
Abstract
In response to traumatic brain injury (TBI), neurons initiate neuroplastic processes through the activation of intracellular signaling pathways. However, the molecular mechanisms underlying neuroplasticity after TBI are poorly understood. To study this, we utilized the fluid-percussion brain injury (FPI) model to investigate alterations in the mammalian target of rapamycin (mTOR) signaling pathways in response to TBI. Mammalian target of rapamycin stimulates mRNA translation through phosphorylation of eukaryotic initiation factor 4E binding protein-1 (4E-BP1), p70 ribosomal S6 kinase (p70S6K), and ribosomal protein S6 (rpS6). These pathways coordinate cell growth and neuroplasticity via dendritic protein synthesis. Rats received sham surgery or moderate parasagittal FPI on the right side of the parietal cortex, followed by 15 mins, 30 mins, 4 h, 24 h, or 72 h of recovery. Using Western blot analysis, we found that mTOR, p70S6K, rpS6, and 4E-BP1 phosphorylation levels were significantly increased in the ipsilateral parietal cortex and hippocampus from 30 mins to 24 h after TBI, whereas total protein levels were unchanged. Using confocal microscopy to localize these changes, we found that rpS6 phosphorylation was increased in the parietal cortex and all subregions of the hippocampus. In accordance with these results, eIF4E, a key, rate-limiting mRNA translation factor, was also phosphorylated by mitogen-activated protein kinase-interacting kinase 1 (Mnk1) 15 mins after TBI. Together, these results suggest that changes in mRNA translation may be one mechanism that neurons use to respond to trauma and may contribute to the neuroplastic changes observed after TBI.
Keywords
Introduction
Traumatic brain injury (TBI) is a serious, debilitating health problem, affecting upwards to 1.4 million people and resulting in 50,000 deaths each year in the United States (Langlois et al, 2004). More than 5.3 million people are coping with disabilities from TBI, which range from mild cognitive deficits to difficulties in accomplishing daily living tasks. A prevalent common complaint after TBI is impairments in the ability to learn and form new memories, as well as difficulties in accessing memories before the TBI incident (Mathias and Mansfield, 2005). However, the molecular and structural neurologic basis for the deficits in learning and memory after TBI is still unknown.
The hippocampus, a structure that is critical for the formation of declarative memories, the knowledge of facts and events, is highly vulnerable to injury during TBI (Maxwell et al, 2003). The hippocampus exhibits a range of molecular, electrophysiological, and structural responses after TBI that may underlie, at least in part, the neurologic basis for memory impairments after TBI (Maxwell et al, 2003; Mathias and Mansfield, 2005; Atkins et al, 2006). In the hippocampus, TBI impairs synaptic plasticity such as long-term potentiation (LTP), induces aberrant axonal sprouting, and decreases synaptic density (Sanders et al, 2000; Santhakumar et al, 2001; Grady et al, 2003; Scheff et al, 2005). Furthermore, the incidence of epilepsy increases in a subset of the population after TBI; this is caused in part by structural and electrophysiological changes in the hippocampus (Golarai et al, 2001; Santhakumar et al, 2001; Garga and Lowenstein, 2006).
Mammalian target of rapamycin (mTOR, also known as FRAP and RAFT-1) is a rapamycin-sensitive serine/threonine protein kinase that plays a major role in controlling mRNA translation initiation and consequently, cell growth and dendritic arborization during hippocampal neuroplasticity (Jaworski et al, 2005; Kumar et al, 2005). Growth factors and nutrients activate the phosphoinositide 3-kinase pathway, leading to the phosphorylation and activation of mTOR on Ser2448. Rapamycin, a highly specific inhibitor, blocks the phosphorylation and activation of mTOR, and inhibits hippocampal LTP through inhibition of mRNA translation (Tang et al, 2002). Mammalian target of rapamycin stimulates protein synthesis in hippocampal neurons by phosphorylating several mRNA translation factors including eukaryotic initiation factor 4E binding protein-1 (4E-BP1) and p70 ribosomal S6 kinase (p70S6K; Haghighat et al, 1995; Holz and Blenis, 2005).
In mammalian cells, mRNA translation is regulated at the initiation 5′ cap via multiple translation factors. The 5′ cap is bound to eukaryotic initiation factor 4E (eIF4E), a key, rate-limiting translation initiation factor. When phosphorylated by mitogen-activated protein kinase-interacting kinase 1/2 (Mnk1/2), which is activated by p44/p42 mitogen-activated protein kinase (MAPK), eIF4E recruits eIF4G to the mRNA. This forms the eIF4F complex and regulates mRNA translation initiation (Duncan et al, 2003). However, 4E-BP1 competes with eIF4G for a binding site on eIF4E. This inhibits formation of the eIF4F complex until 4E-BP1 is phosphorylated by mTOR (Haghighat et al, 1995). When phosphorylated by mTOR, 4E-BP1 dissociates from eIF4E, thus permitting translation of 5′ capped mRNAs related to cell growth and synaptic plasticity (Beretta et al, 1996).
Mammalian target of rapamycin also regulates mRNA translation by phosphorylating p70S6K, which then phosphorylates its downstream substrate, ribosomal S6 protein (rpS6; Ferrari et al, 1991; Holz and Blenis, 2005). Ribosomal S6 protein is an in vivo substrate of p70S6K although it can also be phosphorylated by p90RSK (Pende et al, 2004). Phosphorylation of five clustered serines on the C terminus of rpS6 is temporally correlated with the initiation of protein synthesis. Furthermore, ribosomes with the highest proportion of phospho-rpS6 are most likely to mobilize into polysomes (Ferrari et al, 1991). Ribosomal S6 protein phosphorylation is a critical effector of mTOR in the regulation of cell growth. Mutation of all five phosphorylatable serines to alanines results in decreased cell growth and size (Ruvinsky et al, 2005). Consistently, p70S6K knockout mice or inhibition of mTOR with rapamycin also results in smaller cell and body size (Shima et al, 1998; Fingar et al, 2002). Conversely, overexpression of p70S6K increases cell size (Fingar et al, 2002). Activation of mTOR and phosphorylation of its downstream targets p70S6K and rpS6 controls mRNA translation efficiency of 5′-oligopyrimidine tract-containing mRNAs. These mRNAs encode for translational machinery such as ribosomal proteins and elongation factors to contribute to cell growth and dendritic arborization (Meyuhas, 2000).
Thus, mTOR is an integrally involved in regulating mRNA translation for cell growth, dendritic arborization, and hippocampal synaptic plasticity. Given the deficits in hippocampal synaptic plasticity and synaptic density after TBI, we determined if mTOR and its downstream targets were altered after TBI.
