to protect ischemic brain remains an open question. Halothane anesthesia during focal and global ischemia reduced injury as compared with other anesthetic agents or the ʻawake' state in cats and rodents evaluated from 16 h to 7 days postinsult (Haelewyn et al, 2003; Saito et al, 1997; Warner et al, 1993, 1995; Zausinger et al, 2002). In addition, neurological outcomes improved with halothane exposure (Baughman et al, 1988; Hoffman et al, 1993b; Warner et al, 1993, 1995). However, other studies performed in these same species observed no difference or an exacerbation of ischemic damage and neurological outcome with halothane anesthesia as compared with other anesthetic agents (Browning et al, 1997; Michenfelder and Milde, 1975; Nellgard et al, 2000; Ridenour et al, 1992; Ruta et al, 1991; Warner et al, 1991; Zausinger et al, 2002).
In vitro, primary neuronal—glial cultures and cortical neurons respond to halothane exposure during NMDA-induced toxicity with improved survival as measured by decreased lactate dehydrogenase release (Beirne et al, 1998; Kudo et al, 2001b). However, halothane in rat mixed neuronastrocyte cultures failed to protect against oxidative stress induced by hydrogen peroxide or xanthine—xanthine oxidase yet was protective against iron-induced cytotoxicity (Kudo et al, 2001a). Furthermore, toxic effects were observed in rat corticostriatal brain slices treated with halothane and OGD (Toner et al, 2002). As with isoflurane, halothane is more likely to be neuroprotective at lower doses and in less severe animal or in vitro cerebral ischemia models.
One limitation in the literature is that few studies have examined the effects of halothane administered after an ischemic insult. In a gerbil model of transient forebrain ischemia, halothane anesthesia during the early recirculation period (< 2 h postischemia) abolished motor hyperactivity and significantly decreased CA1 neuronal injury at 48 and 72 h after ischemic injury, respectively (Kuroiwa et al, 1991). No benefits were seen if halothane exposure was performed 2 to 5 h after forebrain ischemia (Kuroiwa et al, 1991). Similarly, in a rat model of transient focal ischemia, postischemic halothane treatment for 5 h after the onset of recirculation had no effect on infarct size and neurological scores 7 days after ischemia (Sarraf-Yazdi et al, 1999). These data suggest that halothane may be a neuroprotectant, but that its therapeutic window is short. Additional studies would be needed to establish if these effects are dose- and insult-dependent. However, regardless of its potential neuroprotective effects, halothane is no longer a clinically relevant anesthetic because of its potential hepatotoxicity and the availability of other inhalational agents with fewer systemic side effects. Primarily for these reasons, halothane is no longer manufactured in the United States. Therefore, it seems unlikely that further experimental studies examining neuroprotection with halothane will be performed.
Sevoflurane
Although sevoflurane has been less well studied than halothane, the available data suggest that sevoflurane may be neuroprotective in the setting of focal or global cerebral ischemia (Pape et al, 2006; Warner et al, 1993; Werner et al, 1995). In addition, protection has been shown when sevoflurane is applied to rat corticostriatal brain slices treated with OGD (Toner et al, 2001). Short- (72 to 96 h) and long-term (28 days) neuroprotection in rats after ischemic insult has also been shown (Pape et al, 2006; Warner et al, 1993; Werner et al, 1995). These initial data must be supplemented with studies to determine optimal dose during mild versus severe ischemia and whether the protection observed in histological and functional outcomes is long lasting.
Desflurane
Desflurane was found to be neuroprotective during focal ischemia in rats (Haelewyn et al, 2003; Tsai et al, 2004) and deep hypothermic circulatory arrest in newborn pigs (Kurth et al, 2001). Neurological outcome was also improved in desflurane-anesthetized rats during incomplete global cerebral ischemia (Engelhard et al, 1999) and in newborn piglets anesthetized with desflurane during low-flow cardiopulmonary bypass (Loepke et al, 2002). While these initial studies with desflurane in ischemic brain show promise, more experimental work is needed before desflurane can be evaluated as a cerebroprotectant in clinical trials.
Nitrous Oxide
In rodents, N2O exposure during incomplete global cerebral ischemia enhanced ischemic damage and resulted in poor neurological outcomes as compared with isoflurane and halothane anesthesia (Baughman et al, 1988, 1989, 1990). Furthermore, N2O (70% N2O/30% oxygen) failed to alter infarct size or behavioral outcomes as assessed at 3 or 14 days after transient middle cerebral artery occlusion (MCAO) and as compared with 70% nitrogen/30% oxygen treatment (Yokoo et al, 2004). While N2O administration (3 h) delivered after transient focal cerebral ischemia can improve cortical histopathology in rat (Abraini et al, 2005; David et al, 2003), N2O administered after global brain ischemia in paralyzed, ventilated primates failed to improve outcome even when administered for 48 h after the initial insult (Gisvold et al, 1984). These results are perhaps explained by the recent observation that even short-term high concentration (hyperbaric) N2O exposure (≤ 3 h) in nonischemic, adult rat brain produces reversible neuronal injury, and prolonged N2O exposure (> 3 h) causes neuronal cell death (Jetovic-Todorovic et al, 2003). In aggregate, N2O alone does not appear to be useful as an ischemic neuroprotectant.
N2O may also suppress the neuroprotective properties of other inhalational anesthetics in ischemic brain when administered as a combination treatment. For example, N2O attenuates isoflurane's neuroprotective effects on neurological outcome, histopathological damage, and degradation of microtubule-associated protein 2, a neuronal cytoskeletal protein sensitive to ischemic injury (Baughman et al, 1989; Sugaya and Kitani, 1997). However, others have documented that isoflurane/N2O combinations reduce or have no effect on ischemic damage when compared with fentanyl/N2O or halothane/N2O combinations (Mackensen et al, 1999; Nellgard et al, 2000).
Other Volatile Anesthetics
Ether, one of the first inhalational anesthetics, can have detrimental effects on experimental stroke outcomes. In a model of bilateral carotid artery occlusion, ether-anesthetized gerbils sustained the poorest neurological outcomes as compared with thiopental, etomidate, ether + thiopental, and ether + etomidate regimens (Hermans et al, 1983). Mortality was also strikingly high in the ether group, with 100% of the animals dead by 1 h after induction of ischemia (Hermans et al, 1983). Xenon is currently being investigated for use as a clinical anesthetic (Hecker et al, 2004). Several studies suggest that xenon may have beneficial effects on neuronal injury and neurological outcomes in rodent cardiopulmonary bypass-associated brain damage (Ma et al, 2003) and other rodent brain injury models (Homi et al, 2003b, Ma et al, 2002; Wilhelm et al, 2002). However, xenon exacerbated ischemic brain damage and aggravated neurological dysfunction in a rat model combining cardiopulmonary bypass and cerebral air emboli (Jungwirth et al, 2006). Post-MCAO treatment with xenon has also been reported to reduce cortical brain damage (Abraini et al, 2005; David et al, 2003). Another agent, enflurane, has been observed to have limited striatal protective effects in vitro (Toner et al, 2002). However, no animal studies have been performed evaluating enflurane's neuroprotective potential during cerebral ischemia. While limited preliminary data would contraindicate the use of ether but would suggest a neuroprotective role for xenon and enflurane, more research is needed to clarify the clinical effects of these agents on perioperative stroke outcomes. This is not likely to occur with ether or enflurane because these agents are no longer available for clinical administration.
