
Abstract
The brain inflammatory response induced by stroke contributes to cell death and impairs neurogenesis. Poly(ADP-ribose) polymerase-1 (PARP-1) is a coactivator of the transcription factor NF-κB and required for NF-κB-mediated inflammatory responses. Here we evaluated PARP inhibition as a means of suppressing post-stroke inflammation and improving outcome after stroke. Rats were subjected to bilateral carotid occlusion-reperfusion, and treatment with the PARP inhibitor N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethylacetamide (PJ34) was begun 48 h later. PJ34 was found to rapidly suppress the ischemia-induced microglial activation and astrogliosis. Behavioral tests performed 6 to 8 weeks after ischemia showed deficits in spatial memory and learning that were lessened by the PJ34 treatment. Immunohistochemical evaluation of hippocampus at 8 weeks after ischemia showed increased neuronal density in CA1 layer of PJ34-treated animals relative to vehicle-treated animals. Bromodeoxyuridine labeling showed formation of new neurons in hippocampal CA1 area in PJ34-treated animals, but not in vehicle-treated animals. Together, these results suggest that treatment with a PARP inhibitor for several days after ischemia enhances long-term neuronal survival and neurogenesis by reducing inflammation.
Introduction
Poly(ADP-ribose) polymerase-1 (PARP-1; also known as poly(ADP-ribose) synthetase or ADP-ribosyl transferase) functions as a sensor of DNA damage. When activated by DNA strand breaks, the enzyme consumes NAD+ to generate ADP-ribose polymers on several nuclear acceptor proteins involved in DNA repair and genome integrity (Jagtap and Szabo, 2005). Extensive activation of PARP-1 can promote cell death in ischemia and other conditions through a process involving NAD+ depletion and mitochondrial release of apoptosis inducing factor (Jagtap and Szabo, 2005). By an entirely separate mechanism, PARP-1 may also contribute to injury after ischemia through its influence on the cellular inflammatory response (Hamby et al, 2007; Moroni, 2008; Oliver et al, 1999).
PARP-1 influences inflammation by interacting with the transcription factor NF-κB. PARP-1 enhances the NF-κB activity at promoter sites by facilitating the binding of NF-κB subunits to DNA and by promoting transcription complex formation (Hassa and Hottiger, 2002; Kraus and Lis, 2003). NFκB drives expression of several pro-inflammatory cytokines and proteases including iNOS, ICAM-1, and TNFα (O'Neill and Kaltschmidt, 1997). The expression of these proteins is down-regulated by PARP-1 inhibition (Chiarugi and Moskowitz, 2003; Oliver et al, 1999; Zingarelli et al, 1998). PARP-1 inhibition has also been shown to suppress NF-κB-dependent gene transcription in microglia (Kauppinen et al, 2008) and to suppress the morphologic changes associated with microglial activation (Chiarugi and Moskowitz, 2003; Kauppinen and Swanson, 2005; Ullrich et al, 2001).
Ischemic stroke induces an inflammatory response, which contributes to cell death and impairs neurogenesis (Ekdahl et al, 2003; Hoehn et al, 2005). Prior studies have identified minocycline as an effective agent for suppressing brain inflammation (Koistinaho et al, 2004). The mechanism of this antiinflammatory effect has not been established but may be due to the potent inhibition of PARP-1 by minocycline (Alano et al, 2006). The aim of the present study was to determine whether a PARP inhibitor, N-(6-oxo-5,6-dihydrophenanthridin-2-yl)- N,N-dimethylacetamide (PJ34), could suppress brain inflammation and thereby enhance long-term recovery and neurogenesis in a rat model of forebrain ischemia. The inflammatory response triggered by brain ischemia takes many hours to develop, thus, providing a relatively long time window in which to initiate a therapeutic intervention (Dirnagl et al, 1999). Here we initiated treatment with PJ34 at 48 h after ischemia, a delay that precluded direct neuroprotective effects. The delayed treatment with PJ34 was found to suppress microglial and astrocyte activation within 24 h, and evaluations performed at 6 to 8 weeks after ischemia showed increased neurogenesis, increased pyramidal neuron density in hippocampal CA1 region, and improved spatial memory in the rats treated with PJ34.
Materials and methods
Materials
Sprague–Dawley rats were obtained from Simonsen Laboratories and were housed two per cage under conditions of constant temperature, light from 6:00 AM to 6:00 PM, and free access to food and water. N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethylacetamide (PJ34) was obtained from Inotek (Beverly, MA). Other materials were obtained from Sigma-Aldrich, except where noted.
Transient Forebrain Ischemia
Transient forebrain ischemia was produced with bilateral common carotid artery occlusion and hypotension as described by Smith et al (1984), under a protocol approved by the San Francisco Veterans Affairs Medical Center animal studies committee. Animals were anesthetized with 2% isoflurane in closed plastic chamber and maintained with 30% O2/70% N2O delivered via face mask. Rectal temperature was monitored and maintained at 37°C±0.5°C throughout the surgical procedure with thermal blanket. The common carotid arteries were exposed through a midline skin incision and carefully freed from the adjacent vagus nerve. The exposed common carotid arteries were occluded by microvascular clips and systemic mean arterial pressure was lowered to 40±5mmHg by withdrawing blood (7 to 10 mL) from the femoral artery into a heparinized syringe maintained at 37°C. Ten minutes later, the clips were removed from the common carotid arteries and the blood was reintroduced to the femoral artery. The incisions were closed, the femoral artery catheter was removed, and anesthesia was discontinued. Animals exhibiting seizures after reperfusion were killed (21 of 120 rats) and not assigned to control or drug-treatment groups.
Treatment Schedule
PJ34 was administered at a dose of 15 mg/kg by intraperitoneal injection, in a volume of 1.0mL of 0.9% saline. Treatment was initiated at 48 h after ischemia and subsequent injections were given at 12-h intervals for 1 to 7 days (Figure 1). The control group received injections of saline vehicle alone. Bromodeoxyuridine (BrdU), a synthetic thymidine analog that incorporates into DNA during the S-phase of the cell cycle, was used to label dividing cells. Bromodeoxyuridine was administered at a dose of 50 mg/kg by intra-peritoneal injection starting at day 5 after ischemia and subsequent injections were given at 12-h intervals until day 9. Rats were killed at the designated time points, and brains were perfusion fixed with 4% formaldehyde, except the rats designated for hematoxylin-eosin staining.