Materials and methods
Traumatic Brain Injury Model
Experiments were performed with male Sprague—Dawley rats weighing 270 to 320 g (Charles River Laboratories, Raleigh, NC, USA). All experimental procedures were in compliance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the University of Miami Animal Care and Use Committee. All feasible measures were taken to reduce animal suffering and the numbers of animals used for these experiments. Animals were maintained for at least 7 days before the experiment in a temperature-regulated room (23°C to 25°C) on a 12 h light/dark cycle. The rats were fasted, but allowed free access to water overnight before the surgery. Moderate TBI was produced with fluid-percussion pressure levels of 2.0 ± 0.2 atmospheres. The surgical preparation for fluid-percussion brain injury (FPI) was performed as previously described (Atkins et al, 2006). Rats were anesthetized with 3.0% halothane in a gas mixture of 70% N2O and 30% O2. The femoral artery was cannulated to deliver pancuronium bromide (0.5 mg/kg, intravenously) every 1 h during the surgical procedure to immobilize the rats. An endotracheal tube was inserted and rats were mechanically ventilated with 70% N2O, 0.5% to 1.5% halothane, and a balance of O2. The animals were then placed in a stereotaxic frame, and a 4.8 mm craniotomy was made over the right parietal cortex (3.8 mm posterior to bregma, 2.5 mm lateral to the midline). A plastic injury tube (18 gauge modified PrecisionGlide needle hub, Becton Dickinson, Franklin Lakes, NJ, USA) was placed over the exposed dura and fixed with dental acrylic. Before and after TBI, blood gases and mean arterial blood pressure were monitored and maintained at physiologic levels. Brain temperature was monitored with a thermistor probe placed in the left temporal muscle, whereas core temperature was determined with a rectal thermometer. Brain temperature was maintained at 37°C with self-adjusting feedback heating lamps. Blood gases, blood glucose, and hematocrit values were monitored 15 mins before TBI, 15 mins after TBI, and then once every hour for up to 4 h after TBI. All animals were maintained within physiologic ranges for mean arterial pressure (120 to 140 mm Hg), pO2 blood gas levels (105 to 170 mm Hg), pCO2 blood gas levels (35 to 45 mm Hg), and blood pH (7.38 to 7.41).
Ten experimental groups were used for these studies. Animals received sham surgery (n = 4) or moderate TBI followed by recovery for 15 mins (n = 5), 30 mins (n = 6), 4 h (n = 6), 24 h (n = 6), or 72 h (n = 6), and then were analyzed by Western blotting. For confocal microscopy analysis, a separate group of animals received sham surgery (n = 4) or moderate TBI and were then perfused at 30 mins (n = 6), 4 h (n = 6), or 24 h (n = 6) after TBI. Sham-operated rats were subjected to identical surgical procedures, but without the fluid-percussion pulse. Animals were maintained under anesthesia for up to 30 mins after TBI during recovery. For longer recovery periods, anesthesia was discontinued and the animals were returned to their cages. To dissect the parietal cortex and hippocampus, animals were anesthetized, tracheotomized, and artificially ventilated at 4, 24, and 72 h after TBI. The animal's breathing was maintained with a respirator while the brain was frozen in situ with liquid nitrogen. The right, injured ipsilateral parietal cortex and hippocampus was dissected in a glove box freezer (–15°C) as previously described (Hu et al, 1999). For confocal microscopy, animals were anesthetized and perfused with ice-cold 4% phosphate-buffered paraformaldehyde while maintaining breathing with a respirator. Brains were sectioned with a vibratome (50 μm, Leica Microsystems, Inc., Exton, PA, USA).
Preparation of Subcellular Fractions
To assess the localization of the changes in mRNA translation factors after TBI, subcellular fractions were prepared from sham-operated rats and rats subjected to moderate TBI followed by 15 mins, 30 mins, 4 h, 24 h, or 72 h of recovery. Hippocampal and cortical tissues were dissected and chopped into small pieces in a glove box freezer (–15°C), and then homogenized on ice with a Dounce homogenizer (35 strokes, 4°C) in 10 volumes of homogenization buffer: 15 mmol/L Tris pH 7.6, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 2.5 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 1.25 μg/mL pepstatin A, 10 μg/mL leupeptin, 2.5 μg/mL aprotinin, 0.5 mmol/L PMSF, 0.1 mmol/L Na3VO4, 50 mmol/L NaF, and 2 mmol/L Na4P2O7. Homogenates were centrifuged at 10,000g at 4°C for 10 mins to obtain pellets and supernatants. The pellets were washed with homogenization buffer containing 1% Triton X-100 (TX-100) and 0.4 mol/L KCl for 30 mins on ice, and then centrifuged at 20,000g for 10 mins at 4°C to obtain pellets (TX-insoluble, denoted P12P) and supernatants (TX-soluble fractions, denoted P12S). The P12P fractions contained insoluble postsynaptic densities, as well as Triton-insoluble nuclear and mitochondrial proteins. The P12S fractions contained soluble, membrane-associated proteins from the synaptic membranes and nuclei (Atkins et al, 2006). The supernatants from the initial centrifugation were centrifuged again at 165,000g at 4°C for 1 h to obtain a supernatant denoted S3 (containing cytosolic proteins) and a pellet denoted P3 (containing microsomes). The TX-insoluble (P12P) and P3 pellets were re-suspended with homogenization buffer containing 0.1% TX-100. Each subcellular fraction was assayed for total protein using the Coomassie Plus assay kit (Bio-Rad Laboratories, Hercules, CA, USA) to load equal protein amounts on the gels.
Western Blot Analysis
Subcellular fractions were electrophoresed on 10% or 12.5% SDS-polyacrylamide gels and then transferred onto Immobilon-P membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 30 mins and then incubated overnight at 4°C with the following primary antibodies (1:1000, Cell Signaling Technology, Beverly, MA, USA): phospho-mTOR Ser2448, total mTOR, phospho-p70S6K Thr389, total p70S6K, phospho-rpS6 Ser235/236, total rpS6, phospho-4E-BP1 Thr37/46, total 4E-BP1, phospho-Mnk1 Thr197/202, phospho-eIF4E Ser209, and total eIF4E. Total protein loading was assessed by actin Western blot analysis (1:2000, Sigma, St Louis, MO, USA). The membranes were then incubated with horseradishperoxidase conjugated anti-rabbit or anti-mouse secondary antibodies for 60 to 120 mins at room temperature (1:1000, Cell Signaling Technology). The blots were developed using enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ, USA) and developed on Kodak Xomat LS or Blue Lite Autorad Film (Eastman Kodak Company, New Haven, CT, USA and ISC BioExpress, Kaysville, UT, USA). Densitometry was performed with Kodak ID image analysis software (Eastman Kodak Company).
Confocal Microscopy
Double-label fluorescence confocal microscopy was performed on coronal brain sections from sham-operated control rats and from rats subjected to moderate TBI followed by 30 mins, 4 h, or 24 h of recovery. At least four different sections prepared from four sham surgery animals and six TBI surgery animals at each time point were imaged. Sections were washed twice in TBS for 5 mins at room temperature and then in 0.01 mol/L sodium citrate buffer pH 6.0 for 5 mins. For antigen retrieval, sections were heated for 10 mins at 100°C. After cooling, sections were placed in TBS containing 0.2% TX-100 for 20 mins. Nonspecific binding was blocked with 3% BSA in TBS and 0.1% TX-100 for 60 mins. Sections were incubated with phospho-rpS6 Ser235/236 or total rpS6 primary antibodies (1:200, Cell Signaling Technology) in TBS, 0.1% TX-100, and 1% BSA overnight at 4°C. Sections were washed three times (10 mins each) in TBS containing 0.1% TX-100, then incubated with fluorescein-labeled anti-rabbit secondary antibody (1:200, Molecular Probes, Carlsbad, CA, USA), and propidium iodide (2 μg/mL, Molecular Probes) in TBS, 0.1% TX-100, and 1% BSA for 1 h at room temperature. Sections were washed two times (10 mins each) in TBS and 0.1% TX-100, mounted (Vectashield mounting medium, Vector Laboratories, Inc., Burlingame, CA, USA), and coverslipped. Sections were analyzed on a Zeiss laser-scanning confocal microscope (Zeiss, Inc., Thornwood, NY, USA).
