
Abstract
Interleukin-18, previously designated interferon γ-inducing factor, is a proinflammatory cytokine structurally related to interleukin-1β and is therefore considered a member of the growing family of interleukin-1–like cytokines. Both interleukin-18 and −1β are synthesized as inactive precursors that necessitate cleavage by caspase-1 for functional activity. In this study, the authors analyzed the expression pattern of interleukin-18, −1β, and caspase-1 in focal brain ischemia induced in rats either by permanent middle cerebral artery occlusion or by photothrombosis of cortical microvessels. Using reverse transcriptase-polymerase chain reaction, they found a delayed increase of interleukin-18 mRNA starting at 48 hours and reaching its peak between 7 and 14 days after ischemia. In contrast, interleukin-1β mRNA peaked within 16 hours and was downregulated thereafter. The time course of caspase-1 mRNA expression paralleled that of interleukin-18, but not of interleukin-1β mRNA. Immunocytochemically, interleukin-18 expression was localized to ED1-positive phagocytic microglia/macrophages infiltrating the necrotic lesion between 3 and 6 days after ischemia. In contrast, interleukin-1β immunoreactivity was expressed by ramified microglia in the infarct border zone and remote ipsilateral cortex during the first 16 hours postlesion. Induction of interleukin-18 was not accompanied by detectable expression of interferon-γ mRNA. Their data show spatial and temporal diversity in interleukin-1 and −18 cytokine family expression in brain ischemia, and suggest a role of the interleukin-18/caspase-1 pathway in late-stage inflammatory responses to focal brain ischemia.
Focal ischemic brain damage elicits a strong inflammatory response that involves both activation of resident microglia and recruitment of blood-derived macrophages and T cells (Arvin et al., 1996; Stoll et al., 1998). Inflammatory mechanisms can contribute to secondary neurotoxicity and the exacerbation of ischemic brain damage (Relton and Rothwell, 1992; Barone et al., 1997), but are also involved in neuroprotection, tissue remodeling, and wound-healing processes (Clark et al., 1993; Wang et al., 1998; Shohami et al., 1999). In rat models of focal ischemia, the early-stage inflammatory response is mainly governed by resident microglia, whereas hematogenous macrophages are recruited with a delay of 2 to 3 days (Schroeter et al., 1997). Active synthesis of proinflammatory cytokines, such as tumor necrosis factor α and interleukin-1β, is restricted to a narrow time window of 12 to 24 hours after lesion induction, and is thereby correlated with the activation of resident glia rather than hematogenous cell infiltration (Liu et al., 1993; Liu et al., 1994; Jander et al., 2000). Therefore, the cytokine profile and the functional implications of late-stage macrophage responses to focal ischemia are currently unknown.
Cytokines of the interleukin-1 family, along with their specific receptors and naturally occurring antagonists, have been critically implicated in mechanisms of acute and chronic neurodegeneration (Rothwell et al., 1997; Touzani et al., 1999). Recently, an additional member of the interleukin-1 cytokine family, interferon-γ–inducing factor, was identified in a mouse model of endotoxic shock and was subsequently designated interleukin-18 (Okamura et al., 1995; Bazan et al., 1996). Similar to interleukin-1β, interleukin-18 is synthesized as an inactive precursor molecule that necessitates cleavage by caspase-1 for functional activation (Ghayur et al., 1997; Gu et al., 1997; Akita et al., 1997). Interleukin-18 is a critical regulator of innate immunity that overall promotes the development of proinflammatory type-1 T cell responses hallmarked by the release of interferon-γ (Kohno and Kurimoto, 1998). Interleukin-18 also exerts proinflammatory effects, such as the upregulation of the intercellular adhesion molecule-1 adhesion receptor independent of the induction of interferon γ (Kohka et al., 1998; Kitching et al., 2000).
In the nervous system, interleukin-18 is induced in prototypic Th1-mediated central and peripheral nervous system diseases such as experimental autoimmune encephalomyelitis (Jander and Stoll, 1998) and neuritis (Jander and Stoll, 2001), respectively, in parallel with caspase-1 messenger RNA (mRNA). Functional studies suggest an important role of the interleukin-18/caspase-1 pathway in the activation of pathogenic T helper cells in the context of central nervous system autoimmunity (Wildbaum et al., 1998; Furlan et al., 1999). Although constitutive expression of interleukin-18 has been described in the brains of normal rats (Culhane et al., 1998) and in glial cell cultures (Conti et al., 1999), its potential role in degenerative and ischemic brain injury is unclear. In the present study, we investigated whether interleukin-18 is induced after focal ischemia in the rat brain. Using reverse-transcriptase polymerase chain reaction (RT-PCR) and immunocytochemistry, we analyzed the time course and cellular localization of interleukin-18 mRNA and protein in two models of permanent cortical ischemia: surgical occlusion of the middle cerebral artery (MCAO) and noninvasive photothrombosis of cortical microvessels. Our data provide evidence for a role of interleukin-18 in late-stage inflammatory responses to focal ischemic brain damage.