Clinical Studies
In contrast to the multitude of experimental animal studies available in the literature, few clinical studies have been performed evaluating inhalational anesthetics, perioperative stroke risk, and neurological outcomes for surgical procedures such as CEA or cerebral aneurysm surgery. One of the first inhalational anesthetics to be considered neuroprotective in CEA patients was cyclopropane (Wells et al, 1963). Desflurane, in contrast to etomidate, has been shown to increase brain tissue oxygen pressure and reduce acidosis in patients subjected to temporary MCAO longer than 15 mins as part of an extracerebral artery to intracerebral artery bypass procedure (Hoffman et al, 1998). These results suggest that desflurane improves tissue metabolic status in ischemic brain regions, most likely through enhanced tissue perfusion (Hoffman et al, 1998).
One prospective and one retrospective study in CEA patients have evaluated the critical regional cerebral blood flow (rCBF), a flow rate when electroencephalographic signs of ischemia are evident (Messick Jr et al, 1987; Michenfelder et al, 1987). These studies determined that the critical rCBF during isoflurane anesthesia was much lower than the critical rCBF during halothane or enflurane anesthesia (Messick Jr et al, 1987; Michenfelder et al, 1987). The incidence of electroencephalogram ischemic changes was also significantly less during isoflurane anesthesia as compared with enflurane and halothane anesthesia, suggesting that isoflurane might offer some cerebral protection during CEA (Michenfelder et al, 1987). However, the retrospective nature of one of these studies and the sequential use of the volatile anesthetic agents evaluated over time may limit the ability to translate the conclusions into prospective clinical practice. For example, it is very likely that surgical and general anesthesia outcomes improved during the time the anesthetic agents were sequentially changed, with halothane used in CEA patients during the first few years of the retrospective analysis followed by studies with enflurane and then isoflurane (Michenfelder et al, 1987). Therefore, the observed positive effects of isoflurane on critical rCBF in this retrospective study may have been due to the effect of time and improved surgical and anesthetic techniques of the investigators rather than to the change in anesthetic.
Additionally, a small randomized prospective preliminary study in coronary artery bypass grafting patients showed that S-100β protein levels, an early cerebral injury marker for neuronal damage and increased blood—brain barrier permeability, were not significantly increased in patients with procedural isoflurane anesthesia as compared with preoperative levels or to propofol anesthesia (Kanbak et al, 2004). However, one retrospective study of patients undergoing middle cerebral artery aneurysm surgery suggests that isoflurane was not as cerebroprotective as propofol, etomidate, or pentobarbital (Lavine et al, 1997). Interestingly, prebypass use of N2O in coronary artery bypass grafting patients did not affect most cerebral spinal fluid markers of central nervous system ischemia as measured 24 h post-coronary artery bypass grafting (Wells et al, 1987).
Many of these studies have used indirect markers of neuroprotection like critical rCBF and S100-β protein levels without any reference or correlation to changes in cognitive function and neuronal damage. As mentioned previously, such determinations may not be as sensitive, predictive, or reproducible as histopathology and cognitive function tests. Sufficiently powered, prospective randomized controlled clinical studies utilizing sensitive and appropriate end points are needed to evaluate inhalational anesthetics and their effects on perioperative stroke and functional outcomes during general and at risk surgical procedures.
Inhalational Anesthetics as Chemical Preconditioning Agents
Brain preconditioning relies on the fact that prior exposure of the brain to relatively minor insults or chemical/pharmacological agents increases tolerance to future, more injurious events. It is thought that this acquired tolerance may be multimechanistic, because benefits can be induced either transiently and rapidly or in a delayed and sustained fashion (Traystman, 2004). Studies evaluating volatile anesthetics may identify new and clinically feasible means of conditioning a brain that will be challenged by ischemia or mechanical injury from surgery and interventional procedures. This section will review studies evaluating the preconditioning effects of inhalational anesthetic agents in experimental ischemic brain models.
Isoflurane
The majority of the work on preconditioning effects of inhalational agents has been performed with isoflurane. The effect of isoflurane preconditioning on cerebral ischemic injury size has been studied primarily in rodent experimental stroke models (Table 2). Very few studies on the effect of isoflurane preconditioning in ischemic brain are currently available in higher order, gyrencephalic animals such as nonhuman primates. Almost universally, isoflurane preconditioning up to 24 h before a permanent or transient focal cerebral ischemic insult significantly reduced brain injury in adult male rodents (Kapinya et al, 2002a, b ; Liu et al, 2006; Zheng and Zuo, 2004). In a canine cardiac arrest model, isoflurane pretreatment significantly reduced neurological deficit scores 20 h postischemia (Blanck et al, 2000). Isoflurane preconditioning 24 h before hypoxia/ischemia also reduced neonatal male and female rat brain cell loss and damage (Zhao and Zuo, 2004). A recent study in mice subjected to transient focal stroke showed neuroprotection in preconditioned young and middle-aged males, but either exacerbation of or no change in ischemic injury in preconditioned young and middle-aged females, respectively, suggesting that gender may modify the response to isoflurane preconditioning preceding ischemic injury (Murphy et al, 2006).