Statistical Analysis
Data are expressed as mean ± standard deviation (n = 4 to 6) as a percentage of sham-operated control levels. Oneway ANOVAs followed by Tukey post hoc t-tests were used for statistical analysis. *P < 0.05, **P < 0.01, and ***P < 0.001 between sham and TBI animals.
Results
Protein synthesis is regulated by multiple signaling pathways; however, a key signaling pathway is through mTOR, which regulates mRNA translation through phosphorylation of p70S6K and 4E-BP1 (Ferrari et al, 1991; Haghighat et al, 1995; Holz and Blenis, 2005). To study alterations in mTOR activation after TBI, subcellular fractions from the ipsilateral injured hippocampi and parietal cortices of animals that received either moderate parasagittal FPI or sham surgery were analyzed by Western blotting. Using antibodies against phospho-mTOR Ser2448, we observed a significant increase in mTOR phosphorylation after TBI in crude synaptosome subcellular fractions (P12S fractions) from the ipsilateral, injured hippocampus and parietal cortex. The increase in phospho-mTOR was significant at 30 mins after TBI, and then returned to basal levels between 4 and 24 h after TBI (Figure 1A). In comparison, total mTOR protein levels were not significantly changed after TBI (Figure 1B). These results indicate that the phosphorylation and activation of mTOR occurs rapidly and transiently in crude synaptosome subcellular fractions after TBI.
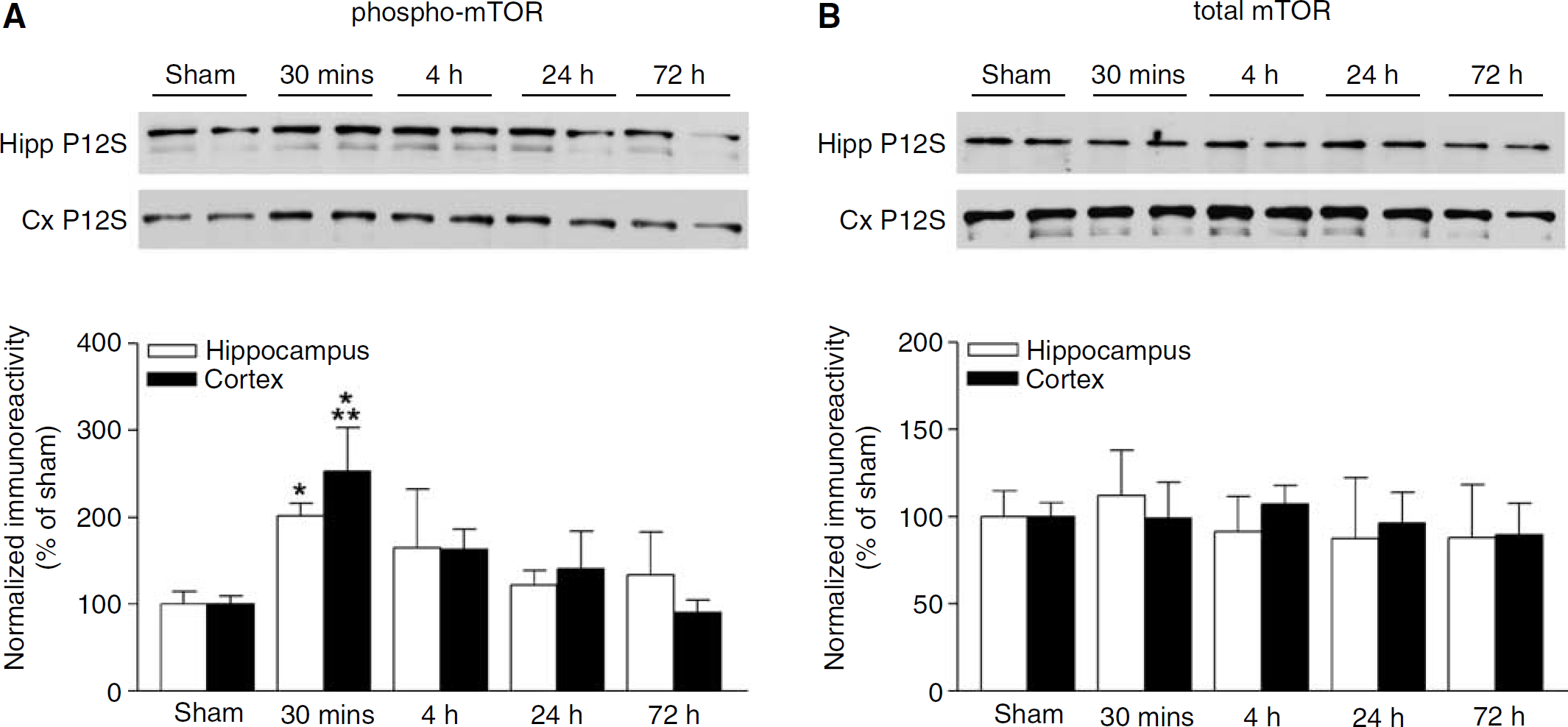
mTOR activation increases after TBI. (
p70 ribosomal S6 kinase and its substrate rpS6 are major downstream effectors of mTOR (Ferrari et al, 1991; Holz and Blenis, 2005). To determine whether this signaling pathway was also altered after TBI, crude synaptosome subcellular fractions (P12S fractions) were analyzed by Western blotting for phosphorylated p70S6K. Consistent with the changes in mTOR activation, we found that p70S6K was significantly activated in both the ipsilateral hippocampus and parietal cortex 30 mins after TBI (Figure 2A). This activation persisted for up to 4 h after TBI, and returned to baseline levels by 24 to 48 h. Like mTOR, total levels of p70S6K were not altered after TBI (Figure 2B).