MATERIALS AND METHODS
Animal experiments
Focal cortical infarctions were induced in adult male Wistar rats either by permanent MCAO occlusion or by photothrombosis of cortical microvessels using inhalation anesthesia [halothane 1.3% in a 1:2 oxygen:nitrogen mixture] as described in detail elsewhere (Schroeter et al., 1994; Jander et al., 1995). All animal experiments were performed in accordance with institutional guidelines.
For MCAO, the skin was cut coronarily between the ear and eye and the temporal muscle was dissected. The temporal skull was opened under microsurgical conditions, the dura was opened with a sterile 27-gauge cannula, and the middle cerebral artery was coagulated twice (at the level of the inferior cerebral vein and distally) and was cut in between. The temporal muscle and the skin was closed in separate layers, which resulted in mainly cortical infarctions that could be macroscopically identified from 4 hours after induction onwards. For RNA isolation and subsequent RT-PCR analysis, infarcted and noninfarcted contralateral cortex was prepared 4, 16, and 24 hours and 3, 6, and 14 days after ischemia (n = 3–4 animals at each time point).
In the photothrombosis model, a fiberoptic bundle of a cold light source was centered stereotactically 4 mm posterior and 4 mm lateral from Bregma on the skull exposed by a dorsal midline incision of the skin. Then, 0.4 mL sterile-filtered Rose Bengal solution (10 g/L) was administered through a femoral vein catheter, and the brain was illuminated for 20 minutes. The skin was sutured, which resulted in a cone-shaped pure cortical infarction that was readily visible 4 hours after induction of ischemia. For PCR analysis, ischemic lesions (A in Fig. 2a) and frontal noninfarcted cortex of the ipsilateral hemisphere (D in Fig. 2a) and contralateral cortex homotopic to the infarct (C in Fig. 2a) were prepared at 4 h, 16 h, 24 h, 2 d, 3 d, 6 d, 14 d, and 28 d (n = 4 per time point). Approximately 30 to 50 mg tissue (wet weight) were obtained from each site.
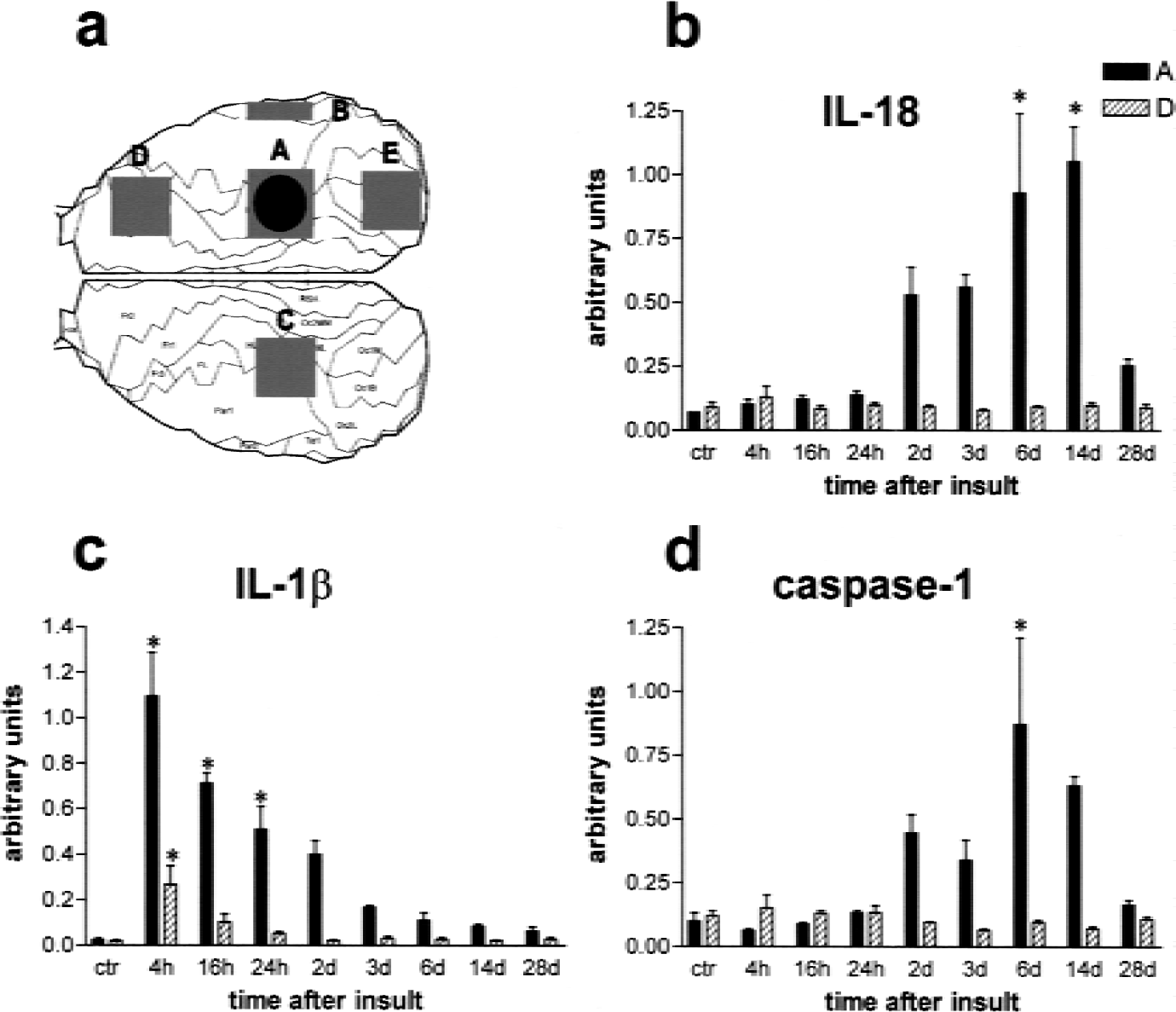
Semiquantitative reverse-transcriptase polymerase chain reaction analysis of interleukin (IL)-18, −1β, and caspase-1 mRNA levels in the photothrombosis model of focal brain ischemia.