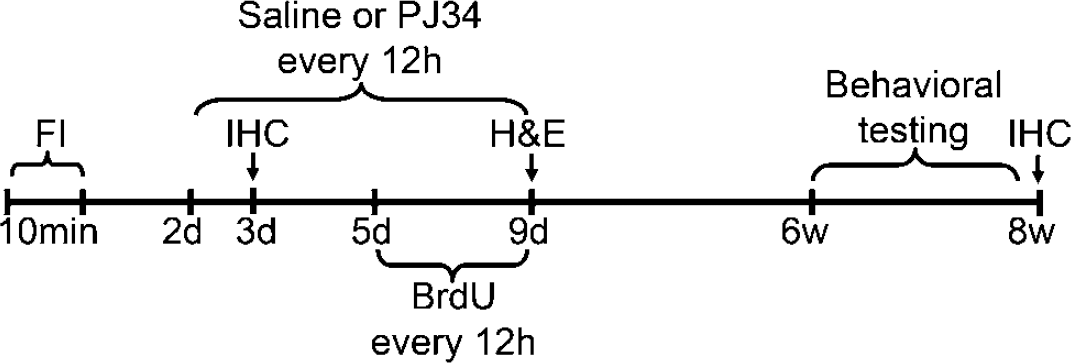
Experimental design. PJ34 and saline i.p. injections were started 48 h after 10 mins forebrain ischemia (FI) and continued every 12 h for 7 days. Bromodeoxyuridine injections were given twice daily between days 5 and 9 after ischemia. Animals were killed 3 days or 8 weeks after ischemia for immunohistochemistry (IHC), or at 9 days after ischemia for hematoxylin-eosin staining (H&E). Behavioral testing was performed at 6 to 8 weeks after ischemia.
Assessment of Neuronal Death
Acute ischemia-induced neuronal death was evaluated 9 days after ischemia. Brains were harvested without perfusion and were frozen with powdered dry ice. Coronal 20 µm frozen sections were prepared, fixed in 70% ethanol, and stained by hematoxylin-eosin staining, as described previously (Hamby et al, 2007). Five coronal sections were analyzed from each animal, beginning 4.0mm posterior to bregma and spaced 200 µm apart. An observer blinded to the treatment conditions counted the number of eosinophilic neurons in the hippocampal CA1 area in both hemispheres of each rat. Data from each animal were expressed as the total number of eosinophilic neurons in both the right and the left CA1 cell fields in the five coronal sections.
Behavioral Tests
The spontaneous open field activity test and the Morris water maze test were performed 6 to 8 weeks after ischemia, as described previously (Morris, 1984). Rats were housed individually in the testing room beginning 3 days before testing to minimize the effects of social influences on behavior. As a first step, spontaneous open field activity was assessed to identify any locomotor differences that could bias the measures of learning and memory. Rats were individually placed in a plexiglass enclosure (40 × 40 inches) equipped with two rows of infrared photocell panels interfaced with a computer. The lower row detected horizontal exploratory activity, and the higher row detected vertical exploratory activity or rearing. Light path breaks were recorded and assessed for measurements of the number of movements, active times, path length, and rearing events. On each of three consecutive days, open field activity was recorded for 10 mins after an initial 1 min adaptation period.
Spatial learning and memory were tested by the Morris Water Maze test, using a circular pool (180cm in diameter, 50cm deep) filled with opaque water of constant temperature (24°C). The rats were trained first to locate a platform with a visible cue (days 1 to 2), and then to locate a hidden platform (days 3 to 5) using large spatial cues in the room. The platform was moved to a new quadrant in each session during the visible platform cue training. The platform remained in the same quadrant throughout all the sessions during hidden platform training. The rats received two training sessions per day for five consecutive days. Each session consisted of three 1-min trials with a 10-min intertrial interval. The interval between the two daily sessions was 3 h. Once the rats located the platform, they were allowed to remain on it for 10 secs. Rats that failed to find the platform within 1 min were manually placed on the platform for 15 secs. Time to reach the platform (latency), distance traveled (path length), and swim speed (velocity) were recorded with an EthoVision video tracking system (Noldus Information Technology, Leesbug, VA) set to analyze two samples per second. Two rats (one from the FI + saline group and one from FI + PJ34 group) were excluded from the analysis, because they never found the platform. These animals also exhibited large infarcts in hippocampus, in contrast to the selective neuronal injury observed in the other postischemic rats.
Immunohistochemistry
The paraformaldehyde-fixed brains were cut in 50 µm cryostat sections through the rostral-caudal extent of the hippocampus, and 5 to 6 evenly spaced sections were evaluated from each brain and analyzed for each histologic ‘n.‘ Immunohistochemistry and double-immunofluorescence staining were performed in floating sections, as described previously (Liu et al, 1998). Primary antibodies used were mouse monoclonal anti-rat CD11b (1:100 dilution) (clone OX-42, Serotec, Oxford, UK), rabbit anti-glial fibrillary acidic protein polyclonal antibody (1:1000 dilution) (Chemicon, Temencula, CA), mouse anti-NeuN monoclonal antibody (1:1000 dilution) (Chemicon), sheep anti-BrdU polyclonal antibody (1:180 dilution) (GeneTex Inc, San Antonio, TX), and mouse anti-PAR monoclonal antibody (1:1000 dilution) (Trevigen Inc, Gaithersburg, MD). Secondary antibodies were mouse, rabbit, or sheep anti-IgG conjugated with Alexa Fluor 594 or 488 (1:500 dilution each) (Molecular Probes Inc, Eugene, OR). Fluorescence signals were visualized by confocal microscopy with appropriate filter sets. Immunostaining was evaluated by using the Zeiss LSM 510 confocal image system with a sequential scanning module by an observer blinded to the experimental conditions. Controls prepared with primary or secondary antibodies omitted showed no detectable fluorescence under the conditions used for confocal photography. Colocalization of BrdU with NeuN was confirmed by collecting stacks of images from consecutive optical slices of 0.77 µm thickness to generate 3 dimensional images of the cells of interest.