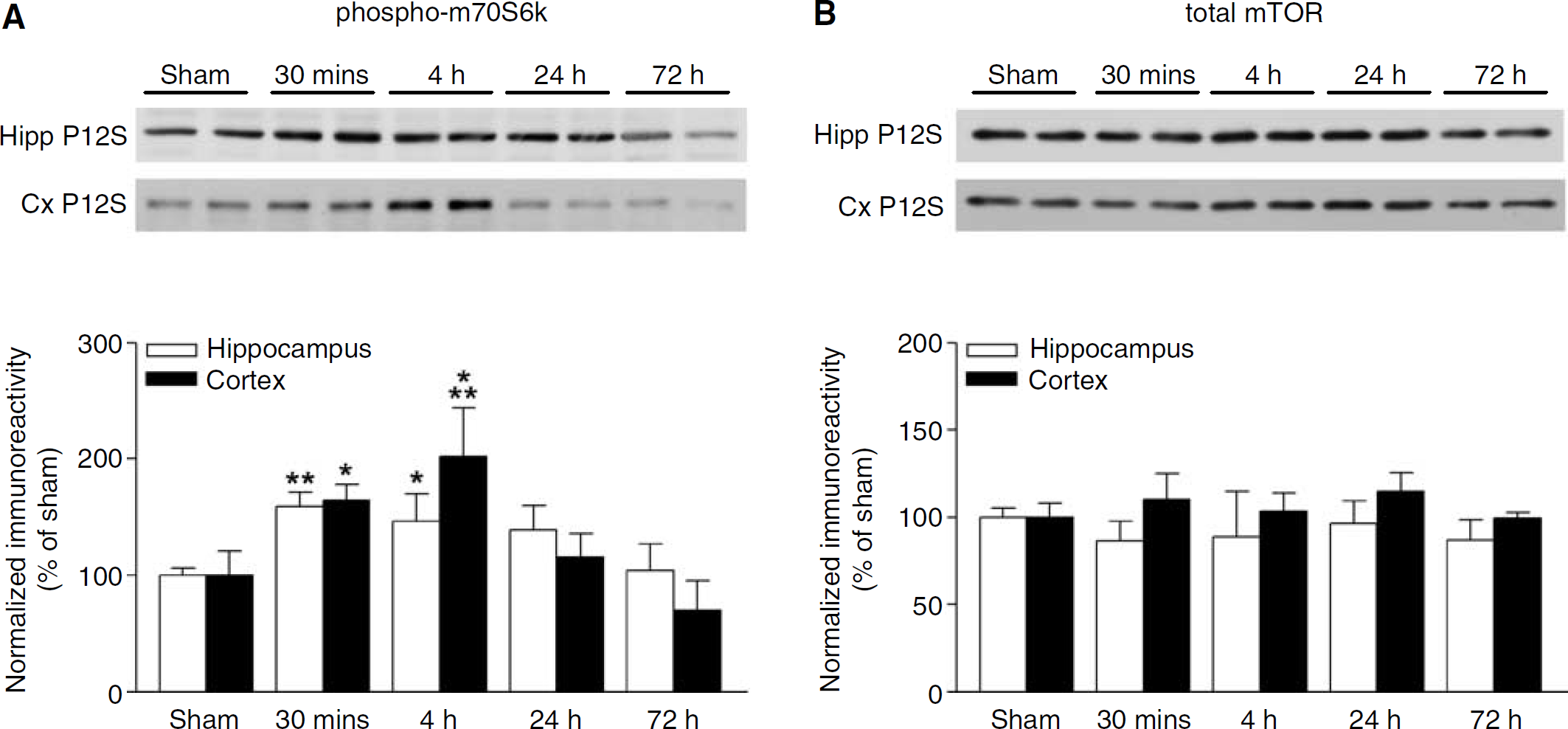
p70S6K phosphorylation increases after TBI. (
Phosphorylation of rpS6 by p70S6K recruits rpS6 into actively translating ribosomes (Ferrari et al, 1991). We therefore analyzed the hippocampus and parietal cortex subcellular fractions by Western blotting with antibodies to phospho-rpS6 Ser235/236 and total rpS6. Similar to ribosomal subcellular distribution, rpS6 was present in crude synaptosome subcellular fractions (P12P and P12S) and the microsomal fraction (Liu et al, 2005). Phosphorylation of rpS6 Ser235/236 increased significantly 30 mins after TBI in crude synaptosomal and microsomal subcellular fractions from the ipsilateral hippocampus (Figure 3A) and parietal cortex (Figure 4A) and persisted for up to 24 h after TBI. There were no changes in total rpS6 levels after TBI (Figures 3B and 4B).
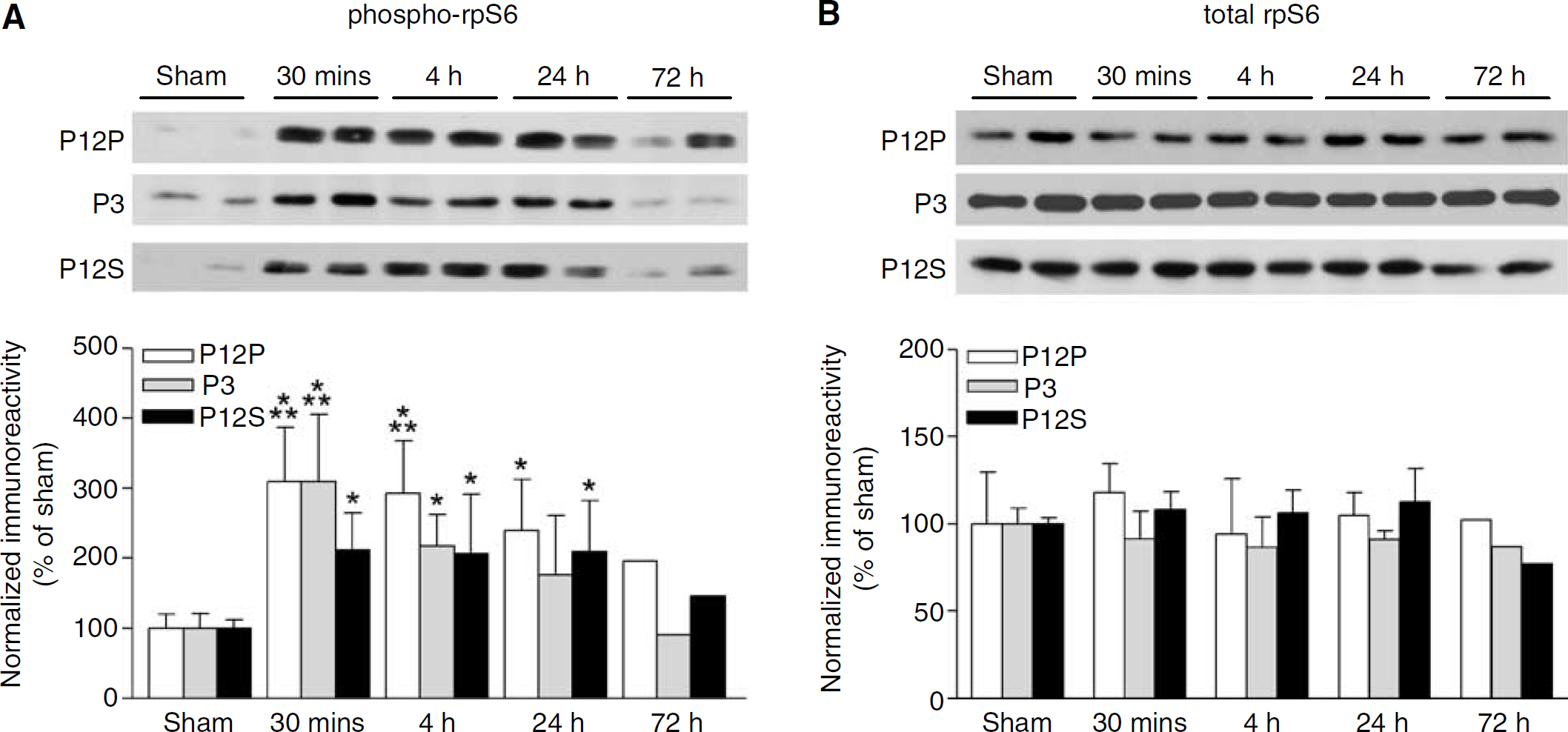
Ribosomal S6 protein phosphorylation increases in the hippocampus after TBI. (
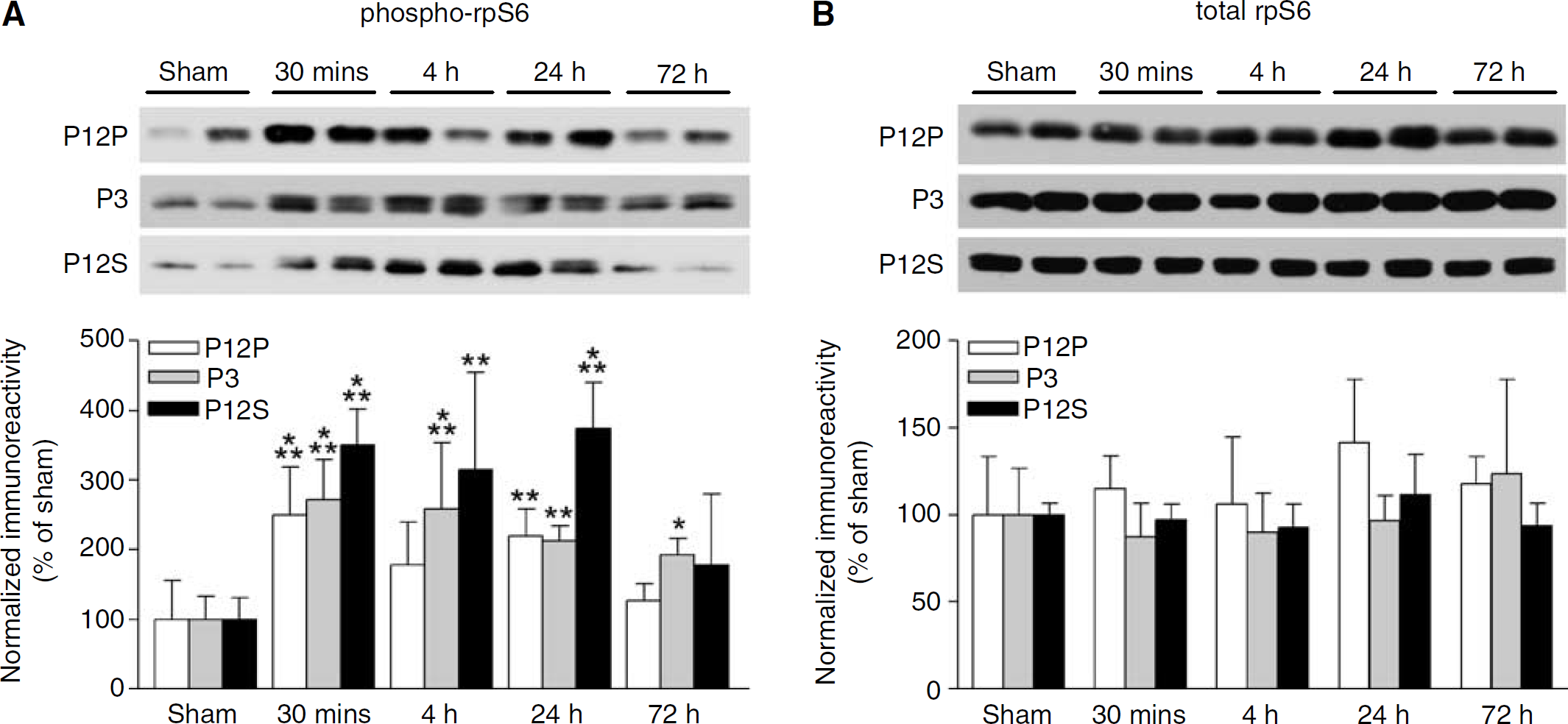
Ribosomal S6 protein phosphorylation increases in the parietal cortex after TBI. (
To study the regional distribution of phosphorylated rpS6 after TBI, we utilized confocal microscopy to image the ipsilateral hippocampus and parietal cortex 30 mins, 4 h, and 24 h after TBI. Sections were probed with antibodies to phospho-rpS6 (Figure 5A) or total rpS6 (Figure 5B), then counterstained with propidium iodide to visualize the nuclei. An increase in phospho-rpS6 immunoreactivity was observed in all subregions of the hippocampus, throughout the parietal cortex, and was localized to neurons.
