
Abstract
The effects of hypoxia-ischemia (HI) on proliferation and differentiation in the immature (postnatal day 9) and juvenile (postnatal day 21) mouse hippocampus were investigated by injecting bromodeoxyuridine (50 mg/kg) daily for 7 days after the insult and evaluating the labeling 5 weeks after HI. Phenotypic differentiation was evaluated using NeuN, Iba1, APC, and S100β as markers of neurons, microglia, oligodendrocytes, and astrocytes, respectively. The basal proliferation, in particular neurogenesis, was higher in the immature than in the juvenile hippocampus. Hypoxia-ischemia did not increase neurogenesis significantly in the immature dentate gyrus (DG), but it increased several-fold in the juvenile brain, reaching the same level as in the normal, noninjured immature brain. This suggests that the immature hippocampus is already working at the top of its proliferative capacity and that even though basal neurogenesis decreased with age, the injury-induced generation of new neurons in the juvenile hippocampus could not increase beyond the basal level of the immature brain. Generation of glial cells of all three types after HI was significantly more pronounced in the cornu ammonis of the hippocampus region of the juvenile hippocampus. In the DG, only microglia production was greater in the juvenile brain. Increased microglia proliferation correlated with increased levels of the proinflammatory cytokines MCP-1 and IL-18 3 days after HI, indicating that the inflammatory response is stronger in the juvenile hippocampus. In summary, contrary to what has been generally assumed, our results indicate that the juvenile brain has a greater capacity for neurogenesis after injury than the immature brain.
Introduction
Perinatal hypoxic-ischemic (HI) brain injury is still a major cause of neurologic disability, including cerebral palsy and epilepsy, as well as cognitive and motor deficits (Johnston et al, 2002). The outcome after brain injury depends on the balance between mechanisms of injury and protection (Walton et al, 1999). In addition, it has recently been shown that the juvenile and adult brain can generate new neurons through proliferation of progenitor cells, and thus might help to repair brain damage (Arvidsson et al, 2002; Bartley et al, 2005; Daval et al, 2004; Eriksson et al, 1998; Plane et al, 2004; Scheepens et al, 2003; Sun et al, 2005). It has been demonstrated that neurogenesis in the mammalian brain continues throughout life but decreases with age (Bondolfi et al, 2004; Gray et al, 2002; Jin et al, 2004; Kuhn et al, 1996; Sun et al, 2005), and this process was shown to be stimulated by various pathologic conditions (Bartley et al, 2005; Daval et al, 2004; Gray et al, 2002; Sun et al, 2005). Neurogenesis induced by brain ischemia implies that deficient neuronal function might be repaired by replacing lost neurons. This possibility offers hope for the development of restorative therapies for neurologic disorders after brain injuries.
Neurogenesis occurs mainly in the subventricular zone of the lateral ventricles and the subgranular zone of the hippocampal dentate gyrus (DG) (Gage, 2000). The hippocampus is involved in learning and memory, and is highly vulnerable to ischemic injuries (Kawai et al, 2004; Liu et al, 1998). Many investigators have shown increased neurogenesis in the adult hippocampus stimulated by brain injury (Arvidsson et al, 2001; Jin et al, 2001; Kawai et al, 2004; Liu et al, 1998; Tonchev and Yamashima 2006; Yagita et al, 2001), but this phenomenon has received much less attention in developing brain. Previous studies have shown that the overall vulnerability to HI increases with brain development, such that the HI time must be shortened to yield the same extent of tissue damage in older animals (Zhu et al, 2005, 2006). There are also developmentally regulated regional differences in the susceptibility to HI injury. For example, the cornu ammonis of the hippocampus (CA) regions of the hippocampus are relatively resistant to HI in the immature brain and do not develop their characteristic vulnerability to ischemia until the brain growth spurt, around P13, whereas the dentate gyrus of the hippocampus (DG) appears to be more vulnerable in the immature brain than in the juvenile or adult brain (Towfighi et al, 1997). We found that apoptotic mechanisms of injury were activated to a greater extent in the immature CA1 and DG, whereas excitotoxic-necrotic mechanisms were activated more in the juvenile and adult hippocampal regions, particularly the CA regions (Zhu et al, 2005).
In the present study, we investigated cell proliferation and differentiation in the mouse hippocampus after a mild HI insult at postnatal day 9 (P9) and 21 (P21), corresponding developmentally to newborn and juvenile human brains, respectively. At P9 the rodent brain is growing rapidly and formation of axons, dendrites, synapses, and myelin is maximal, whereas at P21 brain growth is largely finished and only some myelination remains (Hagberg et al, 1997; Fukuda et al, 2005). At P9 the size of the DG is one third of the size in the adult brain, but at P21 this growth is completed. At P9 the CA region has not yet acquired its characteristic susceptibility to ischemia, but at P21 this vulnerability has reached adult levels (Towfighi et al, 1997). It has generally been presumed that the immature brain would possess a greater capacity for regeneration, but to our knowledge this hypothesis has not been tested at different developmental stages using the same brain injury paradigm.
Materials and methods
Induction of HI
Unilateral HI was induced in C57/BL6 male mice on postnatal day 9 (P9) or 21 (P21) according to the Rice-Vannucci model with slight modification (Rice et al, 1981; Zhu et al, 2005). Animals were anesthetized with isofluorane (5% for induction, 1.5% to 3.0% for maintenance) in a mixture of nitrous oxide and oxygen (1:1), and the duration of anesthesia was < 5 mins. The left common carotid artery was cut between double ligatures of prolene sutures (6.0). After the surgical procedure the wounds were infiltrated with xylocain for anesthesia. The pups were returned to the dam for 1 h and then placed in a chamber perfused with a humidified gas mixture (10% oxygen in nitrogen) for 35 mins (P9) or 30 mins (P21) at 36°C to produce a similar extent of mild brain injury (Zhu et al, 2005). After hypoxic exposure the pups were returned to their biologic dams until killed. Control pups were neither subjected to ligation nor hypoxia. All animal experimentation was approved by the Göteborg committee of the Swedish Animal Welfare Agency (1994–2003).
Bromodeoxyuridine Administration
The thymidine analog bromodeoxyuridine (BrdU) (5 mg/mL, dissolved in 0.9% saline) (Roche, Mannheim, Germany) was injected intraperitoneally at a dose of 50 mg/kg, once daily for 7 days, starting from 24 h after HI. The mice were killed 4 weeks after the last BrdU injection (Figure 1). In separate experiments, BrdU was injected daily for 7 days during the second or third week after HI in P9 mice, to compare proliferation during the first week after HI with the 2 consecutive weeks. All animals were killed 4 weeks after the last BrdU injection.
