
Abstract
The aim of this study was to evaluate the long-term effects of lithium treatment on neonatal hypoxic-ischemic brain injury, inflammation, and neural stem/progenitor cell (NSPC) proliferation and survival. Nine-day-old male rats were subjected to unilateral hypoxia–ischemia (HI) and 2 mmol/kg lithium chloride was injected intraperitoneally immediately after the insult. Additional lithium injections, 1 mmol/kg, were administered at 24-hour intervals for 7 days. Animals were killed 6, 24, 72 hours, or 7 weeks after HI. Lithium reduced total tissue loss by 69%, from 89.4±14.6 mm3 in controls (n = 15) to 27.6 ±6.2 mm3 in lithium-treated animals (n = 14) 7 weeks after HI (P<0.001). Microglia activation was inhibited by lithium treatment, as judged by Iba-1 and galectin-3 immunostaining, and reduced interleukin-1β and CCL2 levels. Lithium increased progenitor, rather than stem cell, proliferation in both nonischemic and ischemic brains, as judged by 5-bromo-2-deoxyuridine labeling 24 and 72 hours as well as by phospho-histone H3 and brain lipid-binding protein labeling 7 weeks after HI. Lithium treatment also promoted survival of newborn NSPCs, without altering the relative levels of neuronal and astroglial differentiation. In summary, lithium conferred impressive, morphological long-term protection against neonatal HI, at least partly by inhibiting inflammation and promoting NSPC proliferation and survival.
Introduction
Perinatal asphyxia-induced brain injury is one of the most common causes of morbidity and mortality, both in term and preterm neonates, accounting for 23% of neonatal deaths globally (Lawn et al, 2005). Survivors of perinatal asphyxia suffer long-term neurologic disability and impairment such as cerebral palsy, mental retardation, and epilepsy. Although many neuroprotective strategies have appeared promising in animal models, most of them were not feasible or effective in human newborns. There are very few randomized controlled clinical trials that have demonstrated improved neurologic outcomes for term neonates, for example hypothermia or erythropoietin (Azzopardi et al, 2009; Zhu et al, 2009b). Thus, there is an urgent need to develop new brain-salvaging strategies that may be used either alone or in combination with hypothermia and/or erythropoietin.
Lithium is a mood stabilizer used in the treatment of bipolar disorder. Recent studies indicate that lithium has robust neuroprotective effects in a variety of paradigms, including ischemic brain injury (Ren et al, 2003; Xu et al, 2003; Yan et al, 2007b). It has been suggested that the therapeutic benefit is mediated by inhibition of glycogen synthase kinase-3β (GSK-3β) activity (Klein and Melton, 1996), which is critical for mitochondrial release of pro-apoptotic proteins (Lee et al, 2007). GSK-3β has multiple roles and inhibiting GSK-3β enhances hippocampal progenitor proliferation (Wexler et al, 2008). Previous studies reported that lithium could increase neural stem/progenitor cell (NSPC) proliferation without affecting differentiation (Son et al, 2003), whereas others found that lithium primarily promoted neuronal differentiation (Chen et al, 2000; Kim et al, 2004) in the adult rodent brain. In the immature brain, however, the basal NSPC proliferation rate is higher and relative differentiation levels are different (Qiu et al, 2007; Zhu et al, 2009c). Furthermore, the immature brain responds differently to injury (Zhu et al, 2005), including the neurogenic regions (Qiu et al, 2007; Zhu et al, 2009c), and modulating neurogenesis in the immature brain greatly affects the outcome of subsequent brain injury (Zhu et al, 2009a). As a result, it is tempting to speculate that lithium could have different effects in the immature than in the adult brain. We previously reported that postinsult lithium treatment after neonatal rat hypoxic-ischemic (HI) brain injury could inhibit neuronal cell death and afford short-term neuroprotection (Li et al, 2010). However, it is still unknown if lithium administration has a long-term effect on neonatal brain injury. In this study, we investigated potential long-term neuroprotective effects as well as effects on inflammation and neurogenesis. We found that postinsult lithium treatment afforded impressive long-term tissue protection. Lithium also promoted hippocampal NSPC proliferation and survival in both control and ischemic brains and reduced microglia proliferation and inflammation after HI. These results strongly suggest that the therapeutic potential of lithium in neonatal HI brain injury should be explored further.
Materials and methods
Hypoxia-ischemia
Postnatal day 9 (P9) male Wistar rat pups were purchased from B&K Universal AB. The rats were randomly assigned to either lithium chloride or vehicle (saline) treatment. Animals were anesthetized with isoflurane (5% for induction, 1.5% to 2.0% for maintenance) in a mixture of nitrous oxide and oxygen (1:1), the duration of anesthesia was < 5 minutes. The left common carotid artery was cut between double ligatures of prolene sutures (6.0). After the surgical procedure, the wound was infiltrated with lidocaine for local analgesia. The pups were returned to their cages for 1 hour and then placed in a chamber perfused with a humidified gas mixture (7.7% oxygen in nitrogen) for 50 minutes at 36°C (Zhu et al, 2003). Following hypoxic exposure, the pups were returned to their cages. Control pups were injected with either lithium chloride or vehicle but were not subjected to HI. All animal experimentation was approved by the Gothenburg Committee of the Swedish Animal Welfare Agency (application no. 145-2008).
Lithium Administration
Lithium chloride (Aldrich, St Louis, MO, USA) was dissolved in normal saline and injected at a dose of 2 mmol/kg intraperitoneally immediately after HI, followed by 1 mmol/kg injections at 24 hours intervals for 6 successive days, maximally. Animals were killed 6, 24, 72 hours, or 7 weeks after HI (Figure 1A).
