
Abstract
Global brain ischemia and reperfusion result in the degradation of the eukaryotic initiation factor (eIF) 4G, which plays a critical role in the attachment of the mRNA to the ribosome. Because eIF-4G is a substrate of calpain, these studies were undertaken to examine whether calpain I activation during global brain ischemia contributes to the degradation of eIF-4G in vivo. Immunoblots with antibodies against calpain I and eIF-4G were prepared from rat brain postmitochondrial supernatant incubated at 37°C with and without the addition of calcium and the calpain inhibitors calpastatin or MDL-28,170. Addition of calcium alone resulted in calpain I activation (as measured by autolysis of the 80-kDa subunit) and degradation of eIF-4G; this effect was blocked by either 1 μmol/L calpastatin or 10 μmol/L MDL-28,170. In rabbits subjected to 20 minutes of cardiac arrest, immunoblots of brain postmitochondrial supernatants showed that the percentage of autolyzed calpain I increased from 1.9% ± 1.1% to 15.8% ± 5.0% and that this was accompanied by a 68% loss of eIF-4G. MDL-28,170 pretreatment (30 mg/kg) decreased ischemia-induced calpain I autolysis 40% and almost completely blocked eIF-4G degradation. We conclude that calpain I degrades eIF-4G during global brain ischemia.
The severe reduction of protein synthesis that occurs. During early reperfusion after global brain ischemia arises from a defect in the formation of the translation-initiation complex (Krause and Tiffany, 1993). One step in this process, the attachment of mRNA to the 40S ribosomal subunit, is coordinated by the eukaryotic initiation factor 4F complex (eIF-4F) comprised of eIF-4A (an ATP-dependent RNA helicase), eIF-4E (which binds the mRNA m7GTP cap), and eIF-4G (which provides docking sites for the aforementioned proteins) (Merrick and Hershey, 1996). Global brain ischemia and reperfusion results in the alteration of several of these initiation factors (Hu and Wieloch, 1993; Burda et al., 1994; Neumar et al., 1995; DeGracia et al., 1996). In particular we reported evidence of proteolysis of eIF-4G (also known as eIF-4γ or p 220) after an ischemic insult (DeGracia et al., 1996). Moreover Etchison et al. (1982) demonstrated that proteolytic loss of eIF-4G during poliovirus infection leads to inhibition of protein synthesis. The identity of the enzymes that degrade eIF-4G as a result of brain ischemia is unknown, but there is evidence that eIF-4G is a substrate of the calcium-activated protease calpain (Wyckoff et al., 1990), and we have shown that calpain I (μ-calpain) activation occurs during brain ischemia (Neumar et al., 1996). Therefore these studies were undertaken to examine whether calpain I activation during global brain ischemia contributes to the degradation of eIF-4G in vivo.
METHODS
Calpain I activation can be detected by the autolytic cleavage of the N-terminus of its 80-kDa catalytic subunit, which produces active fragments with molecular weight by electrophoretic mobility (Mr) of 76 and 78 kDa both in vitro (Croall et al., 1992) and in vivo (Hayashi et al., 1991; Saito et al., 1993; Molinari et al., 1994). Monoclonal antibody against the 80-kDa catalytic subunit of human calpain I (clone B27D8), which reacts with the 76-, 78-, and 80-kDa forms of the calpain I catalytic subunit in rabbits (Neumar et al., 1996), was provided by John Elce (Queens University, Kingston, Ontario) (Samis et al., 1987). In our hands this antibody generated negligible signal with rat specimens. To measure calpain I autolysis in rats, an affinity-purified polyclonal antibody was prepared against a 15-amino acid peptide corresponding to the N-terminal sequence of the rat calpain I catalytic subunit (Sorimachi et al., 1996). This antibody recognizes the 80-kDa catalytic subunit in rats, but has negligible reactivity with the autolyzed forms. An affinity-purified polyclonal anti-eIF-4G antibody was prepared against a conserved 16-amino acid peptide beginning at amino acid 327 in the human sequence (DeGracia et al., 1996). Purified porcine calpain I and recombinant domain I of human calpastatin, which specifically inhibits calpain, were purchased from Calbiochem (La Jolla, CA, U.S.A.). MDL-28,170, a cell-permeant calpain inhibitor (Mehdi et al., 1988), was a gift from Shujaath Mehdi (Marion Merrell Dow Research Institute, Cincinnati, OH, U.S.A.). All other chemicals were reagent grade. All animal experiments were approved by our institutional review board and were conducted following the “Principles of Laboratory Animal Care” (NIH publication No. 86-23, revised 1985).