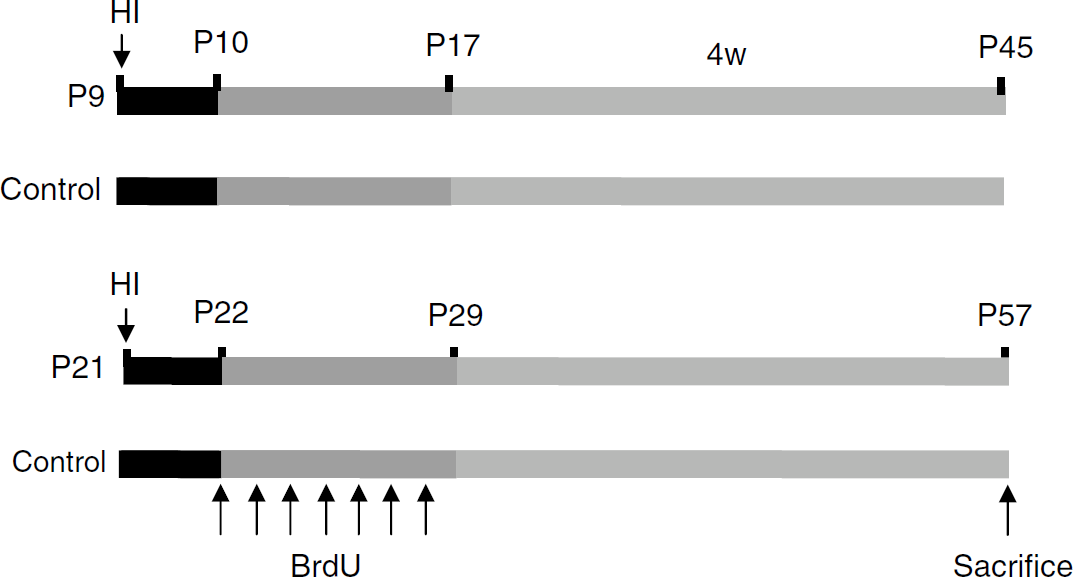
Experimental paradigm. Mice were subjected to hypoxia-ischemia (HI) at postnatal day 9 (P9) or P21 and received BrdU (intraperitoneal, 50 mg/kg) once daily for 7 days starting at 24 h after HI, as indicated. Control animals for each age group received BrdU but were not subjected to HI. All animals were killed 4 weeks after the last BrdU injection.
Immunohistochemistry
The animals were deeply anesthetized with 50 mg/mL phenobarbital and perfusion-fixed with 5% formaldehyde in 0.1 mol/L PBS, followed by immersion fixation in the same fixative for 24 h at 4°C. After dehydration with graded ethanol and xylene, the brains were paraffin-embedded and serial 5 μm coronal sections were cut and mounted on glass slides. Every 50th pair of sections was stained for BrdU. Antigen recovery was performed by boiling the sections in 10 mmol/L citrate buffer (pH 6.0) for 10 mins. Nonspecific binding was blocked for 30 mins with 4% donkey or horse serum in PBS. A monoclonal rat anti-BrdU primary antibody (1:100, 5 μg/mL; clone: BU1/75, Oxford Biotechnology Ltd. Oxfordshire, UK) was applied and incubated at 20°C for 60 mins, followed by a biotinylated donkey anti-rat IgG (H + L) secondary antibody (1:200, 5.5 μg/mL; Jackson ImmunoResearch Lab, PA, USA) for 60 mins at 20°C. To detect inflammation, sections were incubated with antibodies against macrophage/monocyte chemoattractant protein 1 (MCP-1; sc-1785, Santa Cruz Biotechnology, Santa Cruz, CA, USA, 4 μg/mL) or interleukin-18 (IL-18; sc-6179, Santa Cruz Biotechnology, Santa Cruz, CA, USA, 4 μg/mL) at 4°C over night, followed by a biotinylated horse anti-goat IgG (2 μg/mL) for 60 mins. Endogenous peroxidase activity was blocked with 3% H2O2 in PBS for 10 mins. Visualization was performed using Vectastain ABC Elite (Vector Laboratories, Burlingame, CA, USA) with 0.5 mg/mL 3,3′-diaminobenzidine enhanced with 15 mg/mL ammonium nickel sulfate, 2 mg/mL beta-D glucose, 0.4 mg/mL ammonium chloride and 0.01 mg/mL β-glucose oxidase (all from Sigma).
The phenotypes of BrdU-labeled cells were determined using double immunofluorescent staining. Antibodies against NeuN, APC, Iba1, or S100β were used to detect mature neurons, oligodendrocytes, microglia, and astrocytes, respectively. Antigen recovery, blocking and incubation with anti-BrdU were performed as above, followed by one of the following: mouse anti-NeuN monoclonal antibody (1:200, 5 μg/mL; clone: MAB377, Chemicon, Temecula, CA, USA), mouse anti-APC monoclonal antibody (1:100, 1 μg/mL; clone: CC-1; Calbiochem, Darmstadt, Germany), rabbit anti-Iba1 monoclonal antibody (1:1000, 0.5 μg/mL; Wako, Osaka, Japan) or rabbit anti-S100β (1:1000; Swant, Bellinzona, Switzerland) in PBS at 20°C for 60 mins. After washing, the sections were incubated with secondary antibodies, Alexa Fluor 488 donkey anti-rat IgG (H + L) combined with either Alexa Fluor 555 donkey anti-mouse IgG (H + L) or with Alexa Fluor 555 donkey anti-rabbit IgG (H + L) at 20°C for 60 mins. All secondary antibodies were from Molecular Probes, and were diluted 1:150. After washing, the sections were mounted using Vectashield mounting medium.
Cell Counting
Area contours were created, measured and BrdU-positive cells were counted in the CA (the pyramidal cell layer from CA1-4) and DG (the granule cell layer, including the subgranular zone) in every 50th section using the Stereo Investigator software (MicroBrightField, Magdeburg, Germany). Positive cells were expressed as average number per mm3.
Statistics
All data are expressed as mean ± s.d. An unpaired t-test was used when comparing two groups. ANOVA with Fisher's post hoc test was used when comparing more than two groups.
Results
Cell Genesis
Bromodeoxyuridine was used to label dividing cells during the first week after injury. Cells that were generated at the time of injection and survived 5 weeks after HI (4 weeks after the last injection) were detected in the CA and DG of both P9 and P21 control mice. Bromodeoxyuridine-positive cells were sparsely scattered throughout the whole CA area and were clustered in the subgranular zone of the dentate gyrus of the DG (Figures 2A and 2B). The number of BrdU-labeled cells in the CA area of P9 mice was 411 ± 74/mm3 59 ± 13/mm3 in P21 mice, sevenfold higher in P9 mice (P < 0.001) (Figure 3A). The number of BrdU-positive cells in the DG was 1531 ± 256/mm3 311 ± 82/mm3 for P9 and P21 mice, respectively, fivefold higher in the P9 mice (P < 0.001) (Figure 3B). This indicates that cell genesis was several-fold higher in the intact immature brain. After HI, BrdU incorporation was increased in the ipsilateral CA. The number of BrdU-labeled cells was 840 ± 187/mm3 for P9 and 587 ± 159/mm3 for P21, that is, increased approximately twofold and 10-fold in P9 and P21 mice, respectively, compared with age-matched controls (Figures 2C–2D and 3A). There were relatively more BrdU-positive cells distributed in the CA3 area for P9 mice, and in the CA1 area for P21 mice (Figures 2C–2D). In the contralateral, uninjured hemisphere, however, the number of BrdU-positive cells in the CA region was not significantly different from controls at either age (254 ± 29/mm3 in P9 mice and 124 ± 45/mm3 in P21 mice) (Figures 2E–2F and 3A). In the ipsilateral DG, the number of BrdU-labeled cells was 1366 ± 232/mm3 in P9 mice and 1417 ± 447/mm3 in P21 mice, that is no significant change in P9 HI mice compared with P9 controls, but a 4.5-fold increase in P21 HI mice compared with the P21 controls (Figures 2C–2D and 3B). In the contralateral DG, the numbers of BrdU-labeled cells were not significantly different from controls (1045 ± 251/mm3 in P9 mice and 372 ± 66/mm3 in P21 mice) (Figures 2E–2F and 3B). The numbers of BrdU-labeled cells in the CA region after HI at P9 during the first, second and third weeks, respectively, were 411 ± 74, 69 ± 17, and 51 ± 15/mm3 in the control mice, 839 ± 187, 157 ± 38, 88 ± 14/mm3 in the ipsilateral hemispheres and 254 ± 29, 99 ± 20, 50 ± 9/mm3 in the contralateral hemispheres. The numbers of BrdU-labeled cells in the DG after HI at P9 during the first, second and third weeks, respectively, were 1531 ± 256, 449 ± 47 and 266 ± 56/mm3 in the control mice, 1366 ± 232, 912 ± 297, 544 ± 105/mm3 in the ipsilateral hemispheres and 1045 ± 251, 555 ± 86, 299 ± 43/mm3 in the contralateral hemispheres.