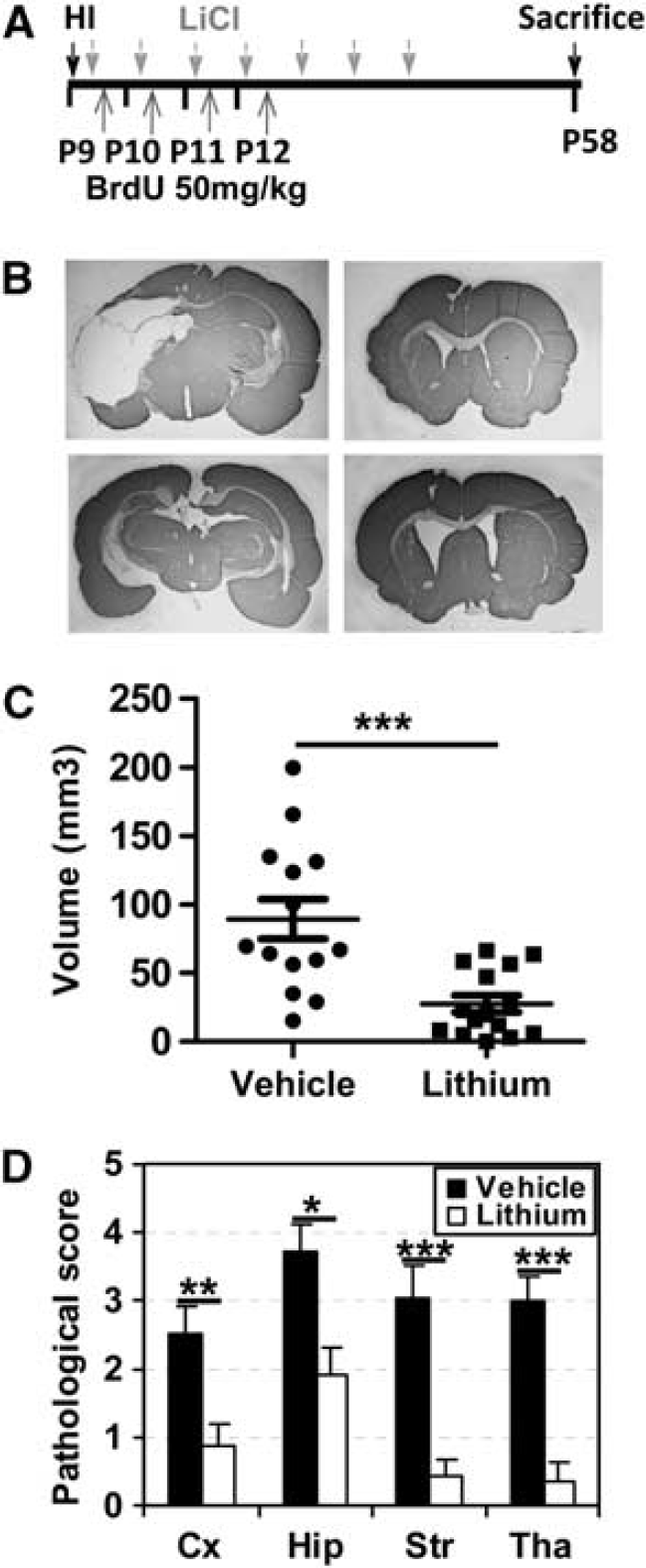
Lithium treatment reduced hypoxia–ischemia (HI) brain injury. (
5-Bromo-2-Deoxyuridine Administration
The thymidine analog 5-bromo-2-deoxyuridine (BrdU) (Roche, Mannheim, Germany, 5 mg/mL, dissolved in 0.9% saline) was freshly prepared before use and injected intraperitoneally (50 mg/kg), once daily for 4 successive days starting from 4 hours after HI.
Immunohistochemistry
The animals were deeply anesthetized with phenobarbital and perfusion fixed with 5% formaldehyde in 0.1 mol/L phosphate-buffered saline, followed by immersion fixation in the same fixative for 24 hours at 4°C. After dehydration with graded ethanol and xylene, the brains were paraffin-embedded, serial cut in 5 μm coronal sections, and mounted on glass slides. On the hippocampus level, every 50th section was stained. Monoclonal rat anti-BrdU (1:100, 5 μg/mL; clone: BU1/75; Oxford Biotechnology Ltd., Oxfordshire, UK), rabbit anti-phospho-histone H3 (PhH3) (serine 10) (1:500, 2 μg/mL; Upstate, Temecula, CA, USA), rabbit anti-Iba-1 antibody (1:1,000, 0.5 μg/mL; Wako, Osaka, Japan), rat anti-galectin-3 (1:100, 5 μg/mL; eBioscience, San Diego, CA, USA), rabbit anti-brain lipid-binding protein (BLBP) (2 μg/mL; Abcam, Cambridge, UK), monoclonal mouse anti-MAP2 (1:1,000, clone HM-2; Sigma, St Louis, MO, USA), primary antibody was applied and incubated at 20°C for 60 minutes, followed by biotinylated horse anti-mouse (1:200; Vector Laboratories, Burlingame, CA, USA), donkey anti-rat IgG (H + L) (1:200, 5.5 μg/mL; Jackson ImmunoResearch Laboratory, West Grove, PA, USA), goat anti-rabbit (1:200; Vector Laboratories) secondary antibody for 60 minutes at 20°C. Endogenous peroxidase activity was blocked with 3% H2O2 in phosphate-buffered saline for 10 minutes. Visualization was performed using Vectastain ABC Elite (Vector Laboratories), with 0.5 mg/mL 3,3′-diaminobenzidine enhanced with 15 mg/mL ammonium nickel sulfate, 2 mg/mL β-D glucose, 0.4mg/mL ammonium chloride, and 0.01 mg/mL β-glucose oxidase (all from Sigma). The number of positive cells was counted on every 50th section in the dentate gyrus (DG) of the hippocampus and at least six sections were counted. The total number of positive cells in each DG was calculated using unbiased stereological counting techniques (StereoInvestigator, Micro-BrightField Inc., Magdeburg, Germany).