As a comparative paradigm of immune-mediated central nervous system inflammation, experimental autoimmune encephalomyelitis (EAE) was induced in 8-week-old female Lewis rats (weight, 180–200 g) by active immunization with a synthetic peptide (25 μg per animal) corresponding to the amino acids 68 to 86 of guinea pig myelin basic protein (MBP68–86), as detailed elsewhere (Jander et al., 1998a). According to the previously defined time course of interferon-γ mRNA expression in this model (Jander et al., 1998a), animals were killed at early (day 11), peak (day 15), and recovery disease stages (day 21) by an overdose of ether (n = 4 rats at each time point). The lumbal and lower thoracic spinal cord (≈ 300 mg wet weight per animal) was rapidly prepared by air insufflation and immediately homogenized in 5 mL TRIzol Reagent (GIBCO BRL, Gaithersburg, MD), and stored at −80°C until RNA isolation.
Semiquantitative reverse transcriptase-polymerase chain reaction
Total RNA was prepared using the TRIzol reagent according to the manufacturer's instructions and was quantified spectrophotometrically. One microgram RNA isolated from each tissue sample was reverse transcribed using oligo(dT)20 primers and SuperscriptII-Reverse Transcriptase (GIBCO BRL) according to the manufacturer's protocol. The cDNA equivalent to 20 ng total RNA was subjected to subsequent PCR analysis, as detailed elsewhere (Jander et al., 1998b), using primer pairs specific for rat interleukin-1β [sense, 5′-AGAAGAGCCCGTCCTCTGTGACTC-3′; antisense, 5′-TCGACAATTGCTGCCTCGTGAC-3′ (Gillen et al., 1998)], interleukin-18 [sense, 5′-ACTGTACAACCGCAGTAATACGG-3′; antisense, 5′-AGTGAACATTACAGATTTATCCC-3′ (Conti et al., 1997)], caspase-1 (Biosource, Camarillo, CA), interferon-γ [sense, 5′-ATCTGGAGGAACTGGCAAAAGGACG-3′; antisense, 5′-CCTTAGGCTAGATTCTGGTGACAGC (Jander et al., 1998a)], and glyceraldehyde-phosphate dehydrogenase (GAPDH) [sense, 5′-CCTTCATTGACCTCAACTACATGGT-3′; antisense, 5′-TCATTGTCATACCAGGAAATGAGCT-3′ (Gillen et al., 1998)], respectively. For each primer combination, linear cycling conditions were determined in preliminary experiments, as described elsewhere (Jander and Stoll, 1998). Cycle numbers were as follows: interleukin-1β = 25, interleukin-18 = 25, caspase-1 = 25, interferon γ = 29, and GAPDH = 18. Controls included RNA subjected to RT-PCR without addition of reverse transcriptase and PCR performed in the absence of cDNA, which yielded negative results. For each PCR product, representative samples were sequenced on an ABI PRISM 310 genetic analyzer (Perkin Elmer, Oak Brook, IL).