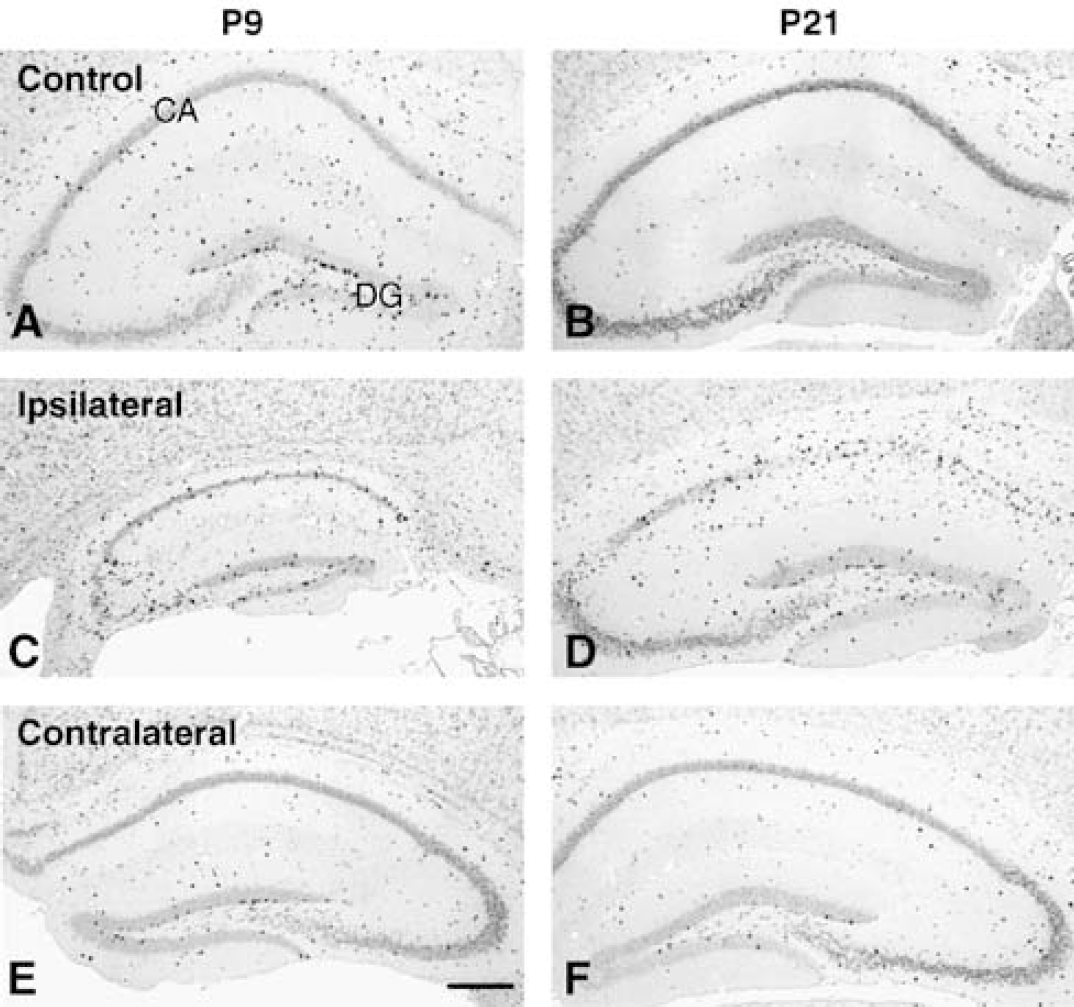
BrdU immunohistochemistry in the hippocampus. Representative BrdU immunostainings from the hippocampus of normal control mice (
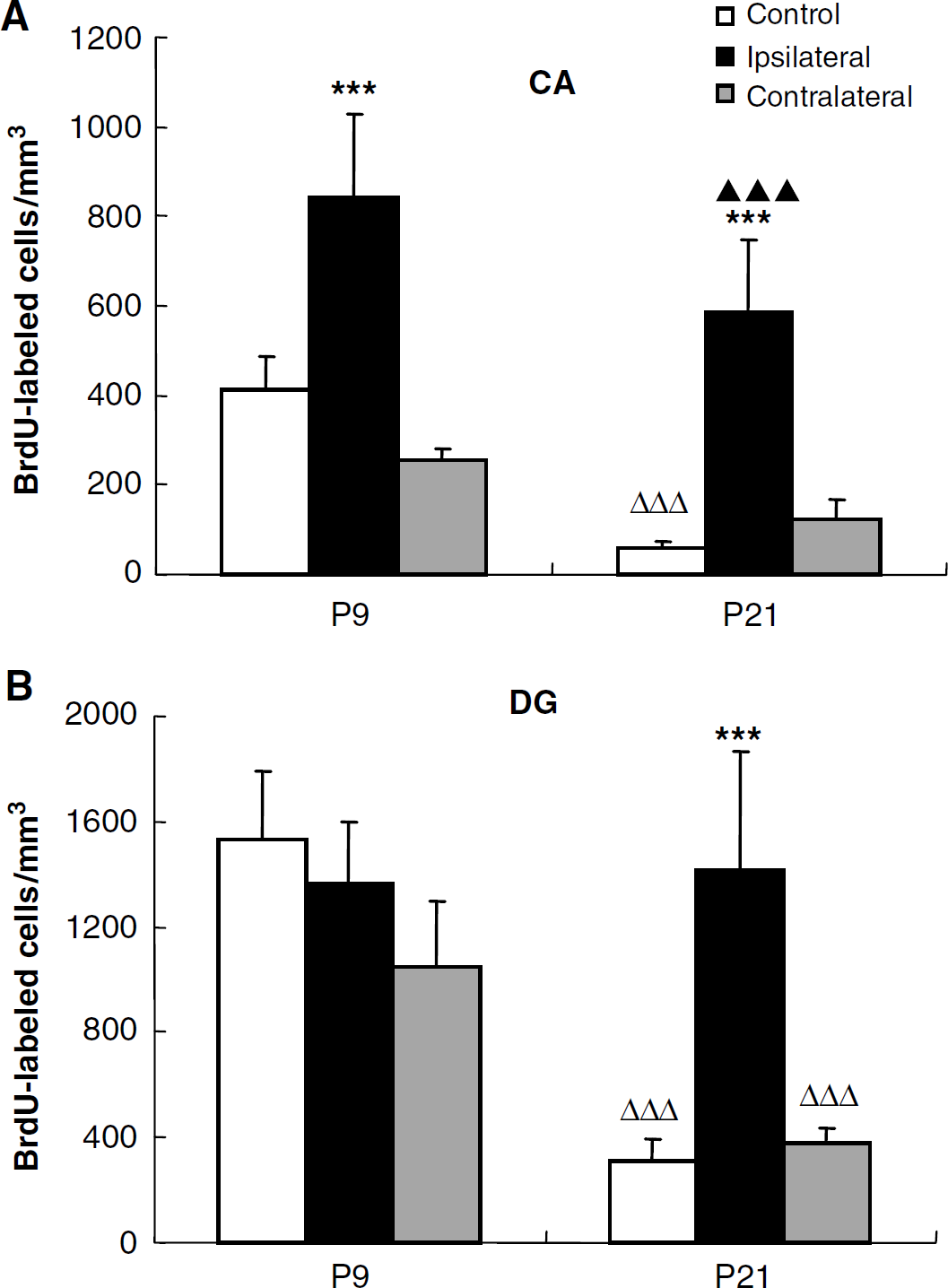
Quantification of BrdU-positive cells. Quantification of BrdU-positive cells in the CA (
Differentiation
To determine the lineage of cells born during the first week after HI, BrdU-labeled cells were also examined for expression of NeuN (marker of mature neurons), Iba1 (marker of microglia), APC (marker of oligodendrocytes) and S100β (marker of astrocytes). The numbers of cells double positive for BrdU and a lineage marker during the second and third weeks after HI at P9 were too few to allow reliable phenotyping.