In vitro calpain I autoproteolysis and eIF-4G degradation were studied in a postmitochondrial supernatant prepared from a male Long-Evans rat anesthetized with 4% halothane. The forebrain was homogenized in 5 volumes of ice-cold buffer A (55 mmol/L Tris, pH 7.4, 165 mmol/L sucrose, 1.1 mmol/L dithiothreitol, 9.0 mmol/L EGTA, 0.25 mmol/L phenylmethyl-sulfonylfluoride [PMSF], 7.7 μg/mL pepstatin, and 1.1 μg/mL aprotinin). The homogenate was centrifuged at 10,000g for 20 minutes at 4°C, and the protein concentration of the supernatant determined by the Folin phenol reagent method. Reactions were immediately run at 37°C in a final volume of 500 μL that included 450 μL supernatant and final concentrations equal to (1) buffer A (2) buffer A plus 10 mmol/L CaCl2, (3) buffer A plus 10 mmol/L CaCl2 and 1 μmol/L calpastatin, and (4) buffer A plus 10 mmol/L CaCl2 and 10 μmol/L MDL-28,170. At 0, 10, and 30 minutes' reaction time, aliquots were withdrawn, and the reactions were stopped by the addition of an equal volume of 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (125 mmol/L Tris, pH 6.8, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol) containing 20 mmol/L EGTA and 3.0 mol/L urea; this solution was immediately boiled for 2 minutes.
For Western blots, 50-μg protein samples were electrophoresed on SDS-polyacrylamide gels (7.5% total acrylamide, 2.5% cross-linker bis acrylamide), transferred to nitrocellulose, immunoblotted with a 1:500 dilution of the antibody against rat calpain I, and developed using enhanced chemiluminescence (ECL, Amersham, Arlington Heights, IL, U.S.A.). The membranes were then stripped (in 100 mmol/L 2-mercaptoethanol, 2% SDS, and 62.5 mmol/L Tris, pH 6.7 at 50°C for 30 minutes) and reblotted with antibody against eIF-4G (1:750).
Ischemia-induced calpain I autoproteolysis and eIF-4G degradation occurring in vivo were studied in female New Zealand white rabbits anesthetized with 4% halothane. Three rabbits were included in each of three treatment groups: (1) control (C), no ischemia plus 1.5 mL/kg drug vehicle (20% ethanol and 80% polyethylene glycol [PEG] 300), (2) 20 minutes' ischemia (20-I) plus 1.5 mL/Kg vehicle; and (3) 20 minutes' ischemia plus 30 mg/kg MDL-28,170 (20-I-MDL). Drug or vehicle was infused intravenously for 10 minutes beginning 20 minutes before the start of the experiment. Cardiac arrest was initiated by intracardiac injection of 5 mEq/kg KCl and confirmed by electrocardiogram, loss of apical pulse, and cessation of spontaneous respiration. At the appropriate time, the forebrains were homogenized in 5 volumes ice-cold buffer (50 mmol/L Tris, pH 7.4, 20 mmol/L EDTA, 20 mmol/L EGTA, 1 mmol/L dithiothreitol, 150 mmol/L KCl, 0.23 mmol/L PMSF, 1 μg/mL aprotinin, 10 μg/mL leupeptin, and 7 μg/mL pepstatin), and then centrifuged at 10,000g for 30 minutes at 4° C. After determination of protein concentration, the supernatant was diluted 1:1 in 2× SDS-PAGE loading buffer containing 3 mol/L urea, and boiled for 2 minutes.
Western blots were performed as described above except that a 1:10,000 dilution of the antibody against human sequence calpain I was used. Immunoblot exposures were adjusted to insure linearity. Relative band densities were quantified by densitometry, and differences between experimental groups were examined for significance by analysis of variance and Fisher's least significant difference posthoc comparison.
RESULTS
In vitro calpain I autolysis and eIF-4G degradation
The antibodies to eIF-4G and the N-terminus of the rat calpain I catalytic subunit identified the expected bands with Mr of 220 kDa and 80 kDa, respectively (Fig. 1); these signals were absent if the primary antibody was omitted or preblocked with the antigenic peptide. As previously stated in the methods section, the calpain I antibody generated from the human sequence protein did not generate adequate signal in our rat brain samples.