Inhalational anesthetic preconditioning effects in experimental stroke models
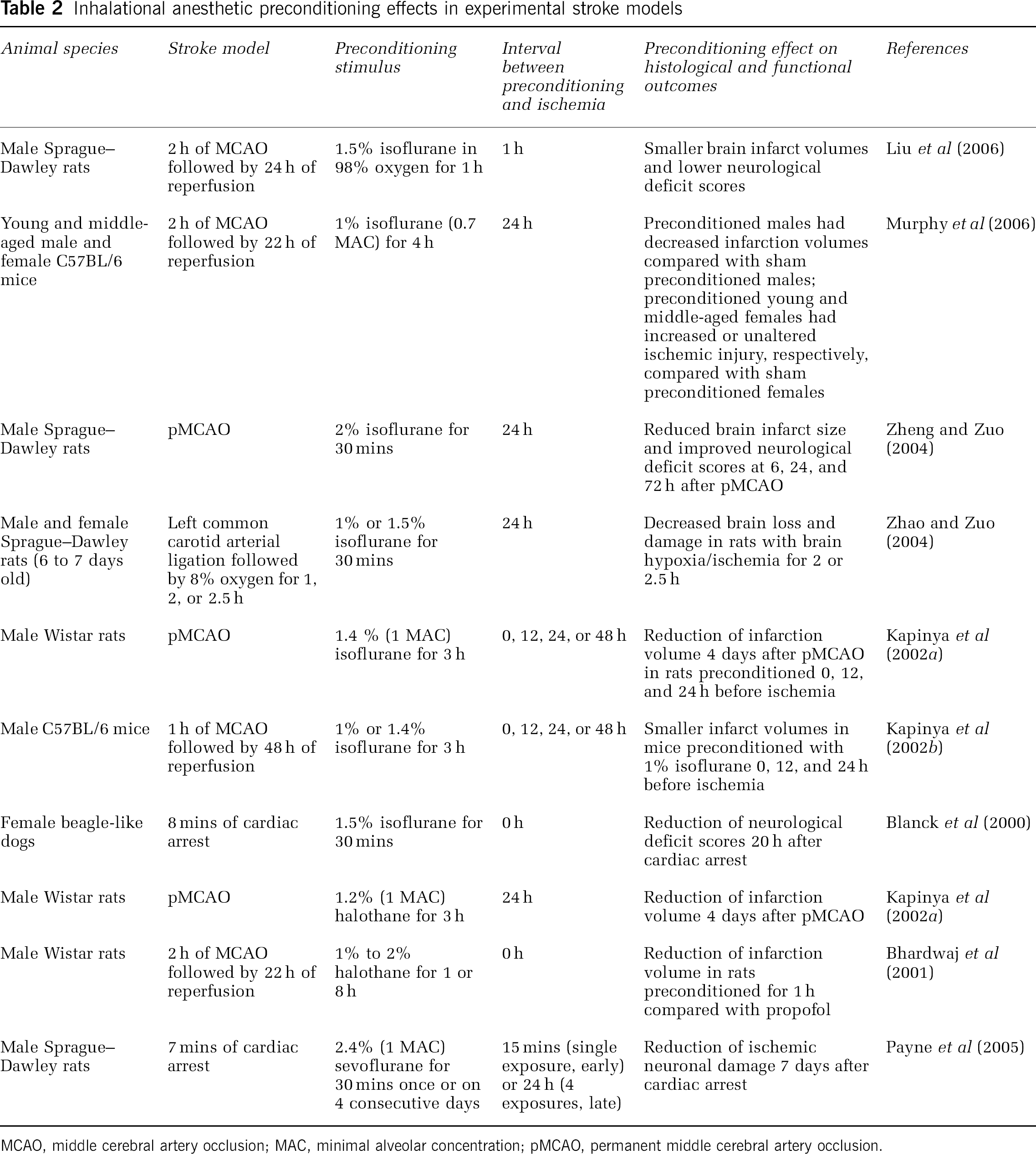
MCAO, middle cerebral artery occlusion; MAC, minimal alveolar concentration; pMCAO, permanent middle cerebral artery occlusion.
Beneficial effects of isoflurane preconditioning before ischemic injury have also been observed in in vitro models. In rat cerebellar and hippocampal slice models of ischemia, isoflurane administered either 15 mins or 24 h before OGD, glutamate-induced delayed cell death, NMDA-induced neurotoxcity or AMPA-induced neurotoxicity reduced or prevented neuronal cell death and degeneration (Bickler et al, 2005; Zheng and Zuo, 2003, 2005). Primary cortical neurons from rats exposed to isoflurane 24 h before OGD showed sustained neuronal viability and reduced lactate dehydrogenase release (Kaneko et al, 2005; Kapinya et al, 2002a). Exposure of primary cortical neuronal cultures to isoflurane before and with continued exposure during OGD also resulted in concentration-dependent attenuation of OGD-induced neuronal apoptosis (Wise-Faberowski et al, 2001).
Halothane
Only a few studies in rat focal stroke models have evaluated the effects of halothane preconditioning on ischemic outcomes (Table 2). Kapinya et al (2002a) observed that in male rats, a 3 h pretreatment with 1.2% (1 MAC) halothane 24 h before permanent MCAO reduced infarct volume measured 4 days after initiation of ischemic injury. In a transient model of focal ischemia in male rats comparing the effects of immediate preischemic exposure of halothane and propofol, a 1 h exposure of 1% to 2% halothane attenuated infarction volume compared with propofol (10 mg/kg bolus, 30 mg/kg per hour infusion) whereas no differences in infarct size were seen between halothane and propofol treatment groups when the preischemic exposure period was extended to 8 h (Bhardwaj et al, 2001). Even fewer in vitro studies have been performed examining halothane preconditioning effects on neuronal injury. Halothane exposure of primary cortical neuronal cultures before and continued treatment during OGD resulted in reduction of OGD-induced neuronal apoptosis (Wise-Faberowski et al, 2001). These preliminary studies would suggest that preconditioning period length and length of the interval between the preconditioning stimulus and ischemia are important factors when determining the potential neuroprotective effects of halothane preconditioning. However, as mentioned previously, the decreased availability of halothane and the accessibility of alternative volatile anesthetics with fewer systemic side effects make it unlikely that additional research evaluating preconditioning with halothane will be pursued.
Sevoflurane
Investigators have recently begun examining whether sevoflurane-induced protection can occur through preconditioning before ischemic brain injury (Table 2). Payne et al (2005) showed that sevoflurane-induced preconditioning 15 mins or 24 h before global cerebral ischemia reduced neuronal damage in rats. In in vitro models, sevoflurane preconditioning in rat hippocampal slices 15 mins before hypoxia and reoxygenation increased recovery of neuronal function after hypoxia in a dose-dependent manner (Kehl et al, 2004).
Desflurane and Other Volatile Anesthetics
At this time, no experimental animal studies have been performed evaluating the preconditioning effects of desflurane or other inhalational anesthetics besides isoflurane, halothane, or sevoflurane before ischemic brain injury.
Mechanisms Under Investigation for Neuroprotection and Preconditioning
Much progress has been made in delineating the molecular and cellular mechanisms of volatile anesthetic action (Mashour et al, 2005). Several of these inhalational anesthetic mechanisms, such as potentiation of GABAergic neurotransmission and antagonism of glutamatergic neurotransmission, may also explain the neuroprotective and preconditioning effects of inhalational anesthetics (Baughman, 2002; Warner, 2004a) as well as of other nonanesthetic neuroprotectants and preconditioning agents under investigation (Danton and Dietrich, 2004). While selected mechanisms have been exclusively studied in the context of either inhalational anesthetic neuroprotection or preconditioning, only a few mechanisms have been evaluated for both phenomena. This section will review mechanisms underlying the neuroprotective and preconditioning effects of inhalational anesthetics (Figure 1).