Evaluation of Brain Microglial and Astrocyte Activation
Microglial activation was scored on the basis of CD11b expression intensity, density of CD11b-expressing cells, and microglial morphology, as detailed in Table 1. The scoring method is slightly modified from 6 that previously published (Kauppinen et al, 2008). Astrogliosis was evaluated by quantifying the intensity of the fluorescence signal from brain sections immunostained for glial fibrillary acidic protein (GFAP), which is upregulated in reactive astrocytes (Eng and Ghirnikar, 1994). All of the brain sections from the different treatment groups were stained in parallel to ensure identical staining conditions. Data were acquired using the NIH Image J program and normalized to the mean expression level observed in sham-operated rats.
Criteria for evaluating microglial activation status
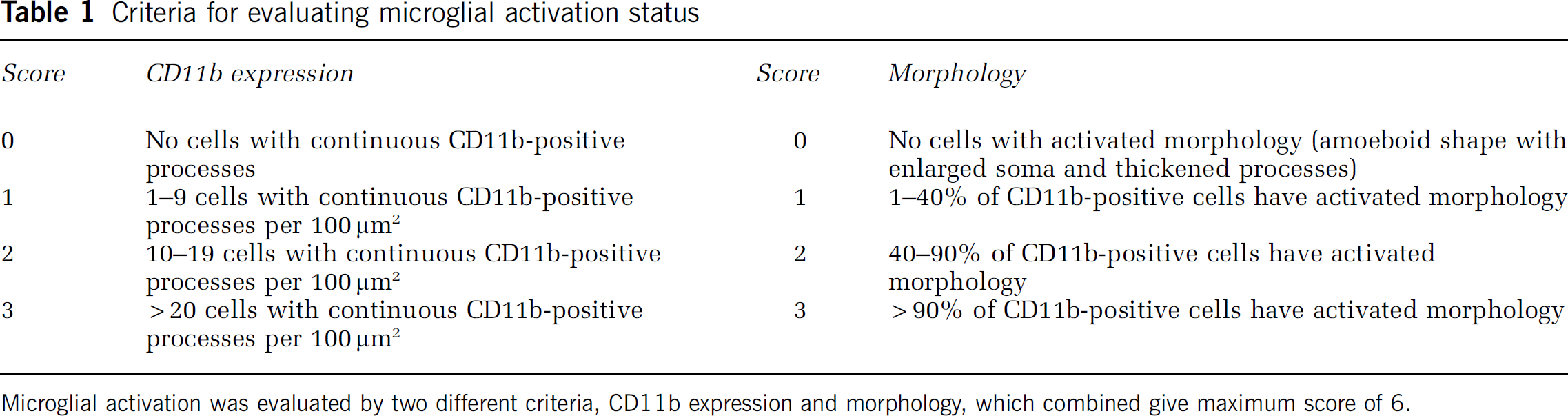
Microglial activation was evaluated by two different criteria, CD11b expression and morphology, which combined give maximum score of 6.
Statistical Analysis
Results are presented as a mean±standard error. Morris water maze path-length data were analyzed with linear mixed model curve fitting using the SAS Proc MIXED procedure (SAS Institute, Cary NC, Ver 9.1.2). The models fit allowed separate trajectories during the testing and training phases as well as animal-specific trends. Immunostaining data were assessed by the nonparametric Kruskal–Wallis one-way analysis of variance test followed by Dunn's test for multiple group comparison. Cell death was assessed by one-way analysis of variance followed by Tukey–Kramer test for multiple comparisons between the treatment groups.
Results
Delayed-Onset Poly(ADP-ribose) Polymerase Inhibition Reduces Microglial Activation and Astrogliosis After Forebrain Ischemia
Activated microglia retract their processes and increase CD11b expression (Kreutzberg, 1996). Brains harvested at serial time points after forebrain ischemia showed microglial activation that was evident within 24 h and peaked approximately 3 days after ischemia (Figure 2A), consistent with prior reports (Hamby et al, 2007; Sugawara et al, 2002). Administration of the PARP inhibitor, PJ34, beginning 48 h after ischemia reduced microglial activation to basal levels within 24 h (Figure 2A and 2B). There was no rebound in microglial activation either during or after the 7-day treatment course with PJ34 (not shown). To further evaluate the role of PARP-1 in microglial activation, immunostaining for poly(ADP-ribose) (abbreviated as PAR), the enzymatic product of PARP-1, was evaluated in PJ34- and vehicle-treated animals at 3 days after ischemia. Ischemia-induced PAR accumulation was seen predominantly in microglia, with little PAR labeling in other cell types (Figure 3), and the PAR formation was completely suppressed by PJ34. Ischemia also induced a robust astrogliosis that peaked at 5 to 7 days (Figure 2C), and treatment with PJ34 also suppressed this astrocyte response (Figure 2C and 2D).