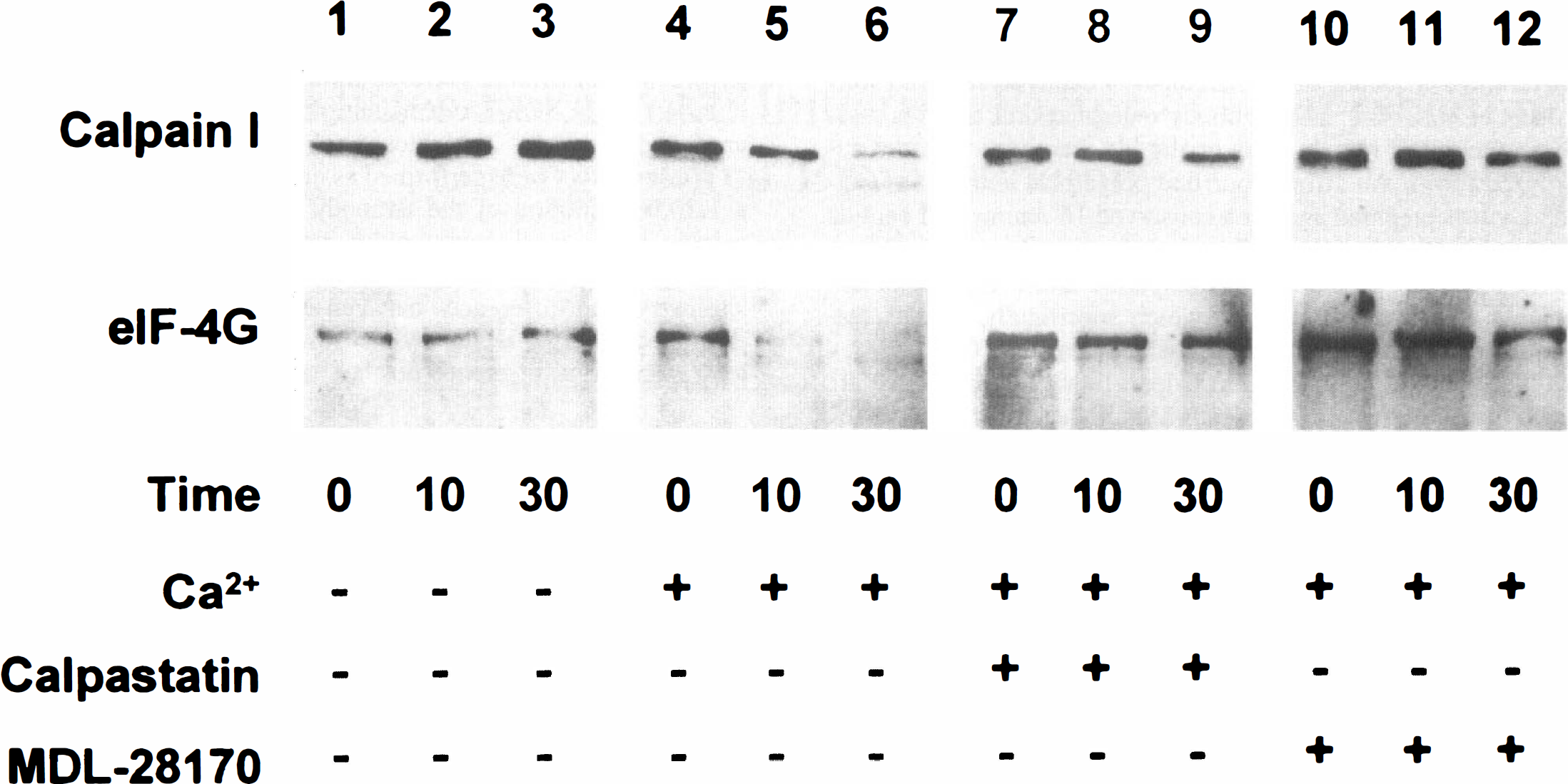
Calpain-mediated degradation of eIF-4G in vitro. Postmitochondrial supernatants were prepared from rat forebrain. Additions were made as shown, and the reactions were incubated for the times indicated (minutes). The immunoblots for calpain I (upper, antibody to rat sequence) or eIF-4G (lower) show no autolysis of calpain I or degradation of eIF-4G in the absence of calcium (lanes 1–3). Although the addition of calcium (1 mmol/L in excess of EGTA) promptly causes the loss of calpain I and eIF-4G (lanes 4–6), this loss is prevented by calpastatin (1 μmol/L, lanes 7–9) or MDL-28,170 (10 μmol/L, lanes 10–12).