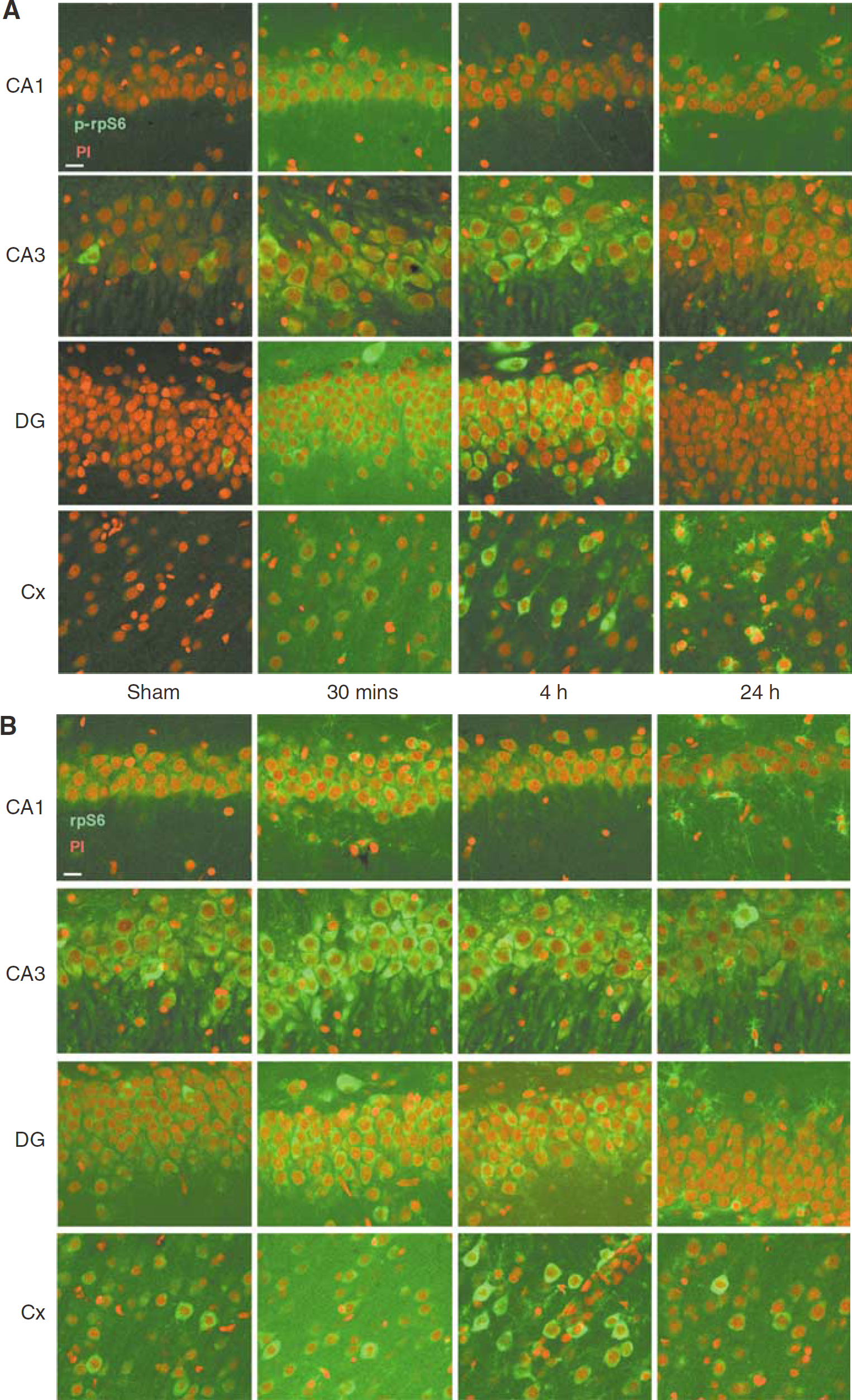
Ribosomal S6 protein activation increases in neurons after TBI. (
In addition to p70S6K, another well-known effector of mTOR signaling is 4E-BP1 (Beretta et al, 1996). Hypophosphorylated 4E-BP1 binds to eIF4E with high affinity, sequestering eIF4E from interacting with eIF4G. When phosphorylated by mTOR, 4E-BP1 releases eIF4E. This allows eIF4E to bind eIF4G and stimulates the translation of 5′ capped mRNAs (Haghighat et al, 1995). Given the activation of mTOR after TBI, we determined if 4E-BP1 phosphorylation was also altered after TBI. Western blots of the cytosolic fractions (S3) for phospho-4E-BP1 revealed a significant increase beginning 1 h after TBI, in both the ipsilateral hippocampus and parietal cortex (Figure 6A). Similar to S6 phosphorylation, 4E-BP1 phosphorylation persisted for up to 24 h after TBI, suggesting mTOR downstream activation lasted longer than did activation of mTOR itself. Total levels of 4E-BP1 did not significantly change after TBI, although a modest, non-significant increase was seen 24 h after TBI (Figure 6B). These results suggest that mTOR signals through both p70S6K and 4E-BP1 to potentially alter mRNA translation after TBI.