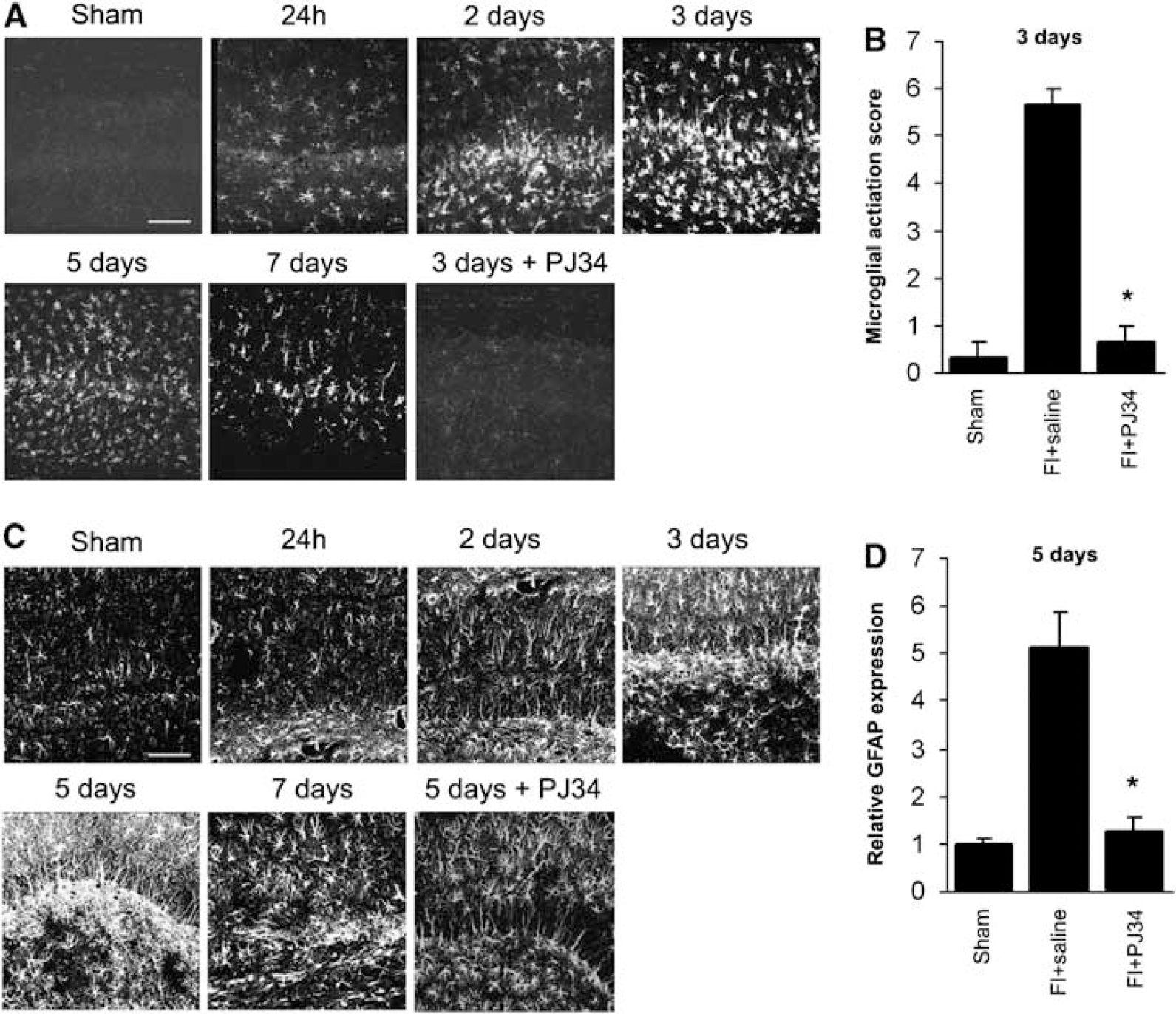
Time course of glial activation in CA1 hippocampus after ischemia. (
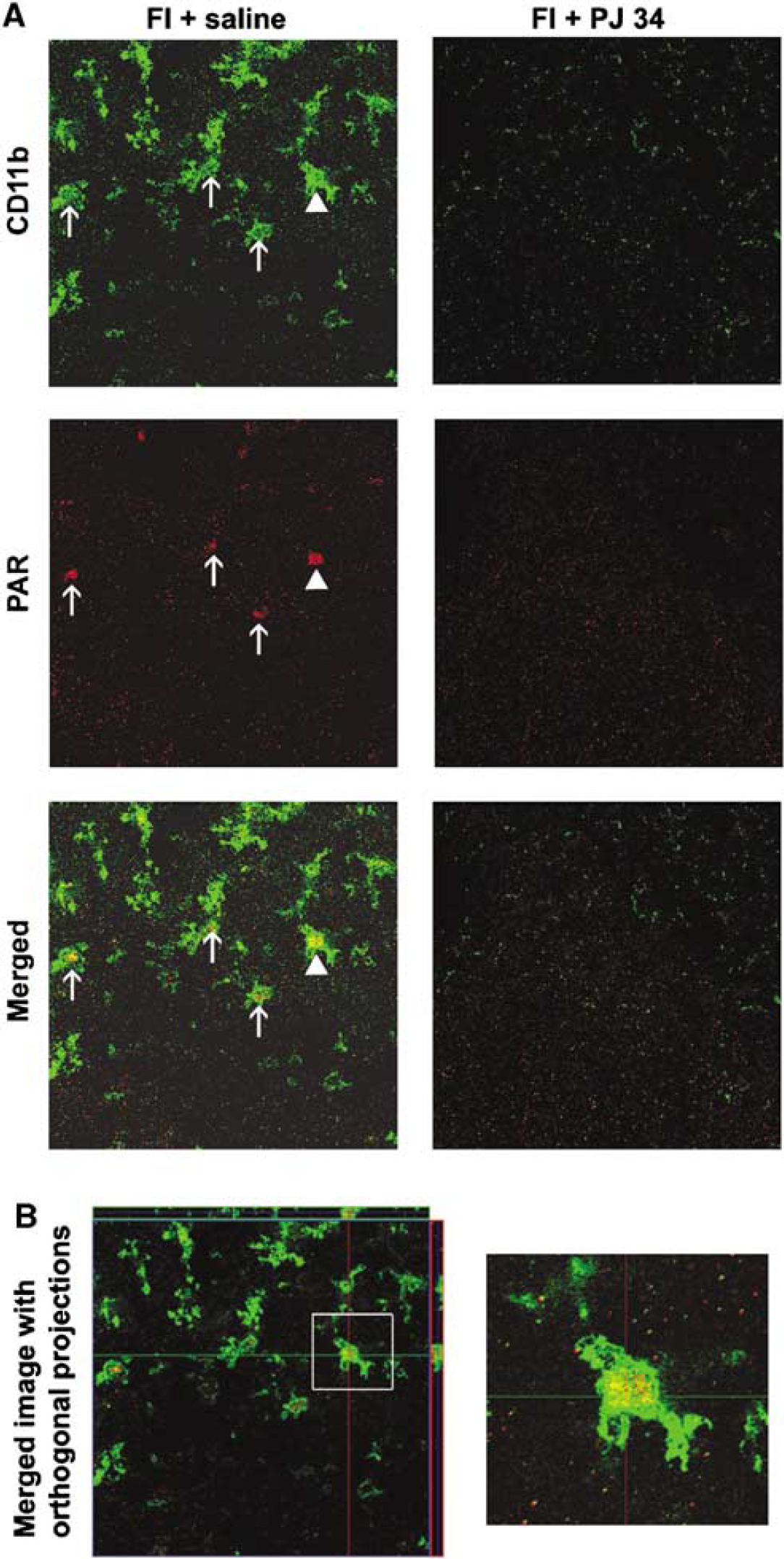
Ischemia induced PARP-1 activation in microglia. (
Prior studies have shown that PARP activation in hippocampal neurons begins within the first hour after ischemia and persists for up to 24 h, and that morphologic evidence of neuronal death is not prominent until after that time (Deshpande et al, 1987; Nagayama et al, 2000). Because PARP inhibition can block acute ischemic neuronal death in addition to the postischemic inflammatory response, we delayed PJ34 treatment until 48 h after ischemia to selectively target the inflammatory response. Hippocampi from brains (n = 5 for each treatment group) harvested 9 days after ischemia were evaluated by hematoxylin-eosin staining to confirm that delayed PJ34 treatment did not affect acute neurodegeneration. There was no decrease in the number of eosinophilic neurons in the rats treated with PJ34 (3,823.2±113.3) beginning at day 2 after ischemia when compared with saline-treated rats (4,013.2±206.8) (Supplementary Figure 1).