The phenotype of BrdU-labeled cells was determined using antibodies against neuronal nuclei (NeuN), S100β, or Iba-1 to detect mature neurons, astrocytes, or microglia, respectively. Antigen recovery was performed as above, followed by incubation with rat anti-BrdU (1:100, 5 μg/mL; clone: BU1/75; Oxford Biotechnology Ltd.) together with mouse anti-NeuN monoclonal antibody (1:200, 5 μg/mL; clone: MAB377; Chemicon, Temecula, CA, USA) and rabbit anti-S100β (1:1,000; Swant, Bellinzona, Switzerland) or rabbit anti-Iba-1 (1:500; Wako) in phosphate-buffered saline at 20°C for 60 minutes. For the double labeling of Iba-1 and galectin-3, the rabbit anti-Iba-1 (1:500; Wako) and rat anti-galectin-3 (1:100; eBioscience) were incubated at 20°C for 60 minutes. After washing, the sections were incubated with secondary antibodies, Alexa Fluor 488 donkey anti-rat IgG (H + L), combined with Alexa 555 donkey anti-mouse IgG (H + L) and Alexa 647 donkey anti-rabbit IgG (H + L) or Alexa 555 donkey anti-rabbit IgG (H + L) at 20°C for 60 minutes. For Iba-1 and galectin-3, Alexa Fluor 488 donkey anti-rat IgG (H + L), combined with Alexa 555 donkey anti-rabbit IgG (H + L) was used. All secondary antibodies were from Jackson ImmunoResearch Laboratory and were diluted 1:1,000. After washing, the sections were mounted using Vectashield mounting medium.
Injury Evaluation
Brain injury was evaluated using total tissue loss volume and neuropathological scoring. The MAP2-positive area in each section was measured using Micro Image (Olympus, Japan). The volume was calculated from the MAP2-positive areas according to the Cavalieri principle, using the following formula: V = ΣA × P × T, where V is the total volume, ΣA is the sum of area measurements, P is the inverse of the sampling fraction, and T is the section thickness. The total tissue loss was calculated as the MAP2-positive volume in the contralateral hemisphere minus the MAP2-positive volume in the ipsilateral hemisphere. The neuropathological score for the cortex, hippocampus, striatum, and thalamus was assessed as described previously (Zhu et al, 2005). Briefly, the cortical injury was graded from 0 to 4, 0 being no observable injury and 4 indicating confluent infarction encompassing most of the cerebral cortex. The damage in the hippocampus, striatum, and thalamus was assessed both with respect to hypotrophy (0 to 3) and observable cell injury/infarction (0 to 3), resulting in a neuropathological score for each brain region (0 to 6).
Cell Counting
In every 50th section, area contours were drawn and measured. To assess proliferation, BrdU, BLBP, and PhH3- positive cells were counted in the subgranular zone. For the late time point (7 weeks after HI), BrdU-positive cells were counted in the granule cell layer (GCL), including the subgranular zone, using the Stereo Investigator software (MicroBrightField). For analysis of newly generated neurons and astrocytes, at least 50 BrdU-positive cells per GCL were analyzed for coexpression of NeuN or S100b using a confocal laser scanning microscope (Leica TCS SP2, Wetzlar, Germany) (Zhu et al, 2010). The ratio was multiplied with the total number of BrdU-positive cells generated from the stereology procedure to calculate the total number of new neurons and astrocytes per DG.
Enzyme-Linked Immunosorbent Assay
Animals were killed by decapitation at 6, 24, or 72 hours after HI. The brains were rapidly dissected out on a bed of ice. The parietal cortex (including the hippocampus) was dissected out from each hemisphere and ice-cold isolation buffer was added (15 mmol/L Tris-HCl, pH 7.6, 320 mmol/L sucrose, 1 mmol/L DTT, 1 mmol/L MgCl2, 3 mmol/L EDTA-K, 0.5% protease inhibitor cocktail (P8340; Sigma), 1% phosphatase inhibitor cocktail 1 (P2850; Sigma), 1% phosphatase inhibitor cocktail 2 (P5726; Sigma), and 2.5 μmol/L cyclosporin A). Homogenization was performed gently by hand in a 2-mL glass/glass homogenizer (Merck Euro Laboratory, Dorset, UK) using, sequentially, two different pestles with a total clearance of 0.12 and 0.05 mm, respectively (10 strokes each). The homogenates were centrifuged at 800 g for 10 minutes at 4°C. The supernatant from the first centrifugation was further centrifuged at 9,200g for 15 minutes at 4°C, producing a crude cytosolic fraction in the supernatant. The supernatants were collected and used for the enzyme-linked immunosorbent assay. For the assay, a rat interleukin (IL)-1β assay kit (RLB00; R&D Systems, Minneapolis, MN, USA) and rat monocyte chemoattractant protein-1 (MCP-1) assay kit (IBL17176; IBL, Gunma, Japan) was used. The standard was diluted in buffer and the assay was performed as recommended by the manufacturer.
Statistics
All data were expressed as mean ± s.e.m. Student's t-test was used when comparing tissue loss, enzyme-linked immunosorbent assay results, or the numbers of immunopositive cells between two different groups. The Mann-Whitney U-test was used for comparing pathological scores. Significance level was assigned at P<0.05.
Results
Lithium Treatment Reduced Hypoxia-Ischemia Brain Injury
In vehicle-treated control rats, brain injury is well detectable 7 weeks after HI (Figure 1B). Lithium treatment initiated shortly after hypoxia was highly efficient in reducing brain injury. The overall tissue loss was 89.4 ± 14.6 mm3 in the vehicle group and 27.6 ±6.2 mm3 in the lithium-treated group, which corresponds to a 69% reduction after lithium treatment (P=0.0005) (Figure 1C). Moreover, lithium treatment reduced signs of injury in all affected brain areas including the cortex, hippocampus, striatum, and thalamus (Figure 1D).