In the absence of added calcium, there was no detectable loss of the calpain I 80-kDa subunit or eIF-4G after 30 minutes' incubation (Fig. 1, lanes 1–3). Addition of calcium without a calpain inhibitor resulted in a rapid decrease of the calpain I 80-kDa subunit and degradation of eIF-4G (Fig. 1, lanes 4–6). Simultaneous addition of calcium and either calpastatin or MDL-28,170 blocked the loss of the calpain I 80-kDa subunit and inhibited the degradation of eIF-4G (Fig. 1, lanes 7–12).
In vivo calpain I autolysis and eIF-4G degradation during complete global cerebral ischemia
During in vivo brain ischemia, the unautolyzed calpain I catalytic subunit is identified as a predominant band with the expected Mr of 80 kDa identical to purified porcine calpain I, and the autolyzed forms appear with Mr of 76 and 78 kDa (Fig. 2, lane 1); this signal was absent if the primary antibody was omitted or preblocked with purified calpain I. The percentage of autolyzed calpain I was estimated by dividing the density of the autolyzed bands (76 kDa + 78 kDa) by the total calpain I band density (76 kDa + 78 kDa + 80 kDa). After 20 minutes' ischemia autolyzed calpain I increased from 1.9% ± 1.1% to 15.8% ± 5.0% (P = 0.001, Fig. 2). Pretreatment with MDL-28170 decreased ischemia-induced calpain I autolysis to 9.5% ± 0.5% (P = 0.04 versus untreated 20-I) (Fig. 2). The anti-eIF-4G immunoblot of this same membrane shows that 20 minutes' ischemia induced a 68% loss of eIF-4G (Fig. 2, lanes 5–7 and right graph, P = 0.004 versus controls) that was almost completely blocked by MDL-28,170 (Fig. 2, lanes 8–10, P = 0.01 versus untreated 20-I).
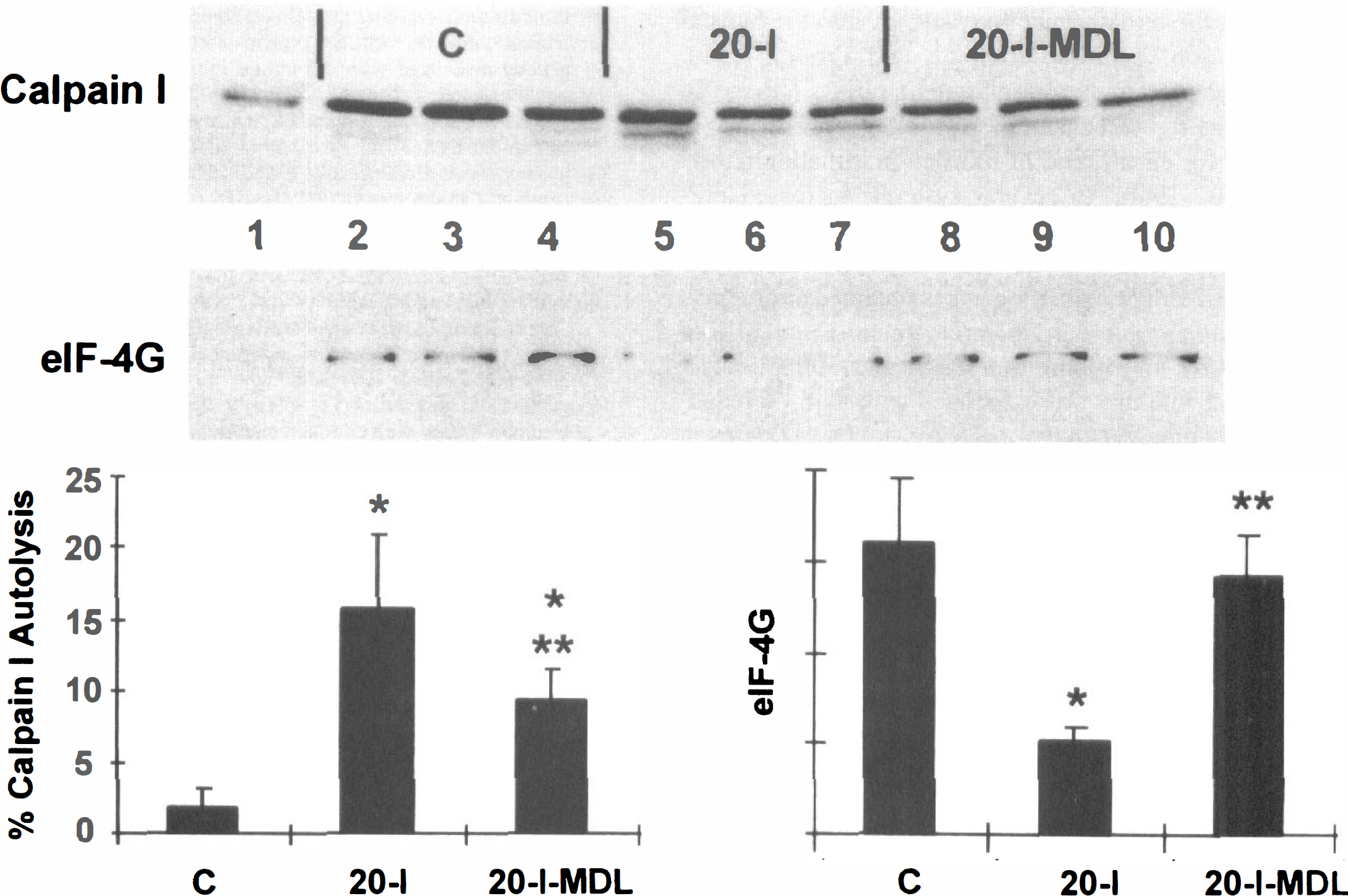
MDL-28,170 blocks in vivo calpain I autolysis and eIF-4G degradation during 20 minutes of cardiac arrest. Rabbits were pretreated with MDL-28,170 (30 mg/kg) or drug vehicle 20 minutes before the onset of 20 minutes of ischemia. Postmitochondrial supernatants were prepared from forebrain and immunoblotted for calpain I (upper, antibody to human sequence) or eIF-4G (lower). Lane 1, 10 ng purified