Delayed Poly(ADP-ribose) Polymerase Inhibition Promotes Neurogenesis in Hippocampal CA1
Bromodeoxyuridine was injected on days 5 through 9 after ischemia to evaluate the influence of PJ34 on neurogenesis (Liu et al, 1998). Brains harvested 8 weeks after ischemia showed nonneuronal BrdU-positive cells in all treatment groups (Figure 4A). Newly produced neurons, as identified by double staining with BrdU and NeuN, were observed in the dentate gyrus of all treatment groups, though number of new neurons was higher in ischemic rats compared with sham animals (not shown). In the CA1 region, BrdU/NeuN double-labeled cells were observed only in the postischemic rats that had been treated with PJ34 (Figure 4B). Conversely, the number of nonneuronal BrdU cells was lower in this treatment group than in the saline-treated postischemic rats.
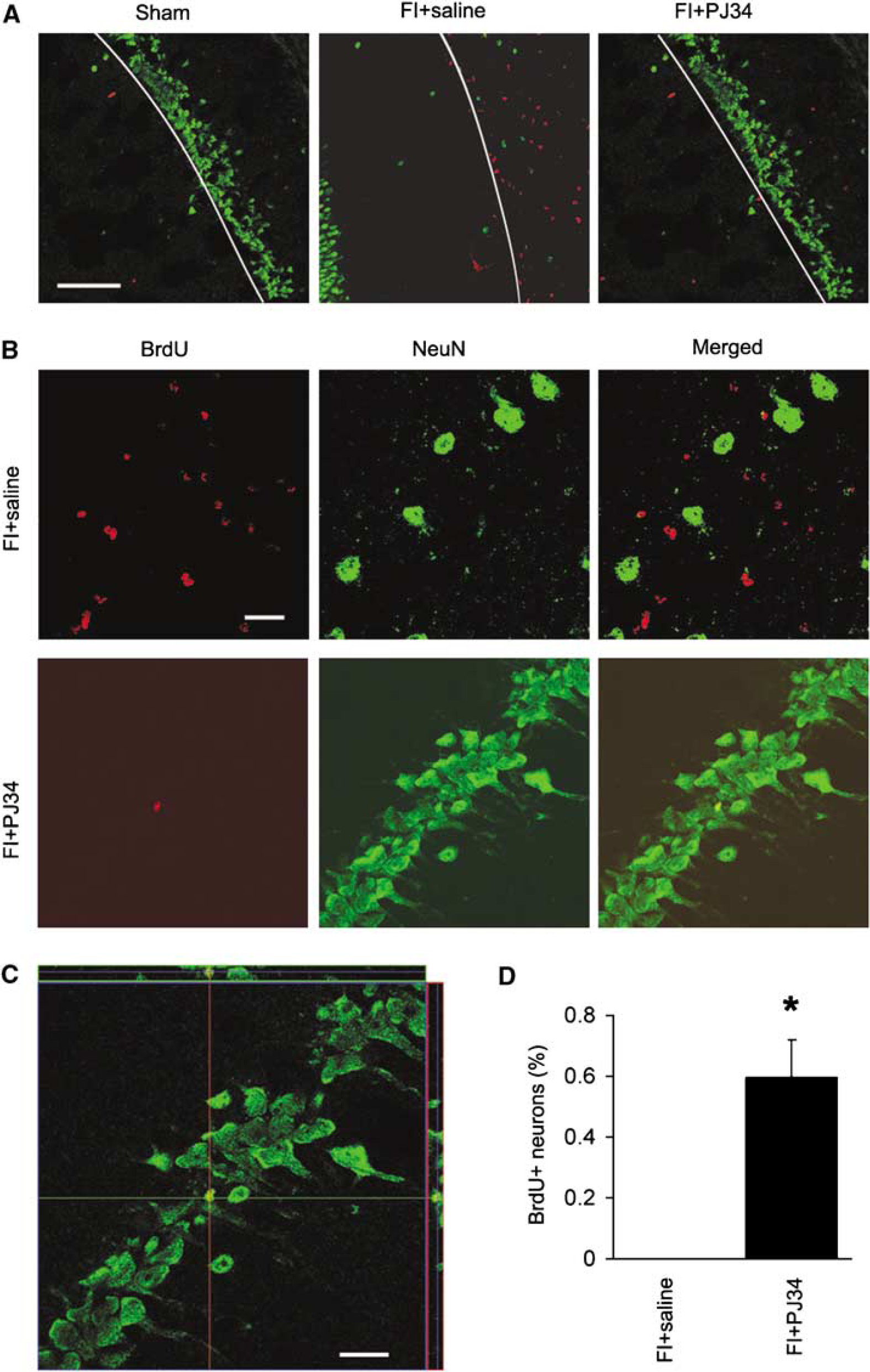
Delayed PARP inhibition promotes neurogenesis in the CA1. (
Delayed Poly(ADP-ribose) Polymerase Inhibition Results in Increased CA1 Pyramidal Neuron Density After Ischemia
NeuN staining in brains harvested 8 weeks after ischemia showed the neuronal density in the CA1 pyramidal layer to be much greater in the PJ34 treated than in saline treated postischemic rats (Figure 5). Neuron density in the PJ34-treated group was 61.2±12.8% of the sham-ischemia group, and neuron density was 17.7±3.4% of the sham-ischemia group (P > 0.01; n = 4).