Lithium Treatment Decreased Microglia Activation and Inflammation After Hypoxia-Ischemia
Under normal conditions, microglia were scattered throughout the brain, as indicated by Iba-1 immunostaining (Figures 2A and 2B), and this pattern was similar in lithium- and vehicle-treated brains in the cortex (Figure 2C) and the hippocampus (Figure 2D). The number of Iba-1-labeled cells in the cortex increased ∼ 10-fold 72 hours after HI, but only fivefold in lithium-treated animals (Figure 2C). After 7 weeks, the number of microglia decreased to about one third of the number seen in the cortex 72 hours after HI. In the cornu ammonis region, however, the number of Iba-1-positive cells was higher after 7 weeks than after 72 hours after HI, suggesting that proliferation of microglia or recruitment of monocytes occurs in this region during late recovery. This increase was more pronounced in the vehicle-treated group than in lithium-treated rats (Figure 2D). Galectin-3, a marker of pathologically activated microglia, was observed only in injured areas 72 hours after HI (Figure 3A). The number of galectin-3-positive cells was lower after lithium treatment in all observed areas, except the cornu ammonis region. Double labeling of Iba-1 and galectin-3 showed that approximately one third (cornu ammonis region) to half (cortex) of all Iba-1-positive cells also were galectin-3-positive 72 hours after HI (Figure 3B). During late recovery, 7 weeks after HI, galectin-3-positive cells could still only be observed in injured areas, displaying a sixfold decrease compared with the numbers observed 72 hours after HI (Figures 3A and 3C). The relative difference was the same 72 hours and 7 weeks after HI, a threefold to fourfold higher number of galectin-3-positive cells in the cortex of vehicle-treated brains (Figure 3C). Based on previous studies of inflammatory markers involved in neonatal HI brain injury (Qiu et al, 2007; Zhu et al, 2009c), we measured the protein concentrations of IL-1β and chemokine (C-C motif) ligand 2 (MCP-1) in the cortex. Both markers were increased as early as 6 hours after HI. Lithium treatment reduced IL-1β and chemokine (C-C motif) ligand 2 protein expression at 6 and 24 hours after HI (Figure 4).
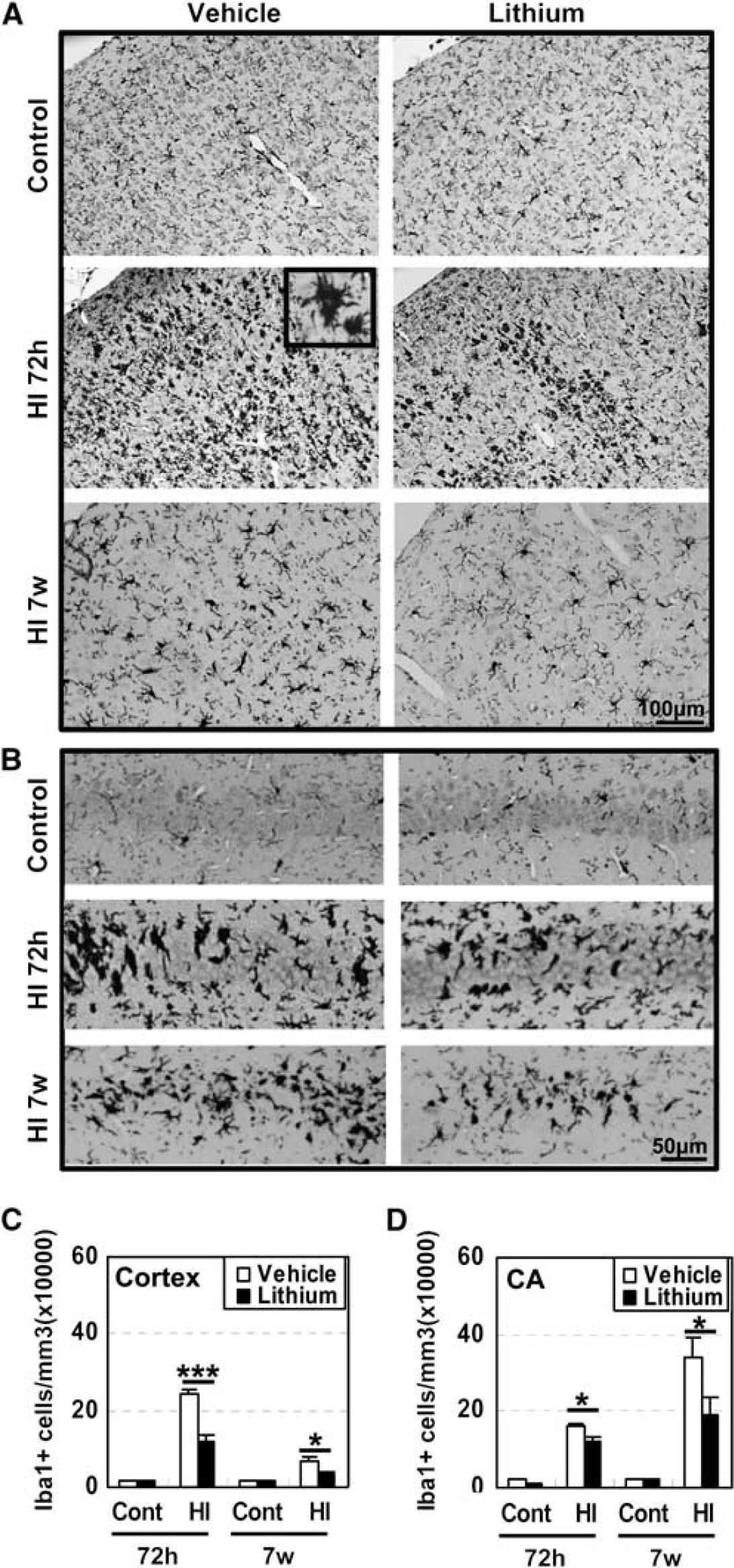
Microglia (Iba-1) immunostaining 72 hours after hypoxia–ischemia (HI). (
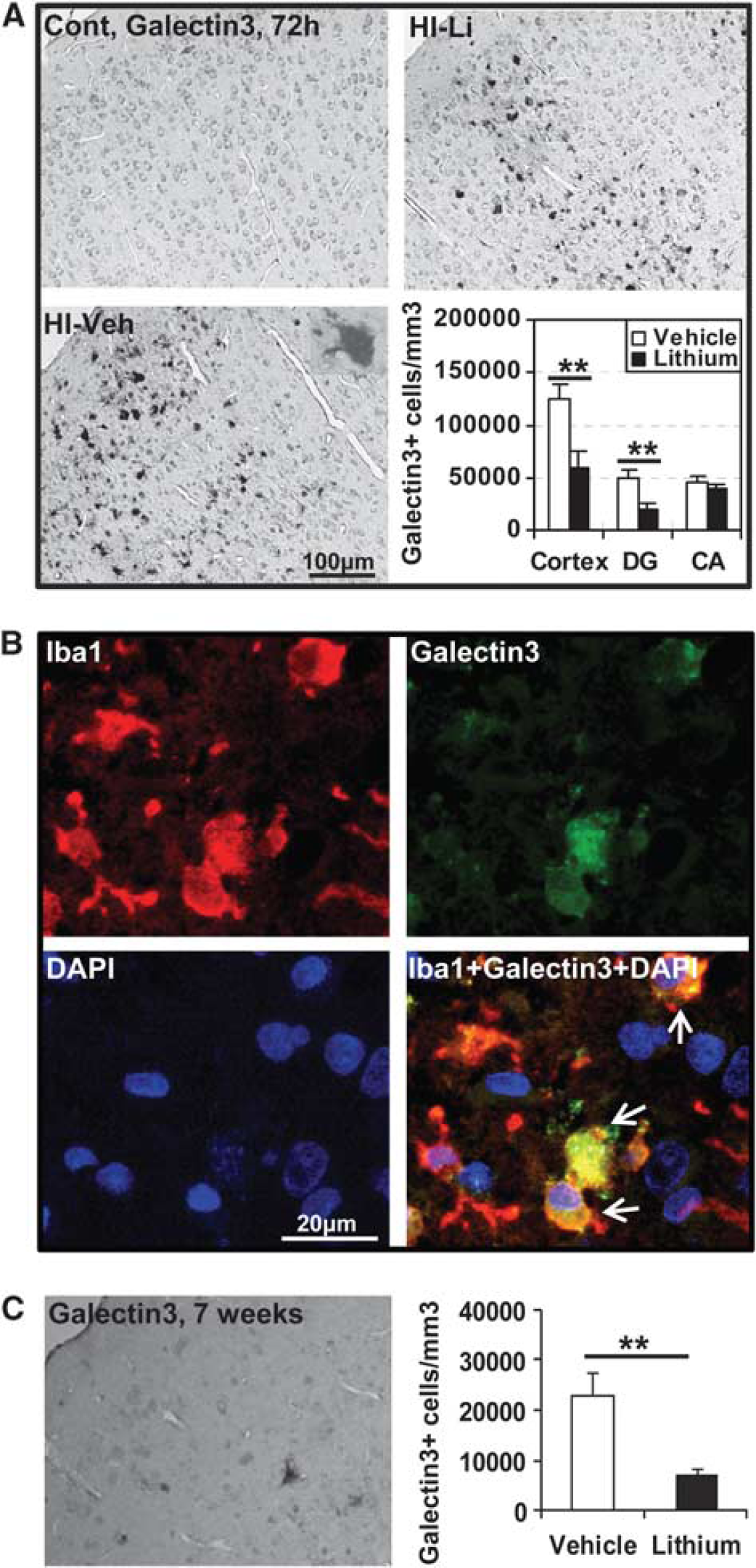
Galectin-3 immunostaining 72 hours after hypoxia-ischemia (HI). (
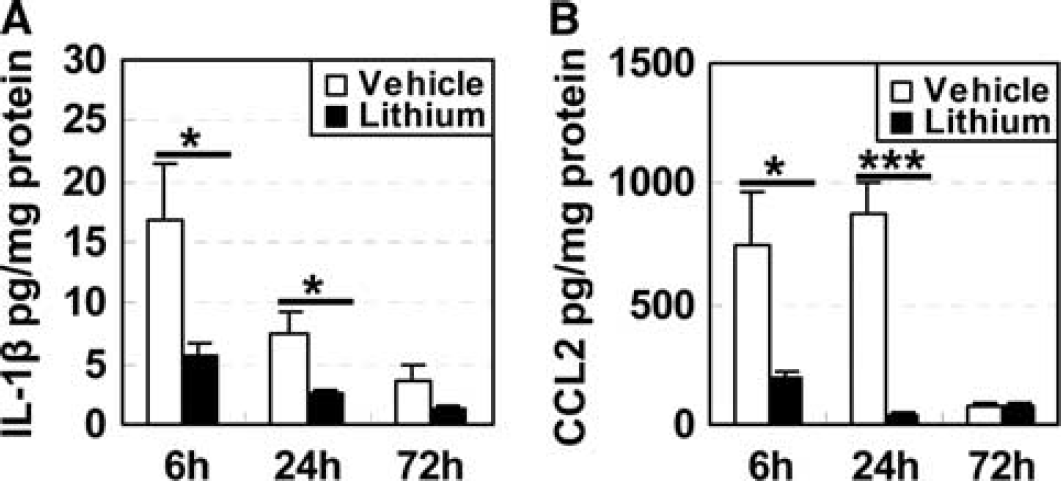
Interleukin (IL)-1 β and MCP-1 enzyme-linked immunosorbent assay (ELISA). (
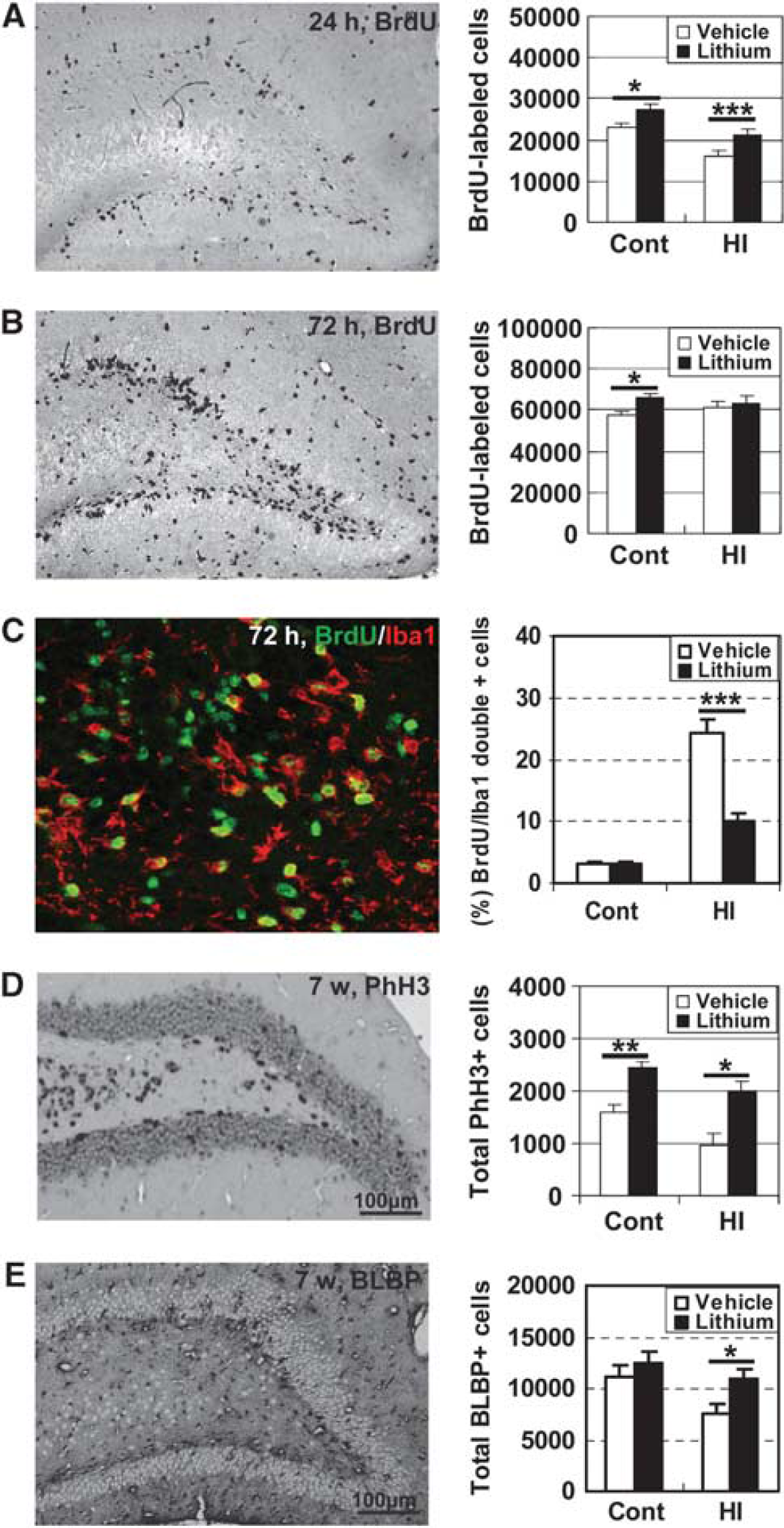
Lithium Treatment Increased Both Proliferation and Survival of Neural Stem and Progenitor Cells
Cell proliferation in the DG was assessed 24 and 72 hours after HI by BrdU labeling and 7 weeks after HI by PhH3 staining. Lithium treatment increased the number of BrdU-labeled cells in the subgranular zone of the DG both in normal control brains and in the ipsilateral hemisphere 24 hours after HI (Figure 5A). Also, 72 hours after HI (after three lithium injections), the number of BrdU-labeled cells was increased in normal control brains, but the number was not different between vehicle- and lithium-treated brains after HI at this time point (Figure 5B), probably because lithium treatment largely prevented the microglial proliferation observed 72 hours after HI (Figure 5C). A long-lasting, even more pronounced, effect on NSPC proliferation by lithium was observed at 7 weeks by PhH3 staining, demonstrating 47% and 104% higher numbers of PhH3-positive cells in the nonischemic and postischemic lithium-treated animals, respectively (Figure 5D). The number of undifferentiated, BLBP-positive cells was not altered in the normal control brains after lithium treatment, but was 44% higher after HI in lithium-treated animals (Figure 5E). This indicates that the pool of undifferentiated cells was unchanged in nonischemic animals but that lithium prevented the HI-induced loss of BLBP-positive cells (Figure 5E).