Quantification and statistical analysis
The PCR products were separated on ethidium bromide-stained agarose gels and quantified densitometrically, as described previously (Jander and Stoll, 1998). Relative mRNA levels were obtained by normalization against the respective GAPDH mRNA level in each sample, and are presented as mean ± SD throughout. To compare mRNA levels at different time points and between different cortical areas, analysis of variance (Tukey multiple comparison test) was performed using GraphPad Prism 3.0 software (San Diego, CA, U.S.A.). To analyze the relation between cytokine and caspase-1 mRNA levels in tissue samples, we performed linear regression analysis using Microcal (Northampton, MA, U.S.A.) Origin 5.0 software.
Immunocytochemistry
At 8 hours and 3, 6, and 14 days after induction of cortical photothrombosis (n = 3 at each time point), rats were deeply anesthetized with ether and perfused with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) through the left ventricle. Whole brains were removed from the scull, postfixed in the same fixative overnight at 4 °C, and cryoprotected by overnight infiltration with 20% sucrose in phosphate buffer at 4°C. Free-floating 50-μm cryostat sections were washed three times in Tris-buffered saline containing 0.05% Triton X-100 (TBS-T). Endogenous peroxidase was blocked by 30-minute incubation in 0.3% hydrogen peroxide in TBS-T. After three washes in TBS-T, sections were incubated with affinity-purified goat polyclonal antibodies against rat interleukin-1β (1:400 dilution, R&D Systems, Minneapolis, MN, U.S.A.) or rat interleukin-18 (1:2000, R&D Systems), respectively, in 2% normal horse serum in TBS-T for 16 hours at 4°C. After three washes in TBS-T, bound antibody was detected using biotinylated horse antigoat immunoglobulin G (Vector Laboratories, Burlingame, CA), respectively, and the ABC Elite kit (Vector) with diaminobenzidine as substrate. Control experiments in which primary antibodies were either omitted, replaced by irrelevant control immunoglobulin G, or preadsorbed with a 10-fold excess of recombinant cytokine proteins (R&D Systems) revealed complete loss of specific immunostaining. Sections were mounted onto gelatin-coated slides, air-dried, dehydrated with ascending series of ethanol, cleared, and coverslipped with Entellan (Merck, Darmstadt, Germany).
Immunofluorescent double-labeling and confocal microscopy
Free-floating sections were pretreated as described previously and incubated for 16 hours at 4°C with goat polyclonal antibody against rat interleukin-18 (1:500, R&D Systems) combined with mouse monoclonal antibody ED1 against phagocytic macrophages (1:2000, Serotec, Oxford, UK). Immunofluorescent detection of bound interleukin-18 antibody was performed with catalyzed reporter deposition (Shindler and Roth, 1996) using horseradish peroxidase-conjugated donkey anti-goat immunoglobulin G (1:500; Jackson Immunoresearch Laboratories, West Grove, PA, U.S.A.) followed by the TSA Plus Fluorescein system (NEN Life Science Products, Zaventem, Belgium) according to the manufacturer's instructions. Bound ED1 mouse monoclonal antibody was detected by conventional immunofluorescence using TexasRed (TXRD)-conjugated donkey antimouse immunoglobulin G (1:100, Jackson Immunoresearch). The horseradish peroxidase-and TXRD-conjugated secondary antibodies were applied simultaneously for 2 hours at room temperature. After completion of staining, sections were mounted using Vectashield mounting medium (Vector Laboratories). Samples were analyzed using a Leica TCS-NT confocal laser scanning system with an argon-krypton laser on a Leica DM IRB inverted microscope. Images were acquired from two channels at 488 and 568 nm. To analyze the localization of different antigens in double-stained samples, images obtained from the appropriate excitation wavelength were collected and merged.
RESULTS
Delayed induction of interleukin-18 and caspase-1 mRNA after focal brain ischemia
We first used semiquantitative RT-PCR to compare the time course of interleukin-18, caspase-1, and interleukin-1β expression in the MCAO model (Fig. 1). Both interleukin-18 and caspase-1 transcripts showed only slight, statistically insignificant increases at early stages (up to day 3) after ischemia. At day 6, however, both interleukin-18 and caspase-1 mRNA exhibited a significant increase compared with the control level. In contrast, interleukin-1β mRNA increased rapidly with an early peak at 4 hours and a decrease thereafter.