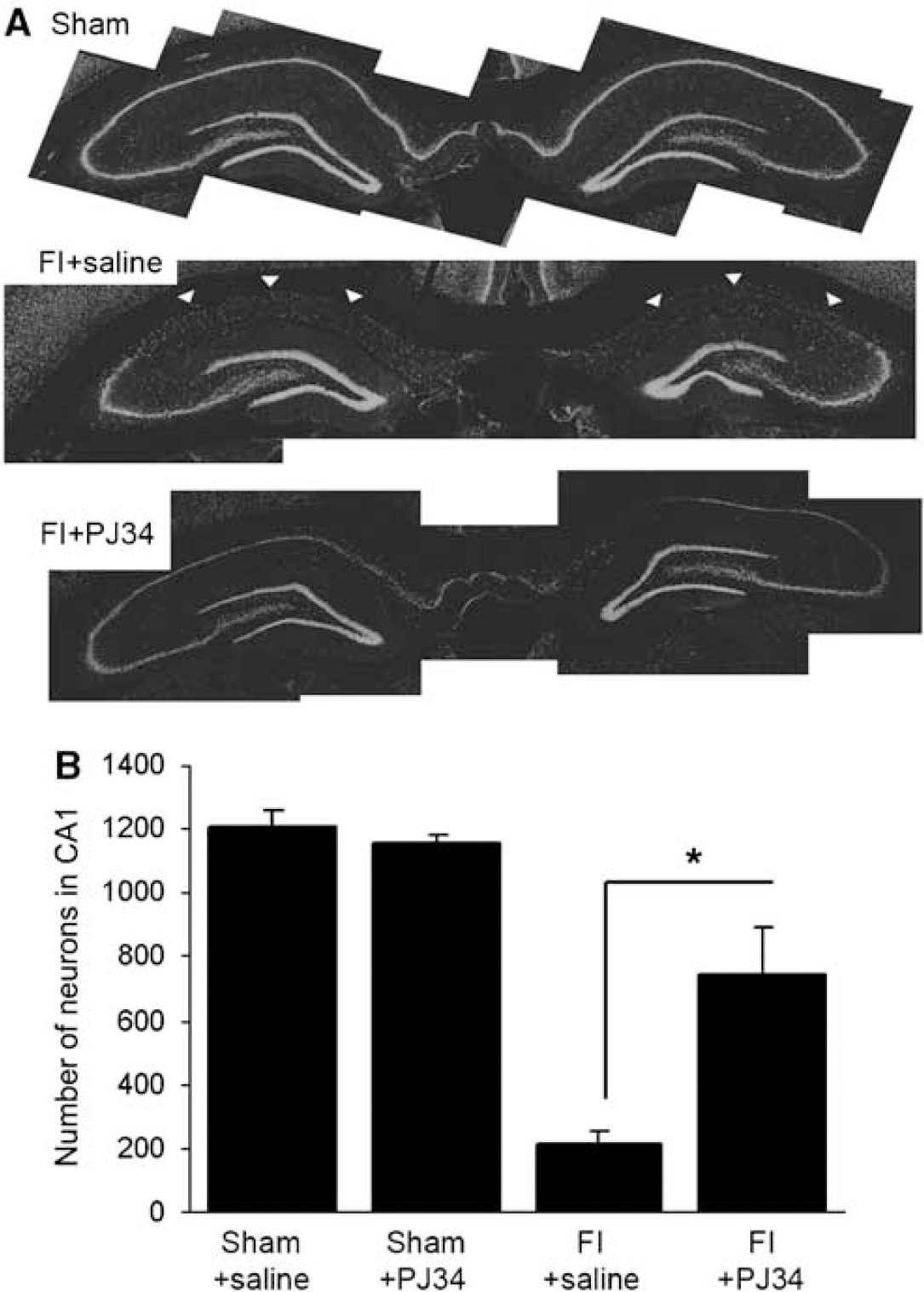
Delayed PARP inhibition leads to increased CA1 pyramidal neuron density in brains evaluated 8 weeks after ischemia. (
Delayed Poly(ADP-ribose) Polymerase Inhibition Improved Spatial Memory After Forebrain Ischemia
There were no significant differences in spontaneous locomotor activity between treatment groups in the open field test, and no significant group differences in swim speed during the Morris water maze test. By contrast, analysis of swim path-length showed significant differences between the treatment groups, indicating differences in spatial learning and memory (Figure 6). In the testing phase (no visual cue on the platform), comparison among all four treatment groups revealed differences in path-length among the groups to be significant at the P = 0.003 level, and direct comparison of PJ34-treated ischemia group with the saline-treated ischemia group showed mean path-length to be longer in the saline-treated ischemia group at P = 0.009. The saline-treated ischemia group also performed worse than the PJ34-treated ischemia group in the training phase sessions (visualcue on the platform; P = 0.049), although the difference between groups when all four treatment groups were compared together was only marginally significant (P = 0.090) in this phase of the test.
Discussion
The results of these studies provide proof of principle that delayed-onset treatment with a PARP inhibitor can effectively suppress brain inflammation and improve recovery after cerebral ischemia. PJ34 administration beginning 48 h after ischemia rapidly suppressed microglial and astrocyte activation, increased neurogenesis in hippocampal CA1, increased neuronal density in CA1, and improved spatial memory and learning. The key aspect of this study is the extended time interval between ischemic insult and the histologic and behavioral outcome measurements. The robust effects of PJ34 treatment observed at 6 to 8 weeks after ischemia indicate that the results of this intervention are long lasting.