Cell proliferation in the dentate gyrus (DG) after hypoxia-ischemia (HI). (
The total number of surviving BrdU-labeled cells per DG was 24,382 ±2,278 after 7 weeks in the nonischemic, lithium-treated group, 36.6% higher than that of the vehicle-treated group (17,846 ± 1,989) (P<0.05). The number of surviving BrdU-labeled cells in the HI animals was only one tenth of the controls (2,333 ± 712), but this increased more than threefold after lithium treatment (7,328 ± 765) (P =0.0023) (Figure 6A). The pheno-typing of newly generated cells surviving 7 weeks later showed that most of them developed into neurons, as judged by BrdU/NeuN double labeling (Figure 6B), and neurogenesis was higher in lithium-treated animals, in both nonischemic control (39%) and HI brains (124%) (Figure 6B). The number of newly generated astrocytes, as indicated by BrdU/S100 double labeling, was also higher in the lithium-treated animals, in both control (71%) and HI brains (176%) (Figure 6B). However, the ratios of neurogenesis and astrogliogenesis were not different between the lithium- and vehicle-treated groups (data not shown).
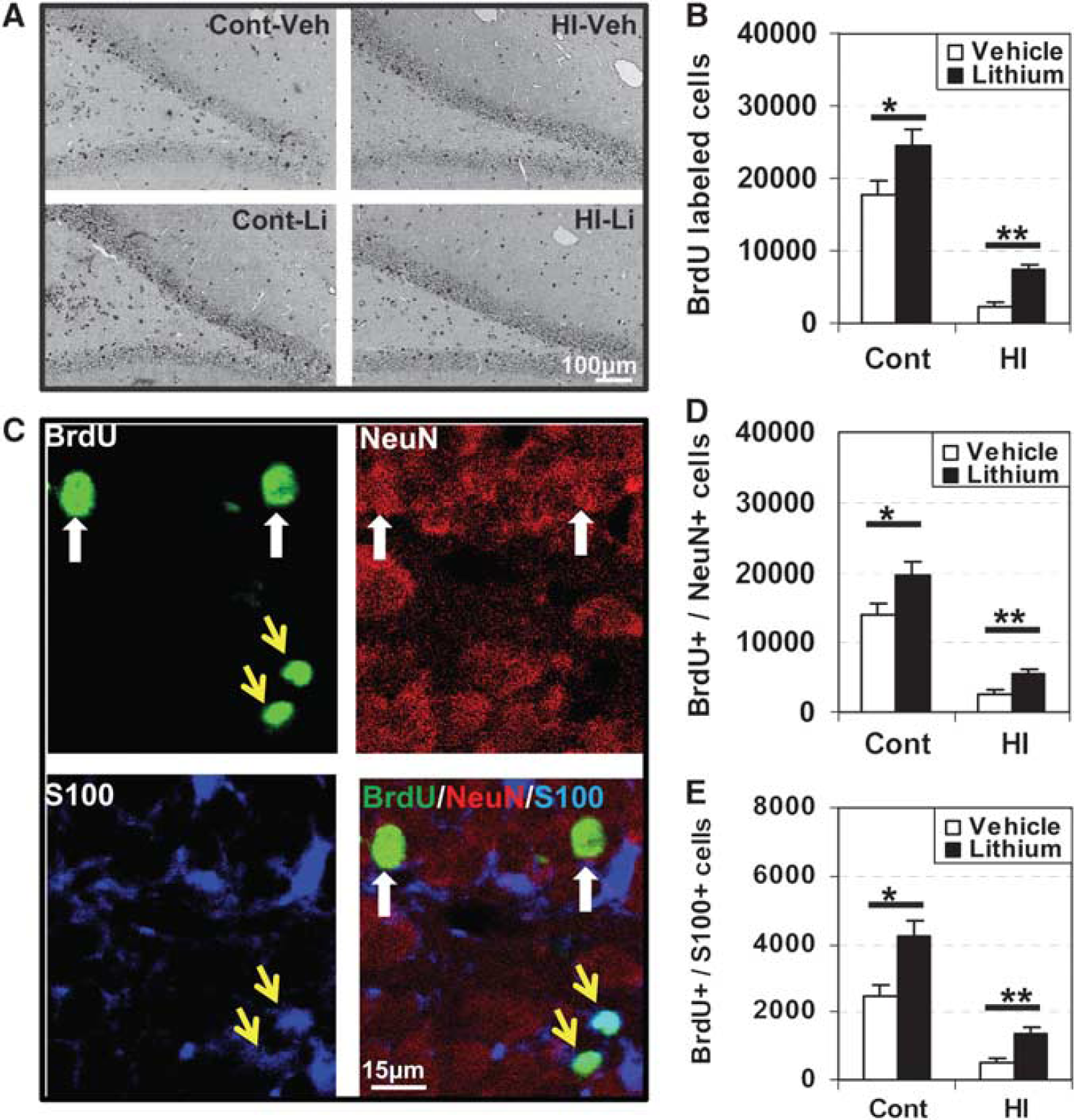
Proliferated cells survival and differentiation in dentate gyrus (DG) at 7 weeks after hypoxia–ischemia (HI). (
Discussion
Lithium is an established drug used in the treatment of bipolar disorder. Recent in vivo and in vitro studies have implied that lithium has neuroprotective properties, reducing ischemic infarction as well as functional deficits (Ren et al, 2003; Xu et al, 2003; Yan et al, 2007b). The cellular mechanisms underlying lithium's protective effects are not completely understood, and two independent aspects may be involved: the prevention of apoptotic cell death (Chen and Chuang, 1999; Li et al, 2010) and the stimulation of compensatory neurogenesis/cell migration (Su et al, 2007). Early after ischemia, lithium may reduce apoptotic death to diminish abnormal behavioral performance (Ren et al, 2003; Xu et al, 2003; Yan et al, 2007b). At later stages, increased generation and survival of newborn cells in the neurogenic regions may also contribute to the beneficial effects of lithium (Su et al, 2007, 2009). Considering that neuronal death in the immature brain, unlike the adult brain, is predominantly apoptotic (Zhu et al, 2005) and that ischemic injury to the immature brain, again unlike the situation in the mature brain, does not increase hippocampal proliferation and neurogenesis (Qiu et al, 2007), lithium appears to have a promising profile for the immature brain. Earlier, we found that lithium treatment reduced apoptosis, autophagy, and brain injury in the immature brain 3 days after HI (Li et al, 2010), but the protection found in this study, 7 weeks after HI, was even more profound (69% compared with 43%), indicating that the long-lasting effects on NSPC proliferation might provide additional benefit.