porcine μ-calpain; lanes 2–4, vehicle-treated nonischemic (C); lanes 5–7, vehicle-treated 20 minutes' ischemia (20-I); lanes 8–10, MDL-28,170 (30 mg/kg intravenously) -treated 20 minutes' ischemia (20-I-MDL). Graphs show the mean and standard deviation for calculated calpain autolysis ([76 kDa + 78 kDa]/[76 kDa + 78 kDa + 80 kDa]) and eIF-4G relative band densities. There is significant autolysis of calpain I (P = 0.001 versus C) and eIF-4G degradation (P = 0.004 versus C) after 20 minutes' ischemia. MDL-28,170 pretreatment partially inhibits calpain I autolysis (P = 0.02 versus C; P = 0.04 versus 20-I) and significantly blocks eIF-4G degradation (P = 0.62 versus C; P = 0.01 versus 20-I) during 20 minutes of complete global ischemia.
DISCUSSION
Calpains have long been suspected to cause proteolysis in the ischemic brain, and we have recently reported direct evidence of calpain I activation during complete global ischemia in vivo (Neumar et al., 1996). The calpains (EC 3.4.22.17) are a family of nonlysosomal, calcium-activated, neutral cysteine proteases, some of which are ubiquitously distributed while others are tissue-specific (Saido et al., 1994; Sorimachi et al., 1994). The ubiquitous forms, calpain I and calpain II, are present in neurons (Fukuda et al., 1990), but no brain-specific isoforms have yet been found (Sorimachi et al., 1994). Calpain I and II share a common 30-kDa regulatory subunit, but each has a unique 80-kDa catalytic subunit. The calpains are absolutely dependent on Ca2+ and are regulated by phospholipids, an endogenous inhibitor (calpastatin), and an activator protein (Croall and DeMartino, 1991). A multitude of regulatory and cytoskeletal proteins known to be in vitro substrates of calpain I are degraded during cerebral ischemia and reperfusion (Kishimoto et al., 1989; Kitagawa et al., 1989; Kuwaki et al., 1989; Seubert et al., 1989; Inuzuka et al., 1990; Onodera et al., 1990; Yanagihara et al., 1990; Wieloch et al., 1991; Tomimoto and Yanagihara, 1992; Yamamoto et al., 1992; Kaku et al., 1993; Neumar et al., 1995).