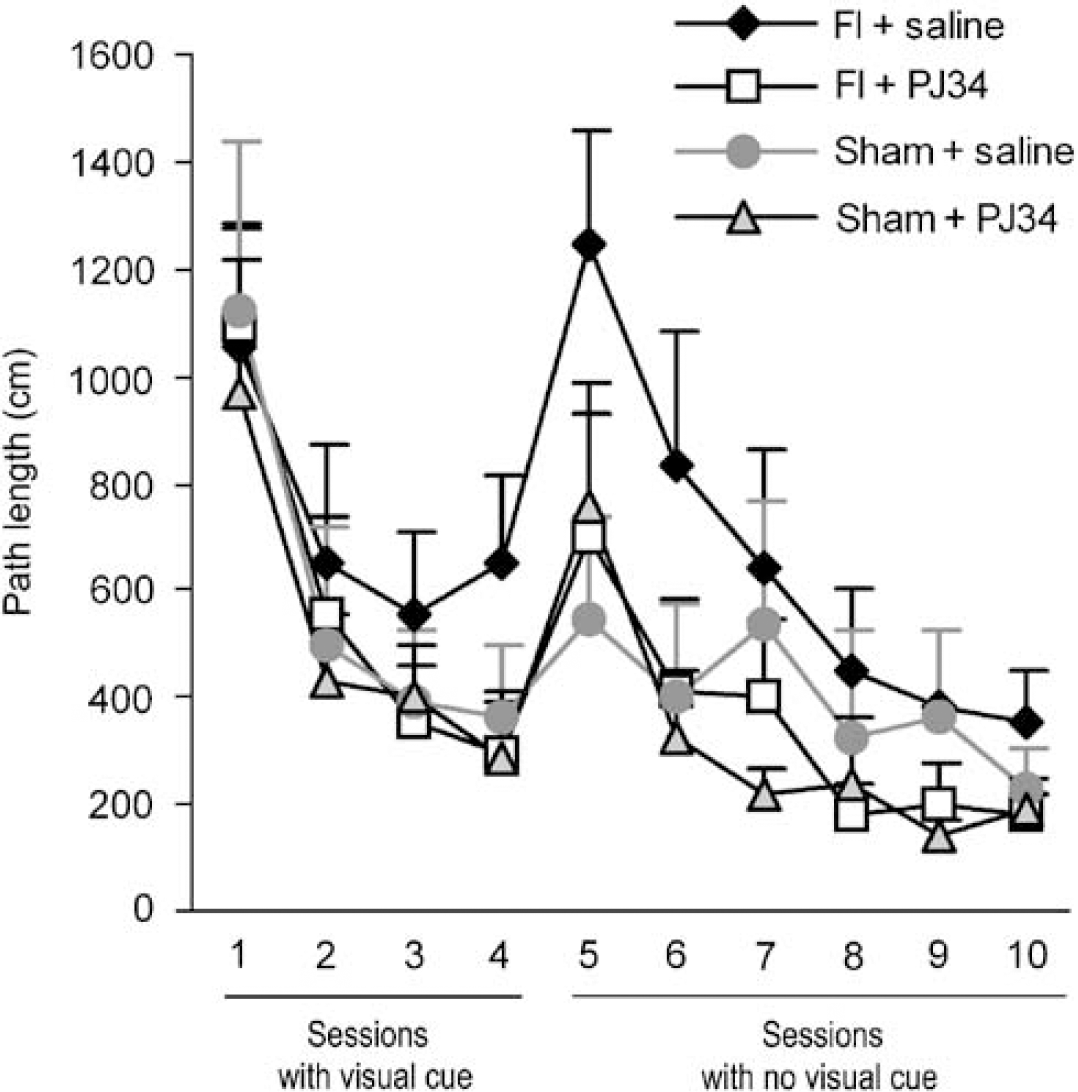
Delayed PARP inhibition improves spatial memory after ischemia. Swim path-length in the Morris water maze test was used to evaluate spatial learning and memory 6 weeks after forebrain ischemia (FI). Sessions 1 to 4 were performed with a visual cue attached to the submerged platform, and sessions 5 to 10 were performed without the platform cue. During the testing phase, rats treated with PJ34 after FI performed better than rats treated with vehicle alone (P=0.009). The saline-treated FI rats also performed worse than sham-operated rats treated with either PJ34 (P=0.003) or saline (P=0.025). n=5 to 6 for each group.
PARP-1 is known to regulate the inflammatory response through its function as a coactivator with NF-κB. PARP-1 facilitates NF-κB activity at promoter sites by enhancing DNA binding and transcription complex formation (Hassa and Hottiger, 2002; Jagtap and Szabo, 2005). There is disagreement as to whether PARP-1 enzymatic activity is required for this interaction with NF-κB. Some reports show that enzymatically nonfunctional PARP-1 can facilitate NF-κB activation (reviewed in Kraus and Lis, 2003), but others have shown that inhibition of enzymatic PARP-1 activity suppresses NF-κB-driven gene transcription in a variety of cell types, including microglia (Chiarugi and Moskowitz, 2003; Kauppinen et al, 2008; Nakajima et al, 2004). It remains possible, however, that the antiinflammatory actions of PJ34 could be mediated by enzymatic inhibition of PARP species other than PARP-1, or by blocking the effect of PARP-1 at transcription sites other than NF-κB (Jagtap and Szabo, 2005). Regardless of mechanism, the antiinflammatory effects of PARP-1 inhibitors are well established (Haddad et al, 2006; Hamby et al, 2007; Kauppinen and Swanson, 2005; Moroni, 2008). PARP inhibition has been shown to reduce ischemia-induced production of TNF, IL-6, and ICAM-1 (Haddad et al, 2006). Moreover, PARP-1 activation can contribute to blood–brain barrier disruption by activating matrix metalloproteinase-9 and promoting cell death of the endothelial and glial cells forming the barrier structure (Haddad et al, 2008; Kauppinen and Swanson, 2005; Lenzser et al, 2007; Moroni, 2008). The present results extend these findings by showing that the PARP inhibitor PJ34 can suppress ischemia-induced microglial activation and astrogliosis in vivo.