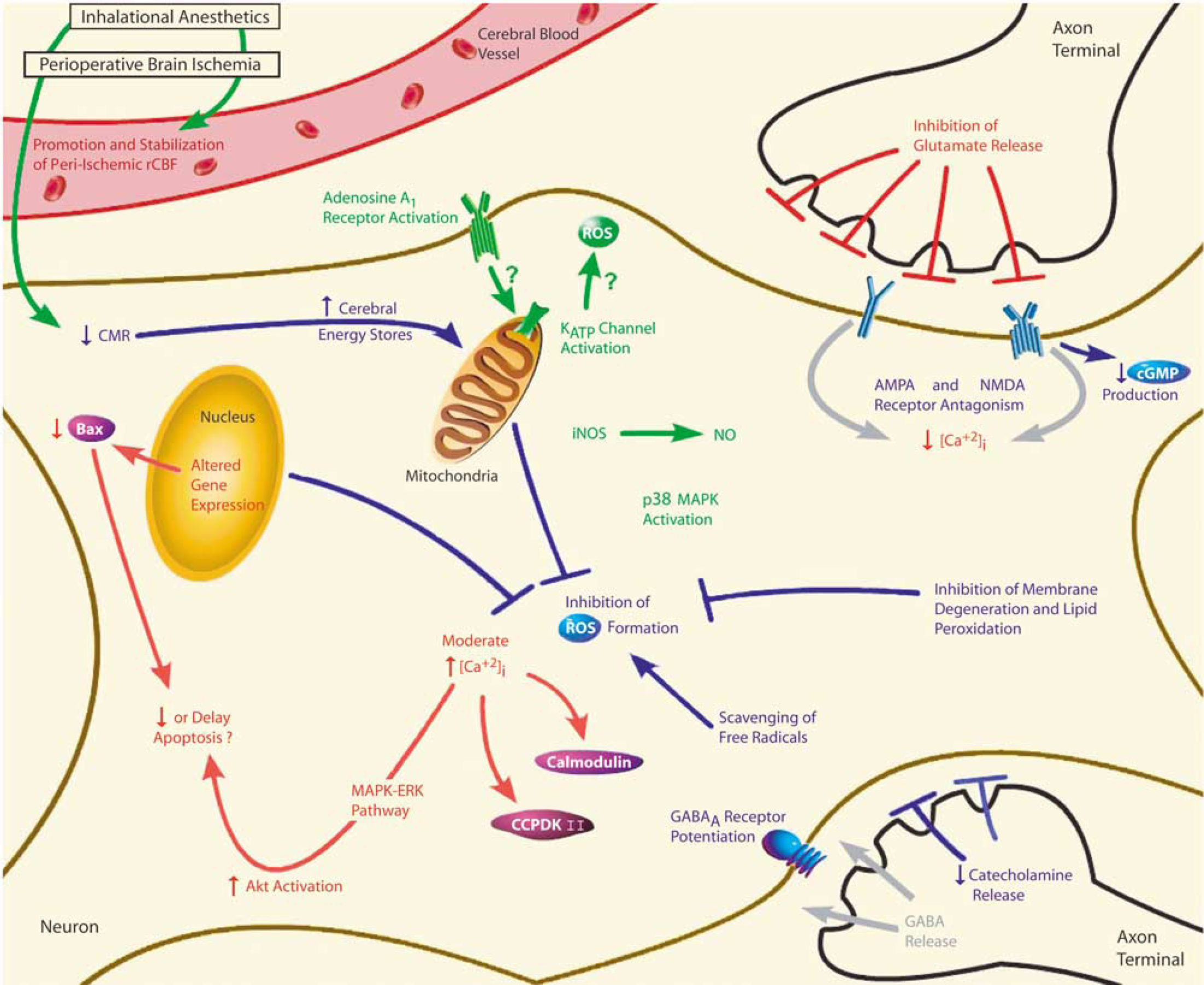
As described in the text, neuroprotective (in blue) and preconditioning (in green) mechanisms currently being explored for inhalational anesthetics in ischemic brain injury are depicted within the neurovascular unit. Mechanisms under investigation for both neuroprotection and preconditioning (in red) are also shown. Inhalational anesthetics administered before (preconditioning) or during (neuroprotection) perioperative brain ischemia are thought to promote and stabilize peri-ischemic rCBF as well as reduce CMR, thus increasing cerebral energy stores in ischemic brain. Neuroprotective effects of inhalational anesthetics in ischemic brain could also occur through inhibition of ROS formation from mitochondrial, nuclear, or cytoplasmic sources; scavenging of free radicals; and inhibition of membrane degeneration and lipid peroxidation. In addition, inhalational anesthetics could modulate ischemic excitotoxicity through attenuation of ischemia-induced catecholamine release, potentiation of GABAergic neurotransmission, and inhibition of glutamergic neurotransmission. Antagonism of AMPA and NMDA receptors for glutamate can subsequently lead to attenuation of ischemia-induced increases in [Ca2+]i and decreases in glutamate-receptor-linked cyclic guanosine monophosphate production. Paradoxically, inhalational anesthetic-induced [Ca2+]i increases may be involved with preconditioning effects in ischemic brain. Such changes in [Ca2+]i can modulate calcium-dependent protective processes in ischemic brain involving calmodulin, CCPDKII, and the MAPK—ERK pathway. Inhalational anesthetics are thought to reduce or delay apoptosis through activation of Akt (protein kinase B), an antiapoptic factor downstream of the MAPK—ERK pathway, and through decreased expression of Bax, an apoptosis-inducing protein. Adenosine A1 receptor activation may also be involved with the preconditioning effects of inhalational anesthetics in ischemic brain. Furthermore, adenosine A1 receptor activation may possibly be a trigger for mitochondrial KATP channel opening and activation, which has been linked to the development of cerebral tolerance. It is speculated that KATP channel activation may alter ROS production, blunt intraischemic mitochondrial calcium accumulation, and improve postischemic mitochondrial energy production. Inducible nitric oxide synthase and the subsequent generation of NO may be important for inhalational anesthetic preconditioning in ischemic brain as well. Lastly, tolerance induced by inhalational anesthetic preconditioning may be mediated through p38 MAPK activation. Abbreviations: α-amino-3-hydroxyl-5-methyl-4-isoxazol propionic acid, AMPA; ATP-sensitive potassium channel, KATP channel; calcium/calmodulin-dependent protein kinase II, CCPDK II; cerebral metabolic rate, CMR; cyclic guanosine monophosphate, cGMP; extraceullar signal-regulated kinase, ERK; γ-amino-butyric acid, GABA; inducible nitric oxide synthase, iNOS; intracellular calcium concentration, [Ca2+]i; mitogen-activated protein kinases, MAPK; nitric oxide, NO; N-methyl-D-aspartate, NMDA; reactive oxygen species, ROS; regional cerebral blood flow, rCBF.
Inhibition of Glutamate Release
Excitotoxicity plays a major role in the initiation and evolution of ischemic brain injury (Bhardwaj et al, 2003). Several research investigators have suggested that inhalational anesthetics given before (preconditioning) or during (neuroprotection) cerebral ischemia are protective through modulation of excitotoxicity (Kawaguchi et al, 2005; Warner, 2004a). One way to alter excitotoxicity is through inhibition of glutamate release (Figure 1).
Results from animal ischemic models are variable regarding the neuroprotective effects of intraischemic inhalational anesthetics on ischemia-induced glutamate release. During global cerebral ischemia in rats, sevoflurane attenuated ischemia-induced increases in cerebral glutamate concentrations (Engelhard et al, 2003). Using in vivo microdialysis, studies in rats and rabbits showed that isoflurane exposure during global ischemia either reduced or had no effect on ischemia-induced glutamate release in cortex or hippocampus as compared with halothane, fentanyl-N2O, or ʻawake' ischemic treatment groups (Illievich et al, 1994; Nakashima and Todd, 1996; Patel et al, 1995). In focal cerebral ischemia, intraischemic isoflurane prevented ischemia-induced efflux of glutamate compared with pentobarbital (Ritz et al, 2006). However, in this same study, isoflurane exposure during transient ischemia did not reduce infarct volume 24 h postischemia, suggesting that inhibition of glutamate release may not be enough to improve brain outcomes after cerebral ischemia (Ritz et al, 2006).