Neuronal Differentiation
Substantial continued postnatal neurogenesis can be detected in the granule cell layer of the dentate gyrus (Figure 4A). In the DG of control mice, the number of BrdU/NeuN double positive cells in P9 mice was 2.5-fold higher than that of P21 mice (539 ± 145/mm3 212 ± 60/mm3 respectively, P < 0.001). After HI, the number of double positive cells did not change significantly in the ipsilateral DG of P9 mice (622 ± 132/mm3), but increased 2.5-fold in P21 mice (527 ± 166/mm3 P < 0.01) compared with controls. No significant changes were observed in the contralateral hemispheres (Figure 5B). As hippocampal pyramidal neurons are generated prenatally, we detected very few BrdU/NeuN double positive cells in the CA region of control mice (9 ± 6/mm3 at P9 and 2 ± 1/mm3 at P21, P < 0.05). After HI, the number was not significantly changed in the ipsilateral CA of P9 mice, but was increased 5.5-fold in P21 mice (11 ± 6/mm3 P < 0.05) compared with normal controls. No significant changes were observed in the contralateral hemispheres (Figure 5A).
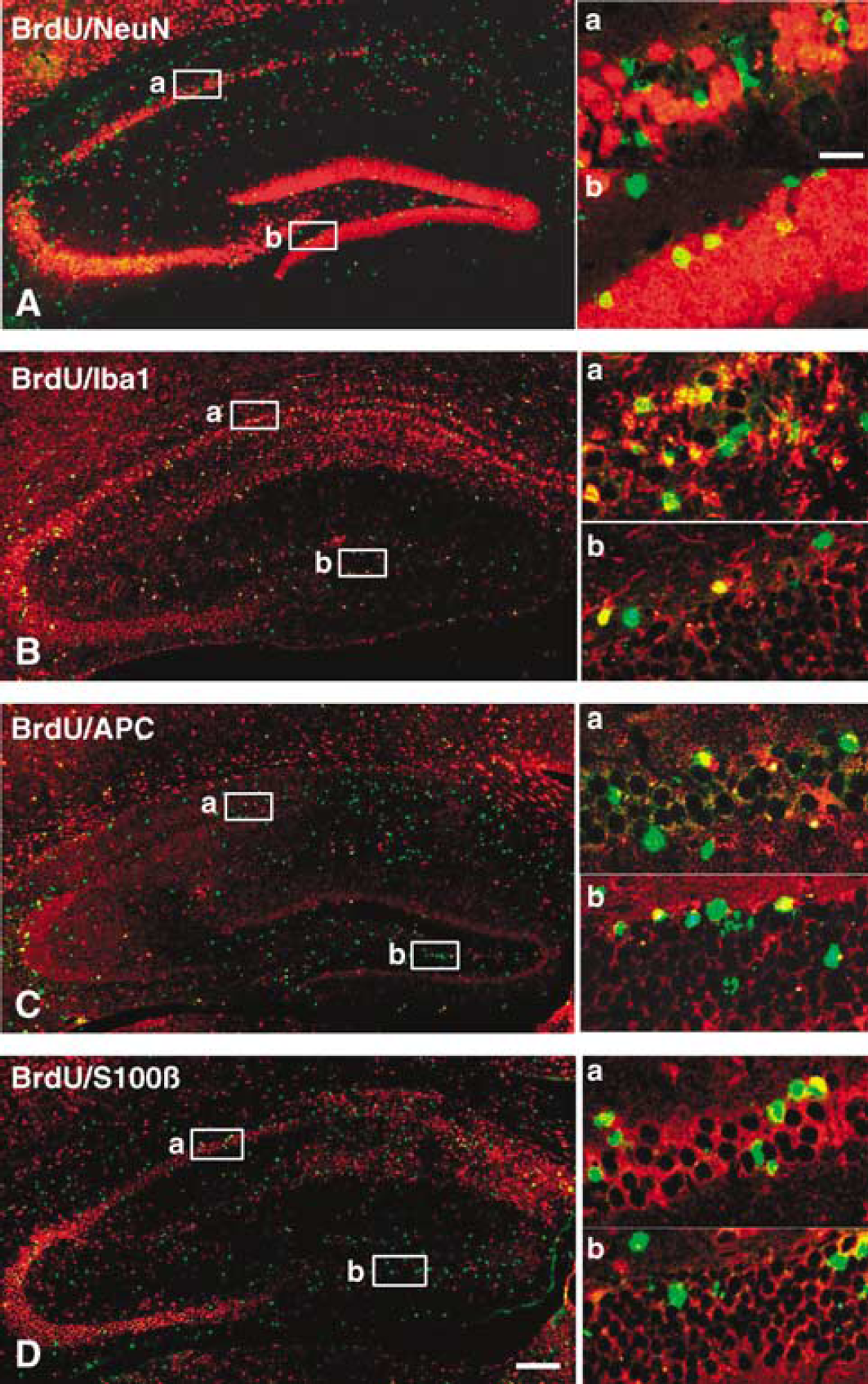
Phenotype of newly generated cells. Representative BrdU (green) and NeuN (
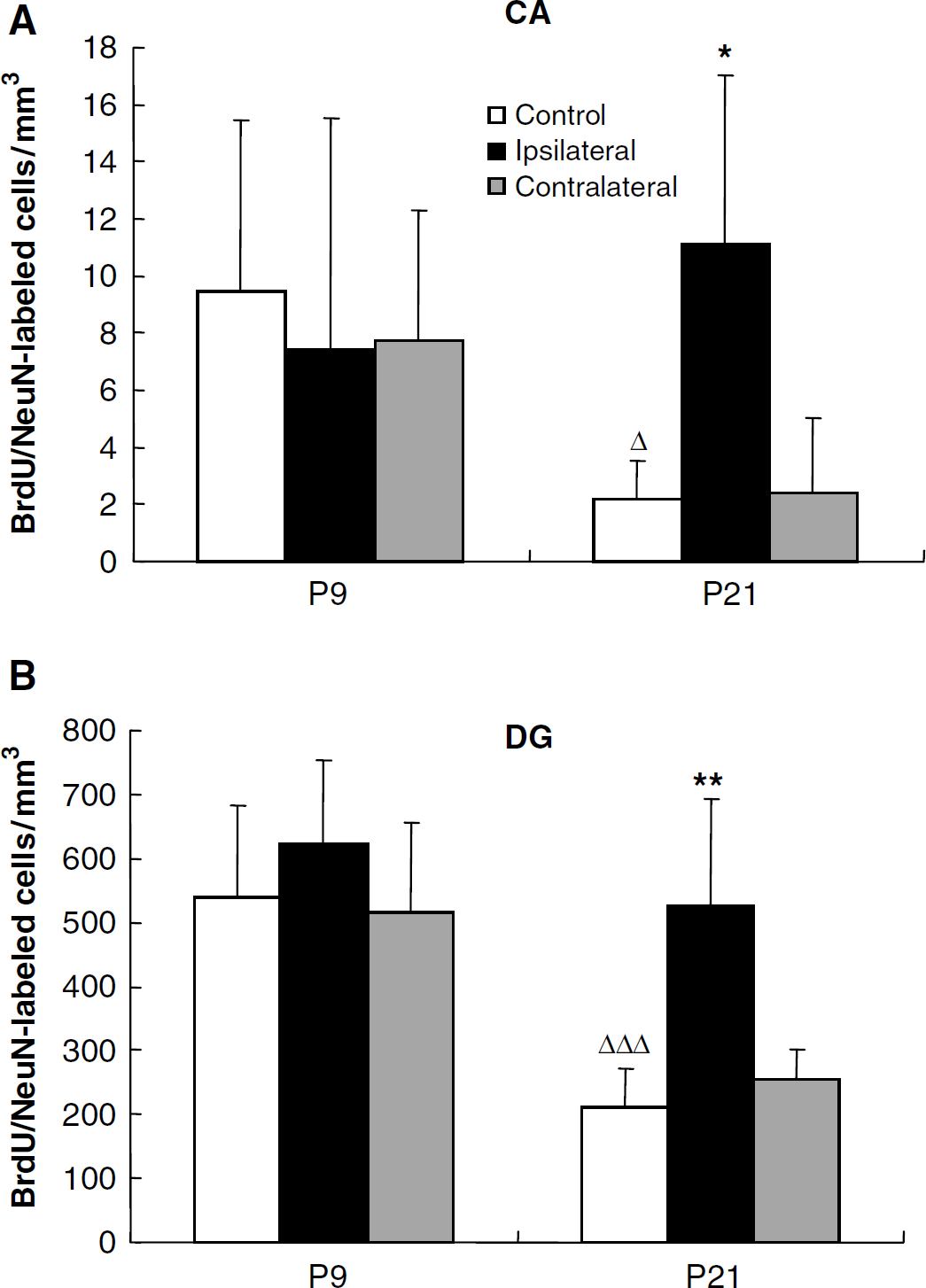