To determine whether calpain I activation during global brain ischemia contributes to the degradation of eIF-4G in vivo, we used the calpain inhibitors calpastatin and MDL-28,170 to demonstrate specificity in vitro and then used MDL-28,170 to show efficacy in vivo. Calpastatin is the specific physiologic inhibitor of calpain. It does not inhibit other cysteine proteases or noncysteine proteases (Dayton et al., 1976; Nishiura et al., 1978; Waxman and Krebs, 1978; Sakon et al., 1982; DeMartino and Croall, 1984; Nakamura et al., 1984; Lepley et al., 1985). MDL-28,170 is a dipeptidyl aldehyde (CbzValPheH; carbamic acid, [1-[[(1-formyl-2-phenylmethyl)amino]carbonyl]-2-methylpropyl]-, phenylmethyl ester that acts as a competitive inhibitor of calpains (Mehdi et al., 1988). It shows inhibitory activity against calpain and cathepsin B but essentially no activity against other proteases (Mehdi, 1991). Hong et al. (1994) have shown that it reduces spectrin (a calpain substrate) degradation and infarct size in a rat model of focal ischemia.
Calpastatin or MDL-28,170 inhibited both eIF-4G degradation and calpain I autolysis in vitro. On the other hand, in vivo MDL-28,170 blocked eIF-4G degradation but had only a partial effect on calpain I autoproteolysis. Inhibition of calpain activity without inhibition of autoproteolysis has previously been reported in cellular studies (Hayashi et al., 1991; Shea et al., 1996; Guttmann et al., 1997). In our case it is likely that the in vivo brain concentrations achieved with 30 mg/kg intravenous MDL-28,170 were significantly less than required to totally inhibit calpain activity. When 50 mg/kg MDL 28,170 is given intraperitoneally, brain levels reach 0.4 μmol/L at 30 minutes (Shujaath Mehdi, personal communication), whereas the concentration to achieve 50% inhibition (IC50) is 1 μmol/L for spectrin degradation in erythrocyte ghosts (Mehdi, 1991). We attempted to increase the dose of MDL-28,170 to 60 mg/kg intravenously, but this dose was lethal, and this effect was not caused by the drug vehicle. This study does not exclude a role for calpain II activity in addition to calpain I. The in vitro calcium concentration for half-maximal proteolytic activity is 7 to 50 μmol/L for calpain I and 300 to 1,000 μmol/L for calpain II (Croall and Demartino, 1984; Inomata et al., 1984; Cong et al., 1989; Edmunds et al., 1991). This suggests that calpain II activation cannot occur in the absence of calpain I activation. Proof that degradation of eIF-4G during brain ischemia is caused exclusively by calpain I awaits the development of a completely specific inhibitor suitable for in vivo studies or an appropriate transgenic animal.
The translation initiation factor eIF-4G plays a central role in orchestrating the attachment of the mRNA to be translated to the 40S ribosomal subunit. It contains binding sites for eIF-4E, eIF-4A, and eIF-3 (Lamphear et al., 1995). In addition, mRNA vary greatly in their binding efficiency to eIF-4G (Merrick and Hershey, 1996). Therefore, depending on the cleavage site, proteolysis of eIF-4G could have complex effects on translation efficiency and message repertoire. Loss of the N-terminal third of eIF-4G shifts translation from capped mRNA to favor uncapped messages (Lamphear et al., 1995; Ohlmann et al., 1995, 1996). However it appears that once eIF-4G and its primary C- and N-terminal cleavage products (i.e., the middle third of eIF-4G [Pestova et al., 1996]) are lost, the cell is unable to translate any messages (Borman et al., 1997).
Eukaryotic initiation factor 4G is not the only initiation factor altered as a consequence of ischemia and reperfusion (Hu and Wieloch, 1993; Burda et al., 1994; Krause et al., 1995; DeGracia et al., 1996, 1997). The overall rate of protein synthesis is dependent on the phosphorylation state of the α-subunit of eIF-2, which introduces the initiator methionyl-tRNA into the translation-initiation complex (Redpath and Proud, 1990). Phosphorylation of the α-subunit of eIF-2, eIF-2α(P), prevents the replenishment of GTP onto eIF-2, which is necessary for each round of translation initiation (Rowlands et al., 1988). In several systems, increased eIF-2α(P) results in decreased protein synthesis (Pain, 1996), and Burda et al. (1994) reported enhanced phosphorylation of eIF-2α in brain homogenates obtained after reperfusion. Studies from our laboratory found about 24% of brain eIF-2α to be phosphorylated after 90 minutes' reperfusion, and this was associated with an 83% reduction in initiation-dependent protein synthesis (DeGracia et al., 1996). Thus, although it may be possible to prevent the loss of eIF-4G by pharmacologic inhibition of calpain, this, by itself, is unlikely to restore protein synthesis.
Footnotes
Acknowledgements
The authors thank John Elce for the kind donation of the human calpain I antibody, Shujaath Mehdi for the gift of MDL-28,170, and Sarah Alousi for her photographic expertise.