Reduction of ischemia-induced glutamate release by inhalational anesthetics given during ischemic, hypoxic, or anoxic injury is also supported by in vitro studies. For example, halothane, isoflurane, sevoflurane, and enflurane decreased glutamate release in rat brain cortical and corticostriatal slices under hypoxic, chemically anoxic, and ischemic (OGD) conditions (Bickler et al, 1995; Eilers and Bickler, 1996; Toner et al, 2001). In addition to their ability to affect presynaptic release of excitatory amino acids, several authors have shown that inhalational anesthetic exposure inhibits postsynaptic excitatory amino-acid pathways. For example, isoflurane and xenon increased tolerance of rat hippocampal brain slices and mouse neuronal—glial cell cocultures to glutamatergic excitotoxicity (Popovic et al, 2000; Wilhelm et al, 2002).
Only a few in vitro studies have evaluated how volatile anesthetic preconditioning may modulate glutamate efflux during ischemia. Zheng and Zuo observed that isoflurane preconditioning in rat cerebellar slices reduced Purkinje neuronal injury from glutamate excitotoxicity (2005) while addition of a specific glutamate transporter inhibitor during OGD blocked any benefits of isoflurane preconditioning (2003). More research is needed clarifying the role of inhibition of ischemic glutamate release in inhalational anesthetic neuroprotection and preconditioning.
Effects on Intracellular Calcium and Calcium-Dependent Processes
Activation of glutamate receptors like the NMDA receptor can lead to increases in intracellular calcium concentration ([Ca2+]i) in the cytoplasm, mitochondria, or the endoplasmic reticulum. When calcium influx becomes excessive, it can then trigger a variety of deleterious intracellular processes within each of these subcellular compartments, leading to eventual neuronal cell death (Bhardwaj et al, 2003; Paschen, 2003). Anesthetics may therefore be protective when administered before or during ischemia through modulatory effects on either [Ca2+]i or glutamate-receptor-mediated calcium influx (Figure 1).
One such effect of intra- or postischemic volatile anesthetics is attenuation of excessive ischemia-induced increases in [Ca2+]i. For example, Bickler et al (1994) showed in rat cortical brain slices that the rate of glutamate and NMDA-mediated calcium influx was reduced during ischemia in the presence of isoflurane. Xenon and N2O decreased NMDA-induced calcium influx in cortical cell cultures when given 15 mins after transient focal cerebral ischemia (Abraini et al, 2005; David et al, 2003). In a gerbil model of forebrain ischemia, intra-ischemic halothane exposure did not alter the calcium reuptake rate as compared with sham operated animals or unanesthetized, nonischemic controls (Racay et al, 2000), suggesting another mechanism for decreasing the ischemia-induced upsurge in [Ca2+]i. Moderate increases in [Ca2+]i may also be neuroprotective. Gray et al (2005) showed that isoflurane exposure during and after hypoxia in rat hippocampal slices was protective through moderate increases in [Ca2+]i and calcium-related signaling pathways involving the mitogen-activated protein kinases (MAPK)-extracellular signal-regulated kinase (ERK) pathway and activation of the antiapoptotic factor, Akt.
Interestingly, inhalational anesthetic-induced [Ca2+]i increases may also be involved with preconditioning effects in ischemic brain. Isoflurane preconditioning in rat hippocampal slice cultures subjected to OGD increased basal [Ca2+]i and protected hippocampal neurons through calcium-dependent processes involving the calcium binding protein calmodulin and activation of the neuroprotective MAPK—ERK pathway (Bickler et al, 2005). In a canine cardiac arrest model, isoflurane pretreatment also preserved the content and activity of hippocampal calcium/calmodulin-dependent protein kinase II, a protein involved in neuronal signaling and neurotransmitter release (Blanck et al, 2000).
Antiapoptotic Mechanisms
Neuronal apoptosis plays a role in the pathogenesis of cerebral ischemia (Bhardwaj et al, 2003). Few investigators have examined the effects of volatile anesthetics on ischemia-induced neuronal apoptosis as a possible neuroprotective or preconditioning mechanism (Figure 1). More is known about the effects of intraischemic inhalational anesthetics on apoptosis in ischemic brain injury (Kawaguchi et al, 2005). For example, isoflurane exposure during rat forebrain ischemia increased apoptosis 2 days after ischemia compared with animals administered propofol (Kodaka et al, 2000). However, the results of this study may be questionable as apoptosis was determined using terminal deoxy transferase uridine triphosphate nick end-labeling (TUNEL) staining. In cerebral ischemia and other pathological conditions, TUNEL staining is nonspecific for apoptosis as both apoptotic and necrotic cells can be TUNEL positive (Graham and Chen, 2001). Therefore, it is more likely that isoflurane exposure in this study increased either apoptosis or necrosis or both after ischemia as compared with propofol.
Evaluation of apoptosis-regulating protein expression can allow an indirect, more qualitative assessment of apoptosis as no staining methods specific for apoptotic cell death are currently available (Graham and Chen, 2001). Sevoflurane exposure during global cerebral ischemia in rats reduced only the expression of the apoptosis-inducing protein Bax but not Bcl-2, p53, or Mdm-2 4 h after ischemia compared with fentanyl-N2O ischemic and sham-operated animals (Engelhard et al, 2004). Kawaguchi et al (2004) examined the effects of isoflurane administration during transient rat focal cerebral ischemia on neuronal apoptosis markers up to 7 days after ischemia. They found that isoflurane exposure initially reduced apoptosis in the first 24 h after ischemia but did not prevent development of apoptosis at 4 and 7 days after ischemia (Kawaguchi et al, 2004). In addition, Inoue et al (2004) found that administration of nonspecific caspase inhibitors pre- and post-ischemia with or without isoflurane exposure during transient rat focal cerebral ischemia led to reduced infarction volume 2 weeks after ischemia, with the combination treatment showing the greatest reduction in infarct injury. Isoflurane alone had no effect on infarction volume compared with ʻawake' ischemic rats at 2 weeks postischemia (Inoue et al, 2004). In rat hippocampal slices, isoflurane exposure during and after hypoxia reduced neuronal cell death and increased activation of Akt, an antiapoptotic factor (Gray et al, 2005).
Modulation of ischemia-induced apoptosis in volatile anesthetic preconditioned brain has only recently been examined as a potential preconditioning mechanism. Only one study has evaluated the preconditioning effect of isoflurane in ischemic brain and its effects on apoptosis. Using rat neuronal cortical cell cultures, Wise-Faberowski et al (2001) showed that halothane or isoflurane pretreatment and continued treatment of rat neuronal cortical cell cultures subjected to OGD led to a concentration-dependent reduction of OGD-induced neuronal apoptosis. However, these results were determined using TUNEL staining and DNA fragmentation and likely reflect changes in both apoptotic and necrotic cell death (Graham and Chen, 2001). More research is clearly needed to evaluate how different inhalational anesthetics given before or during ischemia may affect the initial and continued development of apoptosis after cerebral ischemia.