Quantification of BrdU/NeuN-double positive cells. Quantification of BrdU/NeuN double positive cells in the CA (
Glial Differentiation
Double labeling of BrdU and markers for three glial cell types (astrocytes – S100β, oligodendrocytes – APC, and microglia – Iba1) in the hippocampus is shown in Figures 4B–4D. In the CA region, the numbers of BrdU/Iba1 and BrdU/S100β double positive cells decreased significantly during normal brain development (Figures 4B, 4D, 6A and 6C). After HI at P9, only the numbers of newly generated S100β-positive cells increased in the ipsilateral CA (P < 0.001) compared with control animals (Figure 6). In P21 mice, however, all three types of glial cells increased dramatically in the ipsilateral CA region after HI, 113-fold for Iba1 (274 ± 92/mm3 P < 0.001), 3.7-fold for APC (90 ± 24/mm3 P < 0.001) and 8.5-fold for S100β (57 ± 19/mm3 P < 0.01) (Figure 6). The numbers of Iba1- and APC-positive cells were much higher in the ipsilateral CA region of P21 than P9 mice after HI (Figures 6A and 6B). Among the BrdU-positive cells in the ipsilateral CA of P21 mice, 46.7% expressed Iba1. No significant changes were observed in the contralateral hemispheres (Figure 6).