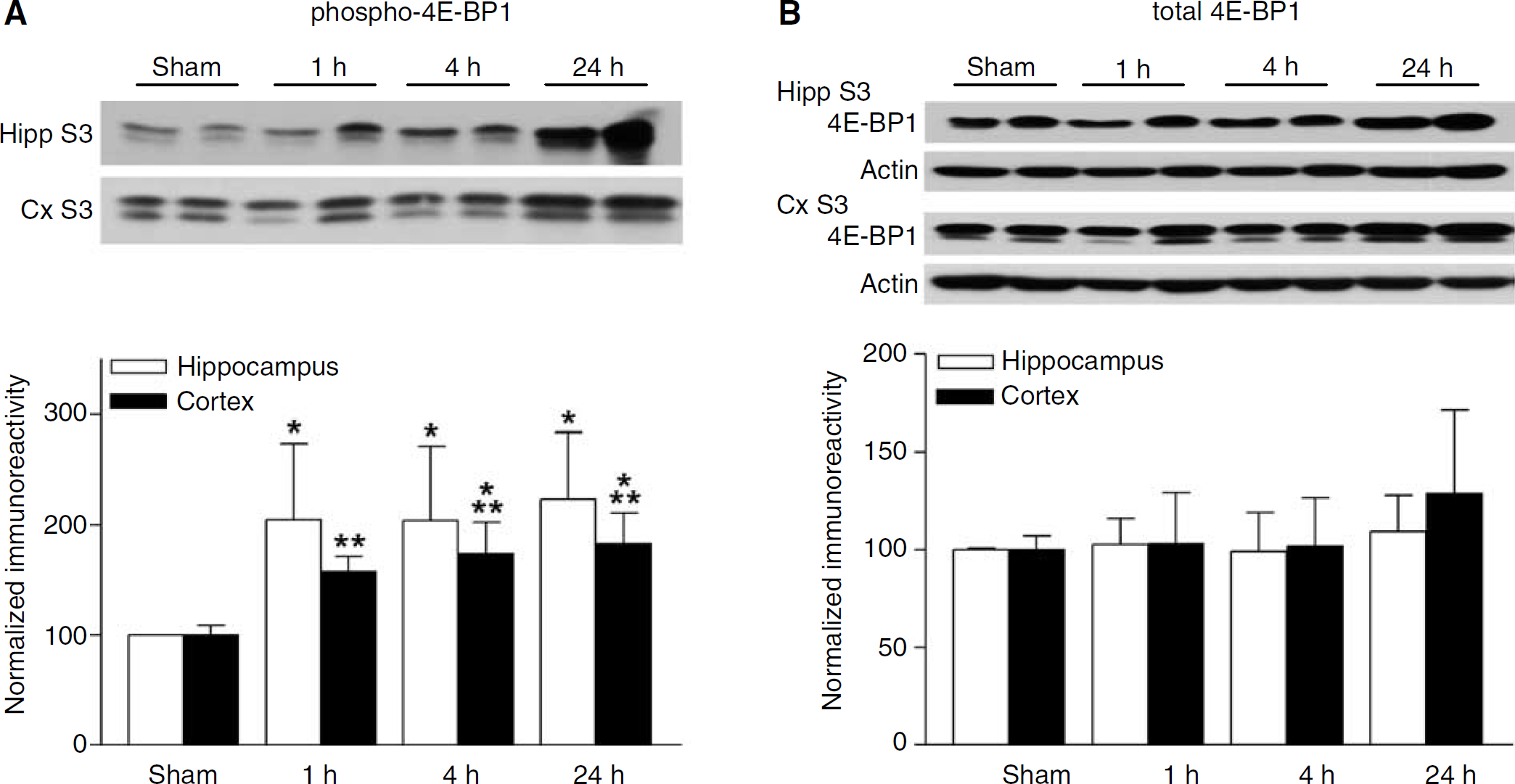
Phosphorylation of 4E-BP1 increases after TBI. (
The translation factor eIF4E, which is inhibited by 4E-BP1, is itself also regulated by phosphorylation. p44/p42 MAPK phosphorylates and activates the protein kinase Mnk1, which phosphorylates and regulates eIF4E (Duncan et al, 2003). Like mTOR, we also found that Mnk1 and eIF4E were rapidly activated by phosphorylation within 15 mins after TBI in total homogenates from the hippocampus (Figure 7A). For Mnk1, this phosphorylation persisted for up to 24 h after TBI, but eIF4E phosphorylation was transient, lasting only 15 to 30 mins (Figure 7B), suggesting differential phosphatase activity on the two molecules. Thus, TBI may result in complex changes in mRNA translation through multiple mTOR and Mnk1 signaling pathways (Figure 8).