Cerebral Blood Flow
Inhalational anesthetic agents given before or after ischemic brain injury can have CBF effects (Warner, 2004a), thus possibly providing protection during ischemia by improving intraischemic or reperfusion blood flow (Figure 1). However, most blood flow studies with intraischemic volatile anesthetics have focused on measuring intraischemic rCBF. Isoflurane administered during rodent forebrain ischemia had no effect on end-ischemic or early reperfusion rCBF compared with fentanyl-N2O (Homi et al, 2003a; Mackensen et al, 2000). However, in focal cerebral ischemia in rats, intraischemic isoflurane led to higher cortical rCBF compared with pentobarbital (Chi et al, 1993). Isoflurane exposure during focal cryogenic brain injury in rabbits also promoted a continuing post-trauma hyperemia leading to increased tissue oxygenation (Murr et al, 1990, 1993). In CEA patients, critical rCBF may be lower for isoflurane than for halothane or enflurane anesthesia (Messick Jr et al, 1987; Michenfelder et al, 1987). However, the CBF threshold for cortical depolarization did not differ between halothane and isoflurane in anesthetized rats subjected to bilateral carotid artery occlusion when these agents were coadministered with N2O (Verhaegen et al, 1992, 1994).
Very little is known about rCBF effects in brains preconditioned with volatile anesthetics. Halothane given either before or during permanent focal cerebral ischemia had little effect on rCBF in the first 2 h of ischemia relative to propofol or α-chloralose anesthesia (Bhardwaj et al, 2001; Saito et al, 1997), but promoted more stable rCBF values 14 h into ischemia compared with the secondary rCBF decreases observed with α-chloralose (Saito et al, 1997). More research is needed to clarify whether inhalational anesthetics given during ischemia are neuroprotective through CBF-dependent mechanisms and whether preconditioning with volatile anesthetics have any beneficial peri-ischemic rCBF effects.
Neuroprotective Mechanisms
Much work has been performed evaluating the mechanisms underlying the neuroprotective properties of inhalational anesthetics in ischemic brain injury. This section will briefly review these neuroprotective mechanisms.
AMPA and NMDA Receptor Antagonism
Glutamate release stimulates AMPA and NMDA receptors. Therefore, inhalational anesthetics could attenuate ischemia-induced excitotoxicity through antagonism of these receptors (Warner, 2004a) (Figure 1). Isoflurane has been shown to reduce AMPA-induced neurotoxicity in rat cerebellar slices when present both during and after AMPA exposure (Li et al, 2002) as well as reduce AMPA receptor-dependent effects in mouse cortical slices (Carla and Moroni, 1992). In rat neuronal cultures, isoflurane also decreased glutamate receptor-linked cyclic guanosine monophosphate production (Zuo et al, 1999). Likewise, halothane reduced excitatory responses mediated primarily by AMPA receptors (Pirot et al, 2000). These data suggest that inhalational anesthetics can reduce neuronal AMPA receptor-mediated responses.
In Xenopus laevis oocytes expressing the human NMDA receptor subtype NR1/NR2B, all of the clinical inhalational anesthetics tested (xenon, cyclopropane, enflurane, isoflurane, desflurane, halothane, and sevoflurane) inhibited NMDA receptors at equianesthetic concentrations (1 MAC) but to different extents, with xenon having the greatest effect while sevoflurane had the least (Solt et al, 2006). Halothane, isoflurane, and sevoflurane can also antagonize NMDA excitotoxicity and NMDA-gated currents in ʻischemic' hippocampal slices, cortical cultures, and mixed cortical neuronal—glial cultures (Beirne et al, 1998; Kudo et al, 2001b; Ming et al, 2002). However, peak effects were sometimes observed at anesthetic concentrations higher than those used clinically (Beirne et al, 1998; Kudo et al, 2001b; Ming et al, 2002). While most of the work on inhalational anesthetics and NMDA/AMPA receptors has been performed in brain slices or cultures, work in rodent brain injury models also supports this neuroprotective mechanism for volatile anesthetics. For example, isoflurane anesthesia in rats decreased cortical excitotoxic injury mediated by AMPA and NMDA (Harada et al, 1999; Kimbro et al, 2000).
Catecholamine Release
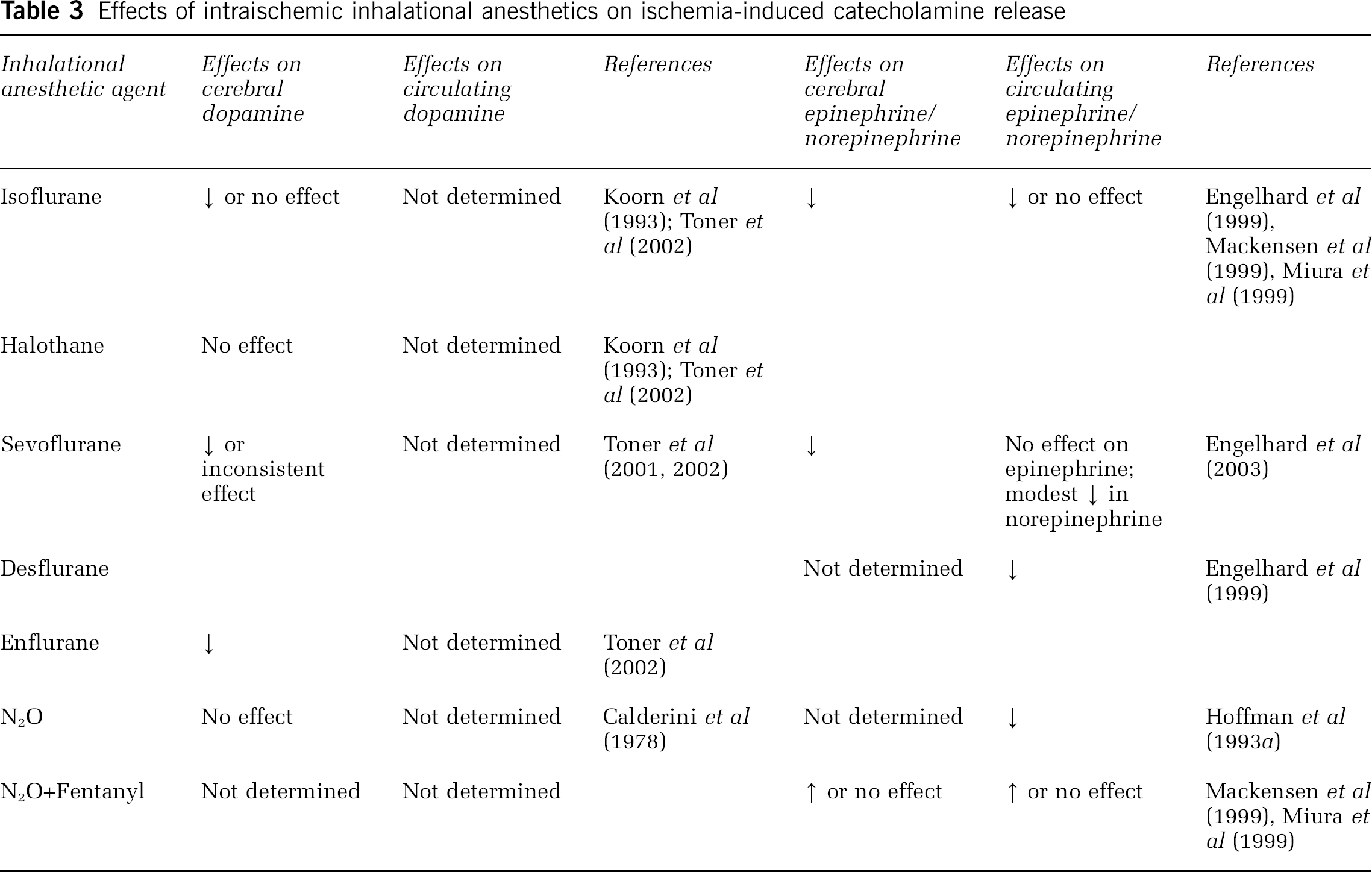
Acute catecholamine release after cerebral ischemia can promote the propagation of brain injury (Bhardwaj et al, 2003). Therefore, attenuation of ischemia-induced catecholamine release could be beneficial (Figure 1). Both cerebral (tissue and extracellular concentrations) and circulating (plasma levels) catecholamines have been implicated in exacerbating ischemic brain injury. Table 3 summarizes the effects of various inhalational anesthetics administered during ischemia and their effects on ischemic-induced cerebral and circulating catecholamine levels. The few studies in which cerebral and circulating catecholamines were concurrently measured while evaluating intraischemic inhalational anesthetic exposure (Engelhard et al, 2003; Miura et al, 1999) suggest a poor correlation between plasma and brain catecholamine concentrations and question the role of circulating catecholamines in the neuroprotective response to inhalational agents during ischemia. More work is needed to determine if the neuroprotective effects of inhalational anesthetic effects are mediated through modulation of cerebral catecholamine release.