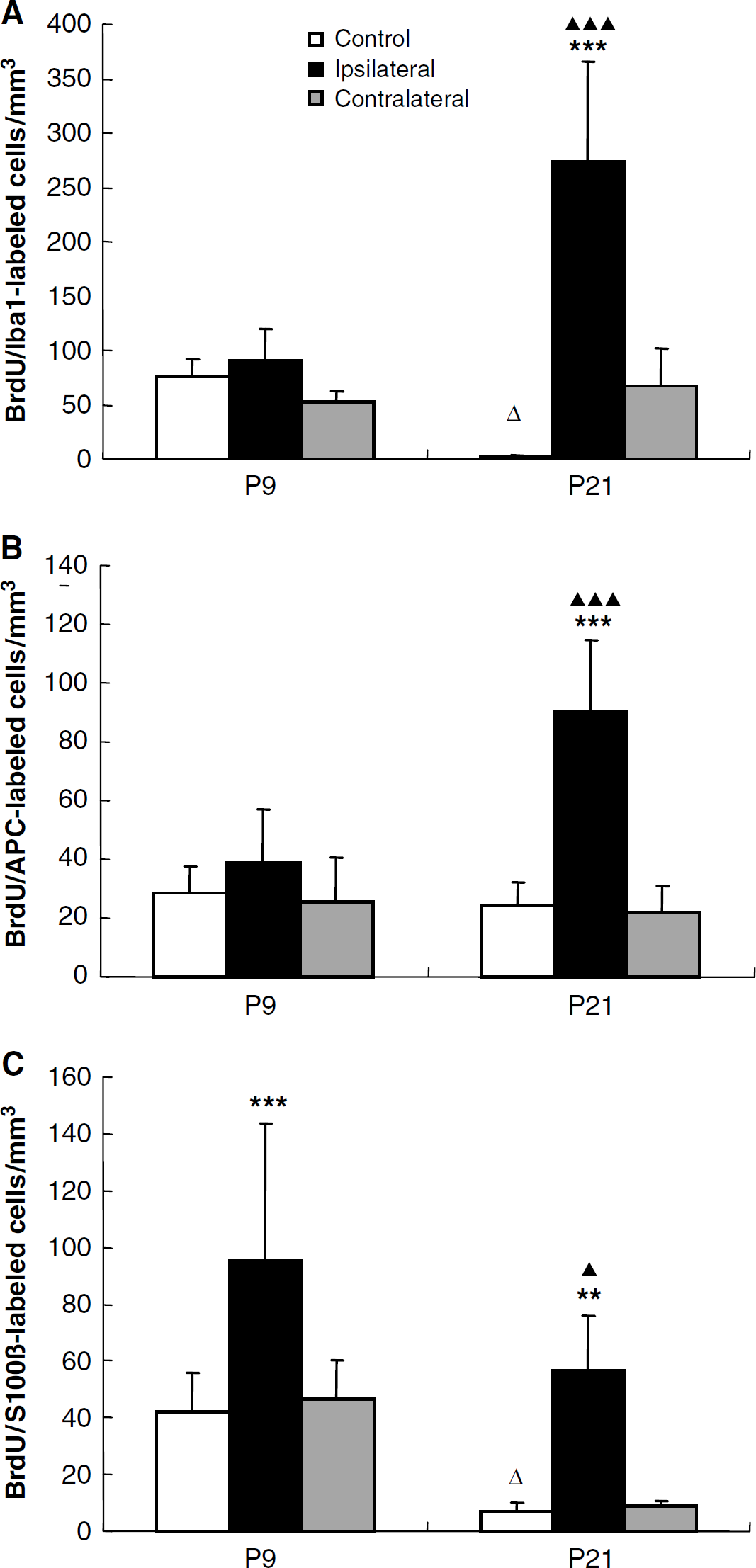
Quantification of BrdU/lba1, APC and S100β double positive cells in the CA region. Quantification of BrdU/lbal (
In the DC, the numbers of BrdU/APC and BrdU/S100β double positive cells decreased significantly during normal brain development (Figures 4 and 7). None of the three glial markers were significantly changed after HI in the P9 mice. In the P21 mice, however, BrdU/Iba1 double positive cells increased 8.2-fold (P < 0.001) compared with control mice (Figure 7A), constituting 15.4% of all BrdU-positive cells in this group. This indicates that generation of microglia is not significantly induced by HI in the immature DG but much more pronounced in the juvenile brain. No S100β double positive cells could be detected in the ipsilateral DG of P21 mice, indicating a dramatic decrease in the formation of new astrocytes in the DG during the first week after HI in P21 mice, unlike P9 mice, where there was no significant difference between control and ipsilateral DG (Figure 7C).
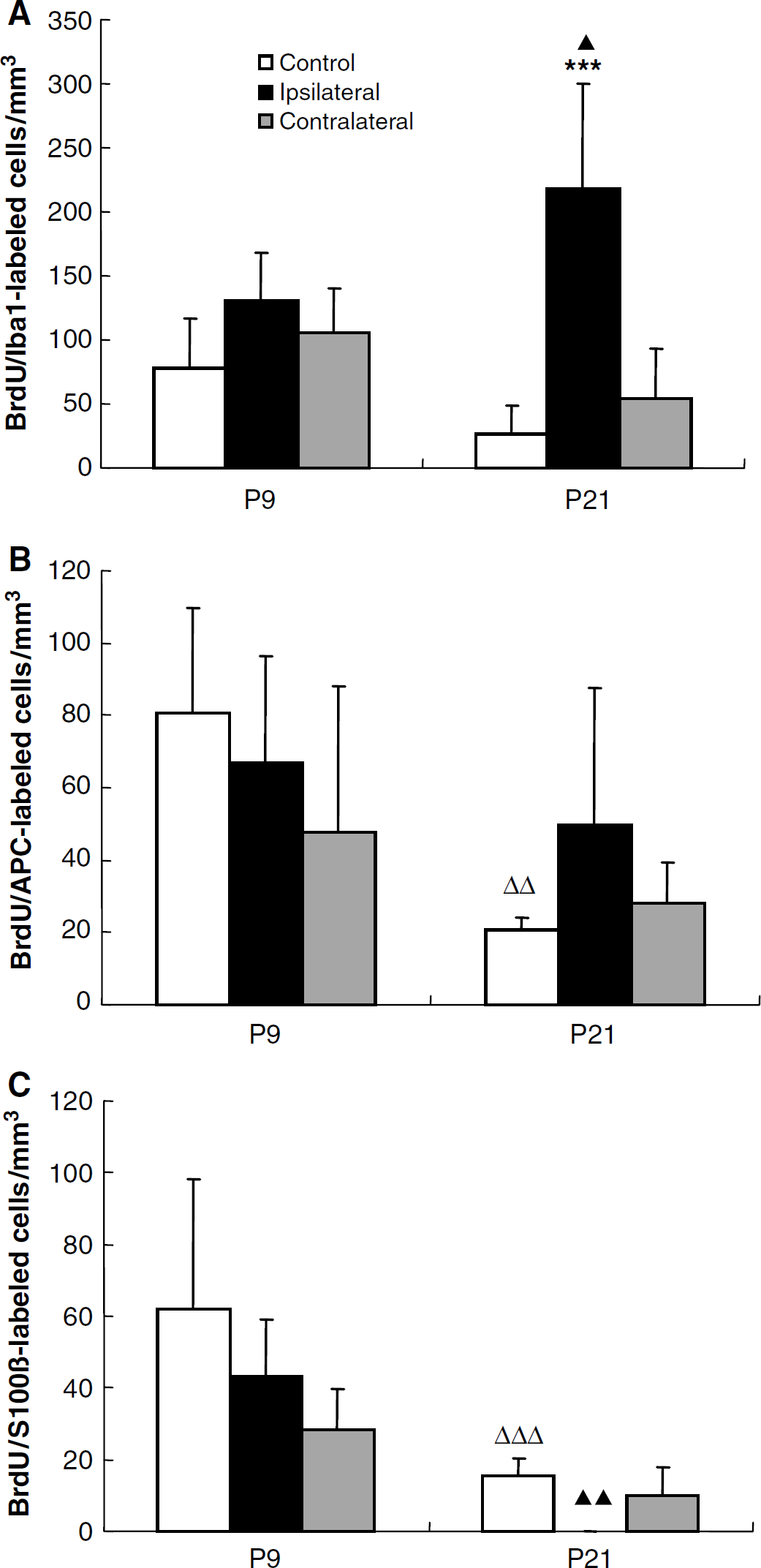
Quantification of BrdU/lbal, APC and S100β double positive cells in the DG. Quantification of BrdU/lba1 (
Cornu Ammonis of the Hippocampus and Dentate Gyrus of the Hippocampus Volumes Enable Estimation of the Total Numbers of Labeled Cells from Densities
The DG is still growing at P9 and has virtually reached its adult size by P21 (Fukuda et al, 2005; Schlessinger et al, 1975), whereas the CA is more or less fully developed already by P9 (Minkwitz and Holz 1975). When the growing DG is subjected to HI, the size reduction is the result of the combined effects of the acute injury and the lack of subsequent growth. When HI is induced at P9 the DG volume seems to be more affected than the CA volume, and the DG size reduction is more pronounced after HI at P9 than P21. The volumes of the CA and DG 5 weeks after HI are indicated in Table 1. When transforming the densities observed (cells per mm3) in Figure 5, for example, into total numbers of cells the unchanged neurogenesis in the P9 mice after HI actually turns into a decrease, whereas the increased neurogenesis observed in the P21 mice after HI still remains virtually unchanged.