A potential confounding factor in these studies is the fact that PARP inhibitors are also directly neuroprotective and have been shown to prevent CA1 neuronal death when administered within a few hours after transient forebrain ischemia (Hamby et al, 2007; Moroni, 2008; Strosznajder et al, 2003). As the brain inflammatory response is influenced by the extent of brain injury, a reduction in acute neuronal death by PJ34 could itself lead to a reduced postischemic inflammatory response. Because of this dynamic, treatment with the PARP inhibitor was not begun until 48 h after ischemia-reperfusion. Prior reports indicate that PARP inhibition does not have an acute neuroprotective effect when initiated at this time point (Hamby et al, 2007), and this was confirmed here by examining the effect of PJ34 treatment on neuronal death in rats killed 9 days after ischemia (7 days after beginning treatment with PJ34). Thus, the effects of PJ34 in this study cannot be attributed to a reduction in acute neuronal death.
Rats treated with PJ34 after ischemia showed increased neurogenesis and increased neuronal density in the CA1 region. As in a prior report (Bendel et al, 2005), these changes correlated with improved spatial learning and memory. However, the newly formed (BrdU positive) neurons identified in the CA1 region of PJ34-treated rats were uncommon (>1% of the neurons), and smaller than most of the neighboring pyramidal neurons. It is possible that these neurons would continue to enlarge over time. Alternatively, these new cells may be interneurons, rather than new pyramidal neurons. Interneurons in the CA1 are inhibitory, limiting glutamatergic input to pyramidal neurons (Klausberger and Somogyi, 2008) and could thereby promote pyramidal neuron survival after ischemic damage to hippocampal circuitry.
In addition to the increase in neurogenesis, rats treated with PJ34 were found to have a much higher number of CA1 neurons when examined 8 weeks after ischemia. The percent of BrdU-labeled neurons in CA1 appears to be far too low to account for this increase, even accounting for the fact that BrdU was used at a conservative dose that may not label all dividing cells (Iwai et al, 2001) and was injected only during the estimated peak neurogenesis period of days 5 to 9 after ischemia. The increased CA1 neuronal density in the PJ34-treated rats might alternatively result from a reduction in delayed neuronal death (death occurring after day 9) in the PJ34-treated rats, due to reduced inflammation or other factors.
Newly formed neurons in the dentate gyrus originate in the subgranular layer of the dentate, whereas newly formed CA1 neurons are derived from progenitor cells of the posterior periventricular zone (Kokaia and Lindvall, 2003). PJ34 was found to increase the number of new neurons in the CA1 hippocampus, but not in the dentate gyrus. The reason for this difference is not established here but could result from the much greater cell death in the CA1 region induced by forebrain ischemia. Transient forebrain ischemia has previously been shown to induce neurogenesis in the CA1 pyramidal cell field (Bendel et al, 2005; Nakatomi et al, 2002; Schmidt and Reymann, 2002). An association between inflammation and impaired neurogenesis has been noted in several reports (Ekdahl et al, 2003; Monje et al, 2003), and a causal relationship between these events is supported by studies showing that indomethacin administered after radiation-induced brain injury enhances neurogenesis in concert with its antiinflammatory effect (Monje et al, 2003). Rats treated with indomethacin after focal cerebral ischemia showed reduced microglial activation and increased neurogenesis (Hoehn et al, 2005), similar to the results obtained here with PJ34 in the rat forebrain ischemia model. In other studies, the suppression of microglial activation with minocycline was associated with enhanced neurogenesis after status epilepticus or lipopolysaccharide injection in rats (Ekdahl et al, 2003). The present results thus provide additional support for inflammation as a key factor influencing neurogenesis after ischemia, and further support PARP inhibition as the mechanism by which minocycline exerts its antiinflammatory effects (Alano et al, 2006).
The microglial inflammatory response is a graded and multifaceted process, involving morphologic transformation, proliferation, migration to the injury site, increased expression of adhesion molecules, MHC antigens and surface markers, and increased secretion of cytokines, chemokines, free radicals, and proteases (Kreutzberg, 1996). Astrocyte activation is characterized by increased glial fibrillary acidic protein expression, cellular hypertrophy, process extension and interdigitation, and proliferation (Eng and Ghirnikar, 1994). The relationships between microglia activation and astrocyte activation have not been established, but it is likely that activation of either cell type can induce activation of the other (Kauppinen and Swanson, 2006; Kreutzberg, 1996). Microglia and astrocyte activation do not, however, have solely deleterious effects after brain injury (reviewed in Kauppinen and Swanson, 2006; Kreutzberg, 1996). Activated microglia increase expression of several neurotrophic factors (Lehrmann et al, 1998), and may direct migration and differentiation of neuronal precursor cells (Aarum et al, 2003). Similarly, reactive astrocytes increase release of nerve growth factor, basic fibroblast growth factor, BDNF, neuregulins, and other factors thought to promote plasticity after brain injury (Kauppinen and Swanson, 2006; Liberto et al, 2004). These potentially beneficial effects of glial activation are not known to be regulated by NF-κB, and thus, may be unaffected by PARP inhibition.
Treatment with PJ34 was not begun in this study until 48 h after ischemia to isolate the antiinflammatory effects from the neuroprotective effects of PARP inhibition; however, the beneficial effects obtained with this delayed treatment strategy may also have clinical implications. A treatment approach for stroke that is effective within a 48-h time window would greatly increase the number of patients eligible for therapy. The stroke model used here, transient forebrain ischemia, was chosen because it induces extensive loss of hippocampal CA1 neurons (Deshpande et al, 1987; Smith et al, 1984), thus permitting the use of hippocampal neurogenesis and spatial memory as experimental outcome measures. An issue to be established in future studies is whether the beneficial effects of PARP inhibition after transient forebrain ischemia are also evident after focal ischemia, which is a more common clinical event.
Footnotes
Acknowledgements
We thank Dr Charles McCulloch, UCSF Epidemiology and Biostatistics Department, for advice with the Morris water maze data, and Mrs Michelle Hong (BS) for help with behavioral tests and BrdU immunostaining. This work was supported by the American Heart Association (TMK) and by U.S. Department of Defense and Department of Veterans Affairs (RAS).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.