Effects of intraischemic inhalational anesthetics on ischemia-induced catecholamine release
Antioxidant Mechanisms
Cerebral ischemia can lead to the production of reactive oxygen species (ROS) that can initiate further damage directly and indirectly to neurons (Crack and Taylor, 2005; Warner, 2004a; Warner et al, 2004). Inhalational anesthetics could potentially be neuroprotective in ischemic brain through inhibition of free radical generation, scavenging of free radicals, and inhibition of lipid peroxidation (Wilson and Gelb, 2002) (Figure 1). Most studies in this area have focused on injectible anesthetics like propofol and thiopental (Wilson and Gelb, 2002). Very few studies have examined whether volatile anesthetics present during cerebral ischemia can have antioxidant effects. Only one in vitro study has examined this question. In primary mixed neuronal—glial cultures, isoflurane exposure during NMDA excitotoxocity failed to protect against cell death because of oxidative stress induced by exposure to hydrogen peroxide or xanthine—xanthine oxidase (Kudo et al, 2001a). There is not enough experimental evidence at this time to support or rule out a role for volatile anesthetics as primary antioxidants in ischemic brain.
Cerebral Metabolic Rate
Volatile anesthetic agents may potentially protect the brain by reducing energy demand in the face of reduced energy supply during ischemia, as almost all inhalational anesthetics reduce CMR (Warner, 2004a) (Figure 1). However, the effect of intraischemic administration of inhalational anesthetics on the relationship between peri-ischemic CMR reduction and reduced histologic damage or improved neurological function is still unclear. For example, in global cerebral ischemia, isoflurane/N2O has been shown to have greater effects on reducing CMR than halothane/N2O (Verhaegen et al, 1992), yet isoflurane ± N2O exposure reduced (Nellgard et al, 2000) or had no effect (Sano et al, 1992) on infarct injury as compared with halothane ± N2O. Although CMR reduction from isoflurane/N2O decreased histological damage compared with halothane/N2O during less severe and more brief periods of global ischemia (Nellgard et al, 2000), such beneficial effects related to decreased CMR were smaller or lost under more severe or prolonged ischemic insult. In contrast to global ischemia, cortical oxygen consumption was found to be higher in isoflurane anesthetized rats subjected to transient MCAO compared with pentobarbital-treated animals (Chi et al, 1993), but isoflurane had no effect on histological injury compared with pentobarbital (Ritz et al, 2006).
Some of these differences in CMR and histological and neurological outcomes in ischemic brain may be partly explained by volatile anesthetic effects on cerebral energy stores such as high-energy phosphates. Dogs exposed to isoflurane during incomplete global ischemia had higher cerebral energy stores of ATP and phosphocreatine and reduced lactate accumulation compared with dogs exposed to N2O, suggesting isoflurane provided some cerebral protection though depression of cerebral metabolism (Newberg and Michenfelder, 1983). Halothane anesthetized rats had significant delays in recovery of ATP and intracellular pH after cerebral ischemia compared with sevoflurane and isoflurane (Nakajima et al, 1997), while there were no differences in ATP and intracellular pH changes between the isoflurane and sevoflurane groups during ischemia and reperfusion (Nakajima et al, 1997). Verhaegen et al (1995) did not find any differences in ischemic cerebral ATP concentrations between isoflurane and pentobarbital-treated rats. All of these results suggest that differences in effects on cerebral energy stores may be species-specific as well as inhalational anesthetic agent-specific (Michenfelder and Milde, 1975). Further work is needed to determine if suppression of CMR is a relevant mechanism by which inhalational anesthetics mediate cerebral protection during ischemia.
γ-Amino-Butyric Acid Receptor Potentiation
Part of isoflurane's neuroprotective effects in ischemic brain may be because of γ-amino-butyric acid (GABA) receptor potentiation (Warner, 2004a) (Figure 1). For example, pharmacological antagonism of GABAA receptors blocked isoflurane neuroprotection in rat hippocampal slices subjected to OGD (Bickler et al, 2003). This would suggest that isoflurane has actions at GABAA receptors as well as at glutamate receptors.
Preconditioning Mechanisms
In contrast to neuroprotection, research focusing on volatile anesthetic preconditioning mechanisms in ischemic brain has been more limited. This section will review what preconditioning mechanisms are currently under examination for inhalational anesthetics. Interestingly, many of the mechanisms described in the previous section for inhalational anesthetic neuroprotection have not yet been investigated as possible preconditioning mechanisms. Just as intriguing, the mechanisms described below for preconditioning with volatile anesthetics have not been tested so far as potential neuroprotective mechanisms. Future research will need to address whether these mechanisms are unique to inhalational anesthetic preconditioning.