The volumes of the cornu ammonis (CA) and dentate gyrus (DG) in the hippocampus of mice subjected to HI at P9 or P21 and killed 5 weeks after the insult
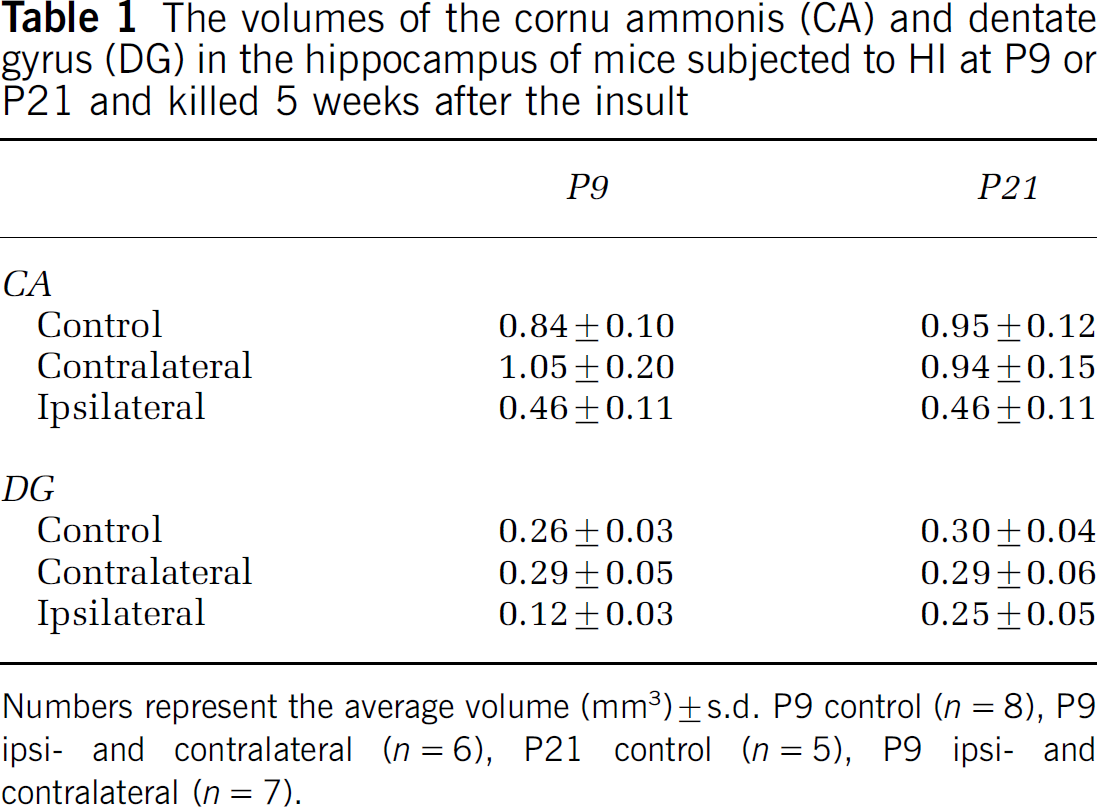
Numbers represent the average volume (mm3) ± s.d. P9 control (n = 8), P9 ipsi- and contralateral (n = 6), P21 control (n = 5), P9 ipsi- and contralateral (n = 7).
Proinflammatory Markers Accompany Microglial Proliferation
To investigate whether the increased microglial proliferation observed in the ipsilateral hemisphere after HI at P21 was accompanied by increased inflammation, the expression of two proinflammatory markers, the chemokines MCP-1 and the cytokine IL-18, was investigated in tissue sections from animals killed 3 days after HI. Animals subjected to HI at P21 displayed intense staining for MCP-1 and, less pronounced, IL-18 in endothelial cells and perivascular areas in the ischemic hippocampus (Figure 8A). Quantification of immunopositive cells in the CA and DG revealed twice as many cells positive for MCP-1 and IL-18 in the ipsilateral hemisphere of mice subjected to HI at P21 (118 and 164, respectively, P < 0.05) compared with P9 (55 and 83, respectively, P < 0.01) (Figure 8B).
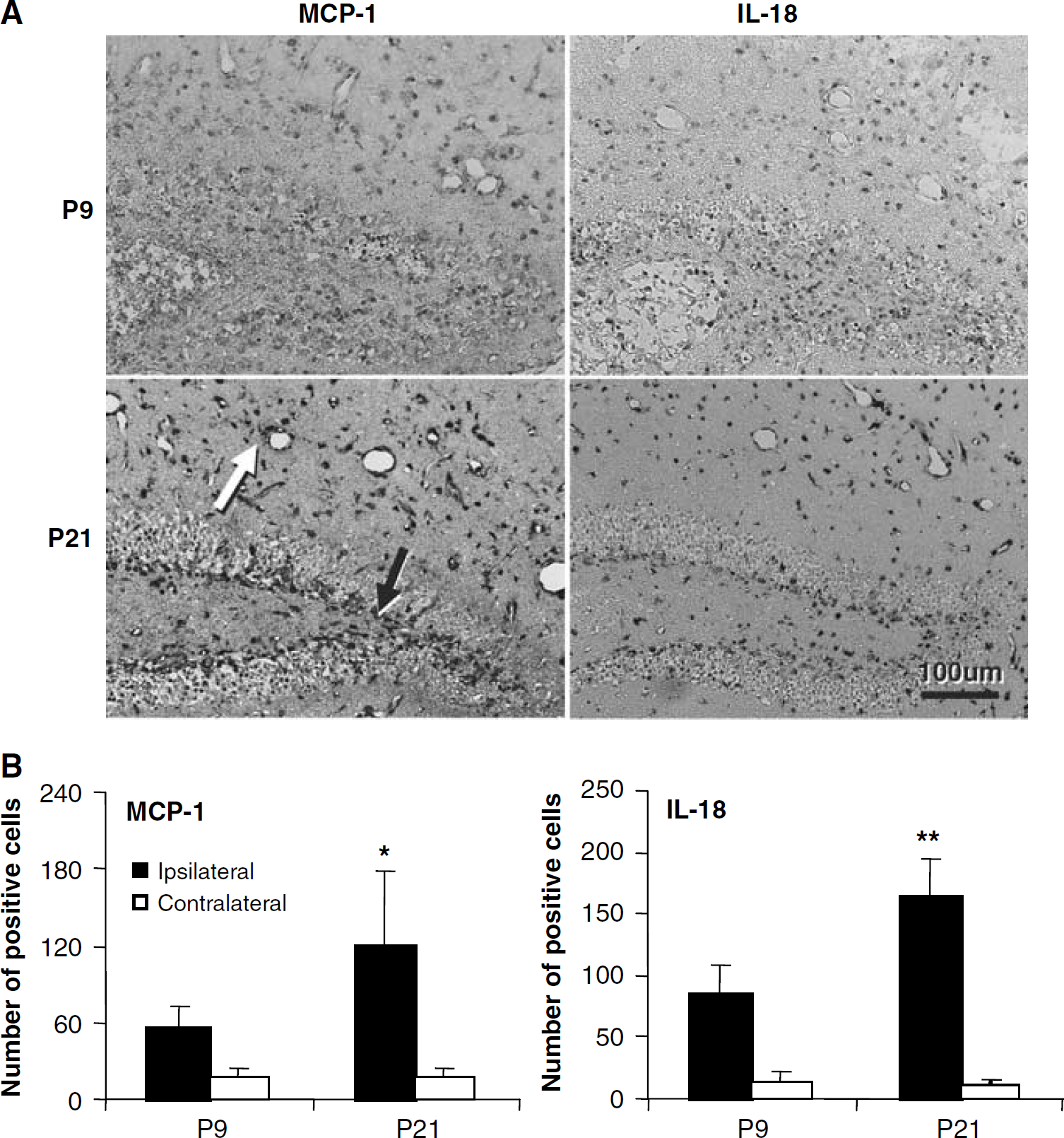
MCP-1 and IL-18 stainings 3 days after HI. (
Discussion
The current study investigated developmental differences in proliferation and differentiation of the mouse hippocampus after an insult using the well-characterized Rice-Vannucci model. This allowed us to evaluate developmental differences under normal conditions (control animals), after hypoxia (contralateral hemispheres) and after hypoxia-ischemia (HI) (ipsilateral hemispheres). We investigated the generation of new cells occurring during the first week after the insult, from P10 to P16 for the immature brain and from P22 to P28 for the juvenile brain. The total number of BrdU-labeled cells was far greater in the immature brain than in the juvenile, consistent with an age-related decline in cell proliferation and neurogenesis (Gray et al, 2002; Kuhn et al, 1996; Sun et al, 2005). To our surprise, the juvenile brain displayed a greater increase in BrdU labeling after HI than the immature brain. This was particularly evident in the DG, where proliferation and neurogenesis was not significantly changed at all in the immature brain but was several-fold increased in the juvenile brain. Interestingly, the increase after HI in the juvenile brains did not reach higher than the control level of the immature brains. This indicates that the immature brains are already working at the top of their proliferative, neurogenic capacity and that even though basal neurogenesis decreased with age, the injury-induced proliferation in the juvenile brains could not increase beyond the basal levels of the immature brain. To allow comparison between the total numbers of newborn cells instead of densities (cells/mm3), the densities shown in the figures can be multiplied with the volumes indicated in Table 1. Doing so, the differences in HI-induced neurogenesis between P9 and P21 is even greater. The unchanged neurogenesis in the DG after HI at P9 turns into a decrease, whereas the several-fold increase in neurogenesis observed after HI at P21 remains, only slightly reduced. Contrary to what has been generally assumed, these results indicate that the juvenile brain has a greater capacity for injury-induced regeneration than the immature brain. It remains to explore what the limiting factors are that prevent neurogenesis beyond this apparently maximal level at both developmental stages.
Other investigators have studied neurogenesis and gliogenesis originating from the subventricular zone after HI in the immature brain of rodents, describing transiently increased neurogenesis, but without long-lasting effects on neuron numbers (Bartley et al, 2005; Plane et al, 2004; Yang and Levison, 2006). These findings are consistent with our findings in the hippocampus. There are a few reports demonstrating increased proliferation (Scheepens et al, 2003), increased generation of oligodendrocytes (Zaidi et al, 2004) and neurons (Bartley et al, 2005) after HI in the immature rodent brain. Increased hippocampal neurogenesis after ischemic insults to the adult brain has been