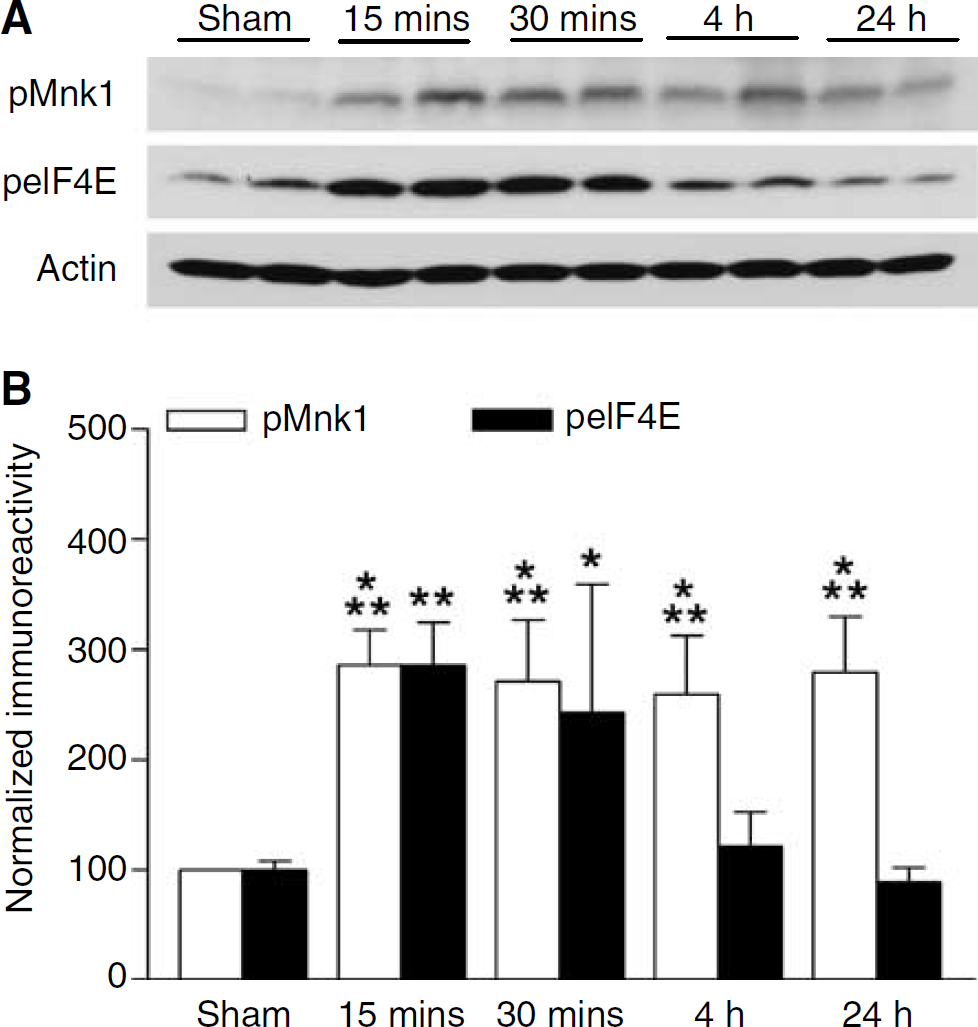
Mnk1 and eIF4E phosphorylation increase after TBI. (
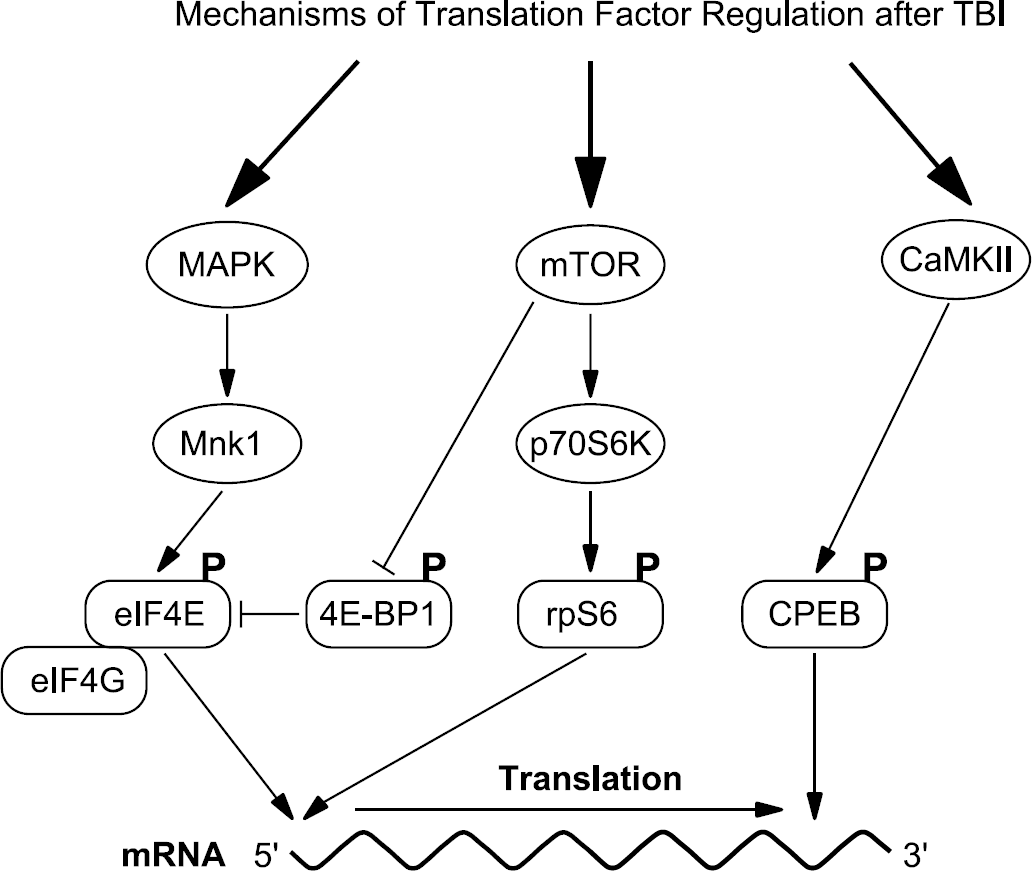
Model of alterations in mRNA translation factors after TBI. Protein synthesis after TBI is regulated by multiple mRNA translation factors. Activation of p42/p44 MAPK phosphorylates Mnk1, which phosphorylates and regulates the translation factor eIF4E. eIF4E binds the 5′ end of capped mRNAs and when stimulated, recruits the translation factor eIF4G to form the eIF4F complex and initiate mRNA translation. Eukaryotic initiation factor 4E is also regulated by 4E-BP1, an inhibitory molecule that competes with eIF4G for binding to eIF4E. When phosphorylated by mTOR, 4E-BP1 releases eIF4E, thus stimulating mRNA translation. mTOR also regulates and stimulates mRNA translation through phosphorylation and activation of p70S6K. p70 ribosomal S6 kinase phosphorylates rpS6, which mobilizes the ribosomes into polyribosomes. Recently, another mechanism to alter mRNA translation after TBI was discovered through the calcium/calmodulin-dependent protein kinase II (CaMKII) pathway. CaMKII and its downstream substrate, the translation factor cytoplasmic polyadenylation element-binding protein (CPEB), are phosphorylated and activated after TBI (Atkins et al, 2006). Cytoplasmic polyadenylation element-binding protein binds the 3′ end of mRNAs and when phosphorylated, CPEB stimulates mRNA polyadenylation and translation. All of these translation factors, eIF4E, 4E-BP1, rpS6, and CPEB, are phosphorylated and activated within 15 mins to 24 h after TBI.
Discussion
Regulation of mRNA translation factors allows neurons to fine-tune the rate of protein synthesis and also to synthesize specific proteins to respond to cell growth, synaptic plasticity, and injury. In the present study, we found several signaling pathways converge to alter mRNA translation after TBI. mTOR, a key regulator of mRNA translation, was transiently activated 30 mins after TBI. The time of onset of mTOR activation was similar to the phosphorylation and activation of its downstream substrates, 4E-BP1, p70S6K, and rpS6, which were also activated within 30 mins after TBI. However, the activation of mTOR after TBI returned to baseline by 4h after TBI, whereas the activation of its downstream targets p70S6K, rpS6, and 4E-BP1 were more prolonged, lasting from 4 to 24 h after TBI. All of these cell signaling changes occurred in both the hippocampus and parietal cortex. We did not see any temporal differences between the parietal cortex and hippocampus for each of the cell signaling molecules assessed; however, the magnitude of the changes was not always equivalent. This suggests that the biochemical response of the parietal cortex and hippocampus is very similar, but not completely identical, after TBI with the FPI model. Additionally, we found that a parallel regulatory pathway, through p44/p42 MAPK and Mnk1, was also activated after TBI in the hippocampus and this stimulated the phosphorylation of eIF4E within a similar time frame. Our previous studies have also demonstrated an activation of the translation factor cytoplasmic polyadenylation element-binding protein after TBI (Atkins et al, 2006). Thus, our current data support the hypothesis that mRNA translation rapidly changes after TBI, possibly in response to changes in ionic homeostasis, neurotransmitter release, and membrane damage (Figure 8).