ATP-Sensitive Potassium Channels
ATP-sensitive potassium (KATP) channels have been shown in the brain (Bajgar et al, 2001) and the cerebral circulation (Rosenblum, 2003). Two types of KATP channels, sarcolemmal and mitochondrial, have been identified (O'Rourke, 2004). While the functions of KATP channels in the brain and in cerebrovascular responses remain unclear, these channels, particularly mitochondrial KATP channels, appear to have an important role in the reduction or delay of ischemic brain cell death (Busija et al, 2004).
Inhalational anesthetic preconditioning is thought to be protective in ischemic brain through KATP channel opening and activation (Figure 1), since several studies utilizing nonspecific (glibenclamide) and specific mitochondrial (5-hydroxydecanoic acid) KATP channel blockers have shown amelioration of any beneficial preconditioning effects with isoflurane or sevoflurane in in vivo and in vitro cerebral, cortical, and hippocampal ischemic and hypoxic models (Kaneko et al, 2005; Kehl et al, 2004; Wang et al, 2006; Xiong et al, 2003). Interestingly, glibenclamide, which inhibits sarcolemmal and mitochondrial KATP channels, had no effect on the protection induced by isoflurane preconditioning in cerebellar slices subjected to OGD, (Zheng and Zuo, 2003), suggesting that there may be regional brain variations in KATP channel type, distribution and activation.
In the literature on ischemic preconditioning and cardioprotection, there is evidence that opening of KATP channels alters ROS production, blunts intraischemic mitochondrial Ca2+ accumulation, and improves postischemic mitochondrial energy production (O'Rourke, 2004). Furthermore, one study examining isoflurane preconditioning mechanisms in rabbit myocardial ischemia suggests that opening of KATP channels acts as a trigger for preconditioning by generating ROS in vivo (Tanaka et al, 2003). These candidate protective mechanisms for ischemic and anesthetic preconditioning in myocardial ischemia could also be applicable to inhalational anesthetic preconditioning in ischemic brain. More research is clearly needed with inhalational anesthetics and their region- and dose-specific effects regarding brain KATP channel activation and its downstream effects in cerebral ischemia.
Nitric Oxide
Nitric Oxide (NO) has several functions in normal brain such as CBF and neurotransmitter regulation (Bhardwaj et al, 2003). However, NO can have both beneficial and detrimental effects in ischemic brain, depending on the amount and source of NO production (Bhardwaj et al, 2003) (Figure 1). Inducible nitric oxide synthase (NOS) may be important for inhalational anesthetic preconditioning-induced neuroprotection in ischemic brain since it plays a role in the protection induced by ischemic preconditioning in brain (Huang, 2004). Two studies evaluating ischemic neuronal injury in rat would suggest that the neuroprotection from isoflurane preconditioning is indeed inducible NOS-dependent (Kapinya et al, 2002a; Zhao and Zuo, 2004). However, administration of a nonspecific NOS inhibitor did not alter ischemic outcomes 2 h post-ischemia in rats exposed to halothane during transient focal cerebral ischemia (Drummond et al, 2005). These results tentatively suggest that NOS activity may be more important for preconditioning effects before ischemia than for volatile anesthetic neuroprotection during ischemia, at least in the early reperfusion period. Unfortunately, research is noticeably lacking on whether endothelial and neuronal NOS play a favorable or unfavorable part in an ischemic brain preconditioned with volatile anesthetics as well as the time course of NOS isoform induction and NO production relative to different inhalational anesthetics.
Adenosine A1 Receptor Activation
Adenosine A1 receptor activation may also be involved with the protective effects of isoflurane preconditioning in rat focal cerebral ischemia (Liu et al, 2006) (Figure 1). A1 receptor activity modified by isoflurane could possibly be a trigger for KATP channel activation leading to the development of cerebral ischemic tolerance. This inhalational anesthetic preconditioning mechanism has been observed in myocardial ischemia (Gassmayr et al, 2003; Roscoe et al, 2000) but is unproven in ischemic brain.
p38 Mitogen-Activated Protein Kinases
Additionally, the tolerance induced by isoflurane preconditioning may be mediated through activation of p38 MAPK, since p38 MAPK inhibition abolishes and p38 MAPK activation mimics the beneficial effects of isoflurane preconditioning in rats subjected to permanent MCAO (Zheng and Zuo, 2004) (Figure 1).
Future Directions
Despite all of the research carried out thus far, the question of whether inhalational anesthetics offer perioperative ischemic brain protection and which agent is the best choice remains controversial. Review of the current literature on inhalational anesthetic neuroprotection and preconditioning effects suggests several avenues for refining research in this area to better answer these questions (see section Research Limitations). Current experimental data on ischemic brain neuroprotection and preconditioning with inhalational anesthetics has been derived primarily from rodent stroke models. Future research on long-term histological and functional outcomes and the neuroprotective and preconditioning mechanisms of volatile anesthetics in ischemic brain needs to expand into stroke models utilizing higher order, gyrencephalic animals such as nonhuman primates if we are to extrapolate better to the human scenario and to improve the direction and design of future clinical studies and trials.
There are very few pending clinical studies evaluating inhalational anesthetics, perioperative stroke risk, and neurological outcomes for surgical procedures such as CEA, coronary artery bypass grafting, or cerebral aneurysm surgery. This scarcity of clinical information makes it difficult to form conclusions about the role of inhalational anesthetics in the morbidity and mortality of human perioperative stroke. Clearly more prospective, well-designed, randomized clinical trials are needed that compare the long-term consequences of different volatile anesthetic agents on perioperative stroke and outcomes for at-risk surgical procedures.
In addition to sorting out inhalational anesthetic- and species-specific effects, investigators also need to keep in mind gender-specific responses to volatile anesthetics. A recent review of randomized and nonrandomized trials evaluating the associations between age and/or sex and procedural stroke risk after CEA concluded that operative stroke risk is increased in women independent of age (Bond et al, 2005). However, experimental studies examining inhalational anesthetic neuroprotective and preconditioning effects in ischemic brain injury have utilized predominantly young male or gender unspecified animals and unsexed cultures, leaving wide open the question of the role of gender and sex steroids. While gender effects are well known to alter experimental ischemic outcomes (Murphy et al, 2004), very few studies have looked at gender effects, choice of inhalational anesthetic agent, and brain outcomes in ischemic and other types of brain injury models. For example, in diffuse traumatic brain injury, female rats do better than male rats but this outcome is dependent on the type of anesthesia (O'Connor et al, 2003). Only one study in mice subjected to transient focal stroke showed exacerbation of or no protection from ischemic injury in isoflurane preconditioned young and middle-aged females, respectively as compared with reduced ischemic injury in isoflurane preconditioned young and middle-aged males (Kitano et al, 2006). These results suggest that gender may modify the response to inhalational anesthetic preconditioning (Kitano et al, 2006). The fundamental basic science for sex differences in volatile anesthetic effects on perioperative stroke risk are unknown and offer a potentially key factor in optimizing anesthetic management of male and female patients undergoing surgical procedures at risk for perioperative stroke.
Footnotes
Acknowledgements
The authors thank Ms Robin Feidelson for her technical expertise and assistance with all manuscript figures.