A number of studies indicate that lithium appears to act on proliferation, but the signal transduction pathway responsible for this effect has not been identified (Su et al, 2007; Yan et al, 2007a). It has been reported that lithium stimulates progenitor proliferation in cultured brain neurons (Hashimoto et al, 2003), shifts the phenotype from predominantly astrocytic to neuronal (Kim et al, 2004), and inhibits apoptosis of mouse NSPCs (Shimomura et al, 2003). In vivo, lithium enhances proliferation of DG progenitor cells (Chen et al, 2000; Son et al, 2003). In this study, we found that lithium increased hippocampal proliferation also in the immature brain and that the effect was long-lasting (at least 7 weeks) as indicated by PhH3 labeling, both in controls and after HI. Cells in the subgranular zone expressing BLBP are undifferentiated, radial glia-like neural stem cells (Suh et al, 2007). The number of BLBP-positive cells was not increased by lithium treatment in control animals, indicating that lithium stimulated proliferation of progenitor cells rather than the asymmetric division of stem cells. Wexler reported that lithium enhances cell proliferation by activating the canonical Wnt pathway (Wexler et al, 2008). GSK-3β is a critical mediator of the Wnt/β-catenin signaling pathway and lithium inhibits the activation of GSK-3β directly and indirectly (Chalecka-Franaszek and Chuang, 1999; Li et al, 2010). It has been reported that lithium upregulates the generation and survival of newborn cells in the hippocampus through the ERK pathway (Yan et al, 2007a). In line with these observations, our previous study shows that lithium prevents dephosphorylation of P-ERK1/2 and P-GSK-3β after HI (Li et al, 2010), but the precise mechanisms whereby lithium increases NSPC proliferation and survival need to be further investigated.
Several studies have reported that lithium not only increases NSPC proliferation, but also increases neuronal differentiation both in vitro and in vivo (Kim et al, 2004; Su et al, 2007). Possible mechanisms favoring neuronal differentiation may involve the BDNF pathway (Su et al, 2009), ERK- and CREB- dependent pathways (Kim et al, 2004), as well as the Wnt pathway (Wexler et al, 2008). However, in this study, the proportion of neuronal and astrocyte differentiation was not different between the lithium and vehicle groups. This is consistent with one report on lithium effects on transient global cerebral ischemia in adult rats (Yan et al, 2007a), but different from another report where lithium increased neurogenesis and decreased astrogliogenesis (Kim et al, 2004). The reason for this discrepancy may depend on differences in the age of the animals, the timing of BrdU injection, and the type or degree of injury.
There is increasing evidence that inflammation has an important role in brain injury (Qiu et al, 2007; Wang et al, 2009; Zhu et al, 2009a, c ). An acute and a prolonged inflammatory response occurred after brain injury, characterized by the production of inflammatory cytokines, leukocyte infiltration in the brain, as well as activation of resident glial cells (Hedtjarn et al, 2004). All these events may contribute to ischemic brain injury (Zhu et al, 2009c). There is substantial evidence demonstrating that activated microglia have the potential of releasing cytotoxic molecules, including proinflammatory cytokines or chemokines, such as IL-1β and CCL2 (MCP-1) (Doverhag et al, 2010). In addition, attenuation of the inflammatory response and microglial activation has conferred neuroprotection in neonatal hypoxic-ischemic brain injury (Doverhag et al, 2010). We have previously demonstrated increased microglia formation after HI in neonatal and juvenile mice, both in the hippocampus (Qiu et al, 2007) as well as in the cortex and striatum (Zhu et al, 2009c), concurrent with increased expression of the pro-inflammatory markers CCL2 and IL-18 (Qiu et al, 2007; Zhu et al, 2009c). In this study, we found that whereas the Iba-1-positive cells were scattered throughout the brain, galectin-3-positive microglia were located only in the injured areas, thereby serving as a marker of pathologically activated microglia, distinguishing them from constitutively activated microglia, which are abundant in the immature, growing, remodeling brain (Kalm et al, 2009). We demonstrate that signs of neuroinflammation, as judged by galectin-3-positive microglia, are present as long as 7 weeks after the insult. Evidence suggests that neuroinflammation is a negative regulator of neurogenesis (Monje et al, 2003). Furthermore, treatment with minocycline after HI reduced microglia activation and injury in the immature brain (Arvin et al, 2002; Cai et al, 2006; Fox et al, 2005) although conflicting reports exist (Tsuji et al, 2004). Further work is needed to elucidate the roles of microglia and consequences of inflammation after HI, but injury-induced inflammation may be an important target for therapeutic interventions. In the current study, 7 days treatment with lithium reduced microglia activation and the expression of pro-inflammatory markers, but it remains to be shown if this was secondary to the reduced injury or an independent, antiinflammatory effect. In either case, it is likely that reduced inflammation helped promote NSPC proliferation and survival.
Lithium has not been approved nor proposed for clinical applications targeting the developing brain. The potentially harmful side effects of lithium depend on the dose, duration of treatment, and age of the patient at the time of treatment. The doses used in this study are in the same range as those used for adult humans (Bowden et al, 2005). We did not find any difference in the body weights of the vehicle- and lithium-treated rats. This suggests that the dose of lithium used is safe for young rats. Lithium has been used for the treatment of bipolar disorder in children (Chang et al, 2005). It is unknown whether lithium at the dose used in this study may have a negative impact on brain development. However, before lithium administration could be recommended as a potential treatment against neonatal brain injury, a more complete safety evaluation is needed.
Footnotes
The authors declare no conflict of interest.