Activation of mTOR after TBI may occur via a number of neurotransmitter receptors. Brain-derived neurotrophic factor is well-established to activate the mTOR signaling pathway and is known to increase after TBI (Truettner et al, 1999; Takei et al, 2004). Brief glutamatergic stimulation of cultured neurons activates mTOR to an extent approaching that elicited by brain-derived neurotrophic factor (Gong et al, 2006). Therefore, glutamate release after TBI may also contribute to the activation of mTOR after TBI (Globus et al, 1995). Activation of mTOR can occur through at least two pathways. Mammalian target of rapamycin is repressed by the GTPase TSC1/2 (Inoki et al, 2002). Phosphoinositide 3-kinase, activated by growth factor receptors, activates AKT (protein kinase B), which phosphorylates TSC1/2, decreasing its GTPase activity. This increases GTP-bound Ras homolog enriched in brain, which then stimulates mTOR (Garami et al, 2003). Furthermore, phosphatidic acid generated from phospholipase D stimulates mTOR as well (Fang et al, 2001). Excessive phospholipase levels have been reported after TBI and may contribute to the activation of mTOR (Phillis and O'Regan, 2003).
The time course of mTOR activation after TBI was shorter in duration than the more prolonged, significant rpS6 activation after TBI. Besides mTOR signaling through p70S6K, the MAPK—p90RSK signaling pathway can also phosphorylate and activate rpS6 (Pende et al, 2004). We have previously observed an activation of MAPK after TBI, suggesting that both pathways may contribute to the increase in rpS6 phosphorylation (Hu et al, 2004). The persistent activation of rpS6 phosphorylation as compared with mTOR and p70S6K phosphorylation may be due to differential phosphatase activity on each of these molecules. Alternatively, amplification of signaling through multiple kinase activation steps, such as through mTOR—p70S6K, can result in an exponential increase in substrate phosphorylation (Ferrell, 1996). Thus, the increase in rpS6 phosphorylation may be a nonlinear readout of the mTOR signaling pathway.
Mammalian target of rapamycin was classically thought to stimulate translation of 5′ oligopyrmidine tract-containing mRNAs through phosphorylation of rpS6, although whether mTOR does so through rpS6 remains controversial (Tang et al, 2001). This subset of mRNAs encodes for proteins involved in the mRNA translation machinery including ribosomal proteins, translation factors, and poly(A)-binding protein (Meyuhas, 2000). This increases the neurons’ capacity for synthesizing proteins necessary for cell growth and dendritic arborization. Traumatic brain injury induces the upregulation of numerous proteins. Microarray analysis of the hippocampus after TBI revealed that 125 mRNAs are increased in the hippocampus and 242 mRNAs are increased in the frontal cortex after TBI (Rall et al, 2003). Thus, activation of mTOR and rpS6 may be to increase the translational capacity of neurons.
Local increases in dendritic protein synthesis through mTOR signaling pathways are an important component of hippocampal synaptic plasticity such as LTP (Tang et al, 2002; Tsokas et al, 2005). Mammalian target of rapamycin and protein synthesis machinery, such as polysomes, are localized to dendrites. Likewise, mTOR expression overlaps with postsynaptic density 95 (Tang et al, 2002). Local application of brain-derived neurotrophic factor to a dendritic region increases phosphorylation of rpS6 in the dendrite at the area of the puff, as well as dendritic protein synthesis (Schratt et al, 2004; Takei et al, 2004). Similarly, we observed increases in activation of the mTOR signaling pathway in synaptic fractions after TBI and our confocal microscopy results suggest these changes are occurring in neurons. Together, these results suggest that mRNA translation regulation through mTOR signaling may occur in both the somata and dendrites of neurons after TBI.
In the hippocampus, the mTOR signaling pathway was activated in all subregions, but only the CA3 and hilar subregions exhibit cell death with the FPI model in the rat (Grady et al, 2003). Knockdown of the NMDA-type glutamate receptor 1 in a mere 6% to 7% of CA1 neurons in the hippocampus is enough to significantly impair memory formation (Cheli et al, 2006). The FPI model, without any accompanying hypoxia, does not result in substantial neuronal loss throughout the hippocampus with the exception of the CA3 and hilar subregions, but does result in significant hippocampal-dependent memory impairments (Lyeth et al, 1990; Carbonell et al, 1998; Grady et al, 2003). Accordingly, hippocampal LTP is impaired and subtle structural damage is seen in the hippocampus after TBI (Sanders et al, 2000; Golarai et al, 2001; Scheff et al, 2005). We speculate that activation of mTOR—p70S6K and MAPK—Mnk1 pathways after TBI could be involved in these neuroplastic changes, given their prominent roles in hippocampal LTP and dendritic arborization (Jaworski et al, 2005; Kumar et al, 2005; Tsokas et al, 2005).
The mTOR signaling pathway is well-established to regulate synaptic plasticity by increasing the mRNA translational capacity within dendrites. Inhibition of mTOR by rapamycin blocks protein synthesis-dependent hippocampal LTP in the CA1 region (Tang et al, 2002). Rapamycin blocks dendritic growth and branching and is considered to inhibit synaptic plasticity by blocking the translation of mRNAs involved in neuroplasticity (Jaworski et al, 2005; Kumar et al, 2005; Tsokas et al, 2005). In relation to TBI, there are several neuroplastic changes in the hippocampus after TBI. Traumatic brain injury activates the TrkB—MAPK—CREB/Elk pathways in hippocampal mossy fibers during the early phase after TBI and induces aberrant axonal sprouting in hippocampal mossy fiber projections during the late phase after TBI (Golarai et al, 2001; Hu et al, 2004). The mTOR signaling pathways may act in concert with the TrkB—MAPK—CREB/Elk pathways for neurons to respond to changes in ionic homeostasis, neurotransmitter release, and synaptic damage after TBI. Given that both the MAPK and mTOR signaling pathways promote cellular and synaptic growth and repair, one may speculate that activation of these pathways may contribute to the remodeling of synaptic circuits and synaptic plasticity after TBI. Alternatively, activation of these pathways could also induce aberrant sprouting, leading to posttraumatic seizures which are often seen after TBI (Golarai et al, 2001). It is still unclear whether activation of these growth-promoting pathways is beneficial after TBI. Therefore, dissecting the cellular and molecular mechanisms underlying normal synaptic plasticity and epileptogenesis after TBI may provide new therapeutic avenues for promoting re-growth and repair, but prevent the development of posttraumatic epilepsy.
Footnotes
Acknowledgements
We thank the Hu laboratory for helpful discussions and AA Oliva for critical reading of this manuscript.