reported by several investigators in rodents and primates (Arvidsson et al, 2001; Jin et al, 2001; Kawai et al, 2004; Liu et al, 1998; Tonchev and Yamashima 2006; Yagita et al, 2001), but to our knowledge there are no studies making systematic comparison between early developmental levels. One challenge when studying neurogenesis using the Rice-Vannucci model is that the hippocampus is often severely damaged, so the hypoxia time has to be titrated to yield a mild insult. Several studies have indicated that neurogenesis in the hippocampus is restricted to the DG (Eriksson et al, 1998; Gage, 2000; Kuhn et al, 1996). However, other studies demonstrated newly born neurons in the CA1 pyramidal cell layer (Bendel et al, 2005; Schmidt and Reymann, 2002). Consistent with the latter studies, we found a few BrdU/NeuN double labeled in the CA region in both age groups, but the numbers were only a fraction of those found in the DG. The finding that the DG appeared less affected by HI at P21 than P9, as judged volume, might be attributed to decreased vulnerability with age, or lack of growth following the insult, as indicated above. It is also possible that increased neurogenesis after HI at P21 caused an initially decreased DG volume to increase during the weeks following the insult. Alternatively, the difference in volume might be explained by the appearance of other cells, like glia or phagocytes. To clarify this additional, shorter time points of evaluation need to be performed for comparison.
When comparing the numbers of cells labeled during the first, second and third weeks after HI at P9, it was clear that the overall level of proliferation was higher during the first week than the two consecutive weeks. However, the numbers of double-positive cells in the CA and DG labeled during the second and third weeks after HI were generally too few to allow reliable phenotyping. Nevertheless, this strongly indicates that the injury-induced proliferation, including neurogenesis, is most pronounced during the early phase after HI.
To detect the phenotype of newborn glial cells, we used BrdU double labeled with Iba1, APC, or S100β to identify microglia, oligodendrocytes, and astrocytes, respectively. Overall, gliogenesis decreased with age, except for oligodendrocytes in the CA. One of the striking differences was that whereas generation of new astrocytes in the juvenile brains increased more than eightfold in the CA region after HI, astrocyte proliferation was virtually abolished in the DG. The significance of this remains to be elucidated. Another striking finding was that microglia proliferation was much more pronounced in the juvenile than the immature brain after HI, consistent with other studies (Pforte et al, 2005; Zaidi et al, 2004), although systematic comparison using the same injury paradigm is lacking. The massive microglial proliferation occurring during the first week after HI in P21 mice was also accompanied by increased expression of at least two proinflammatory markers, MCP-1 and IL-18, indicating that the postischemic inflammation is more pronounced in juvenile, fully developed hippocampus.
In summary, we found an age-related decline in cell birth, including neurogenesis. Much to our surprise, the juvenile, virtually fully developed hippocampus displayed a greater increase in cell production than the immature hippocampus after HI, particularly in the DG, where proliferation and neurogenesis were not significantly changed at all in the immature brain. Injury-induced proliferation and neurogenesis in the juvenile hippocampus could not increase beyond the basal levels of the immature hippocampus. Contrary to what has been generally assumed, this indicates that the juvenile brain seems to have a greater capacity for regeneration after injury than the immature brain. Interestingly, postischemic inflammation, as judged by microglia proliferation and expression of proinflammatory cytokines, was also much more pronounced in the juvenile hippocampus. It remains to explore what the developmental factors are that regulate neurogenesis and inflammation in the brain, both under physiologic conditions and after HI.
Footnotes
Acknowledgements
The authors are grateful for the skillful technical assistance of Rita Grandér.