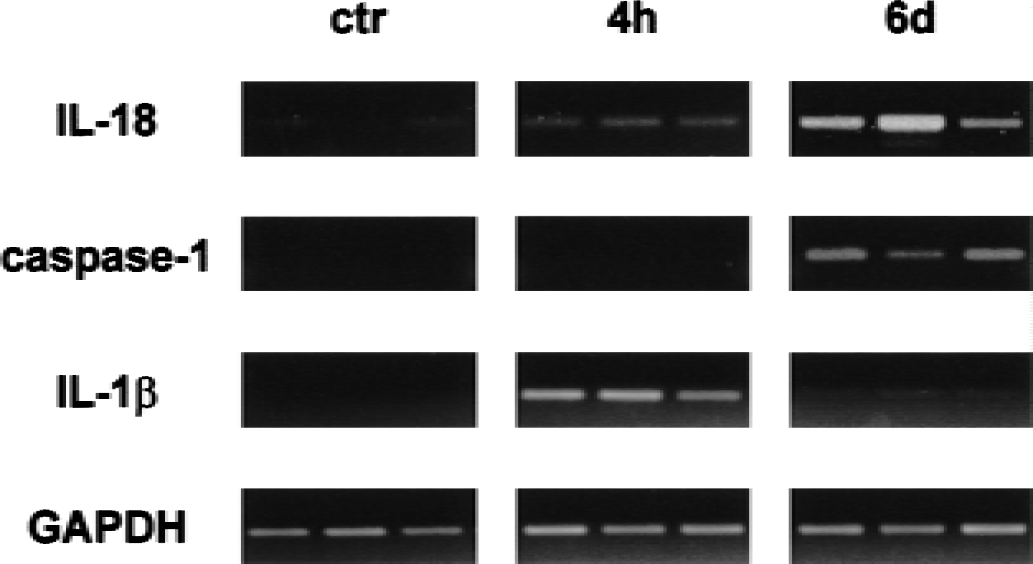
Original reverse-transcriptase polymerase chain reaction findings showing differential induction of interleukin-18, caspase-1, and interleukin-1β mRNA in cortical ischemic lesions induced by permanent occlusion of the middle cerebral artery. Representative results from three animals are shown at each time point. Whereas interleukin-1β mRNA is induced strongly at 4 hours and downregulated at 6 days, both interleukin-18 and caspase-1 mRNA induction are considerably delayed with peak levels reached at 6 days after ischemia. Transcript levels of the housekeeping gene glyceraldehyde-phosphate dehydrogenase (GAPDH) remained constant at all time points.
To study the spatiotemporal distribution of interleukin-18 and caspase-1 mRNA induction in more detail, we used the model of photochemically induced cortical ischemia in the rat (Watson et al., 1985; Jander et al., 1995). With this method, relatively small, sharply demarcated cortical infarcts are generated that, in contrast to the MCAO model, reliably allow the differentiation between ischemic infarct and nonischemic intact surroundings of the ipsilateral hemisphere. The time course of infarct development and inflammation is similar in both models (Stoll et al., 1998; Jander et al., 2000). Using the photothrombosis model, we have shown previously that the induction of inflammatory gene expression after focal ischemia is not restricted to the primary lesion, but likewise involves the entire ipsilateral hemisphere with upregulation of proinflammatory cytokines (Jander et al., 2000) and activation of resident microglia (Schroeter et al., 1999). These remote responses are elicited by cortical spreading depression (Jander et al., 2000; Jander et al., 2001).
In the photochemically induced infarctions, we found an early increase in IL-1β mRNA and a delayed induction of both interleukin-18 and caspase-1 mRNA identical to the MCAO model (Fig. 2). Semiquantitative analysis revealed that the increase of interleukin-18 (Fig. 2B) and caspase-1 mRNA (Fig. 2D) was restricted to the evolving infarct, whereas interleukin-1β (Fig. 2C) additionally showed a significant increase in the nonischemic cortex of the ipsilateral, but not contralateral, hemisphere. We subsequently related the transcript levels of caspase-1 in each tissue sample to those of interleukin-18 and −1β, respectively (Fig. 3). This correlative analysis revealed a highly significant association of caspase-1 mRNA induction with interleukin-18 (r = 0.91, P < 0.0001, Fig. 3A), but not with interleukin-1β mRNA levels (r = −0.07, P < 0.54, Fig. 3B). Thus, although caspase-1 has been implicated in the processing of both interleukin-1β and −18 into their active forms, the spatiotemporal pattern of caspase-1 expression paralleled that of interleukin-18, but not of interleukin-1β mRNA.
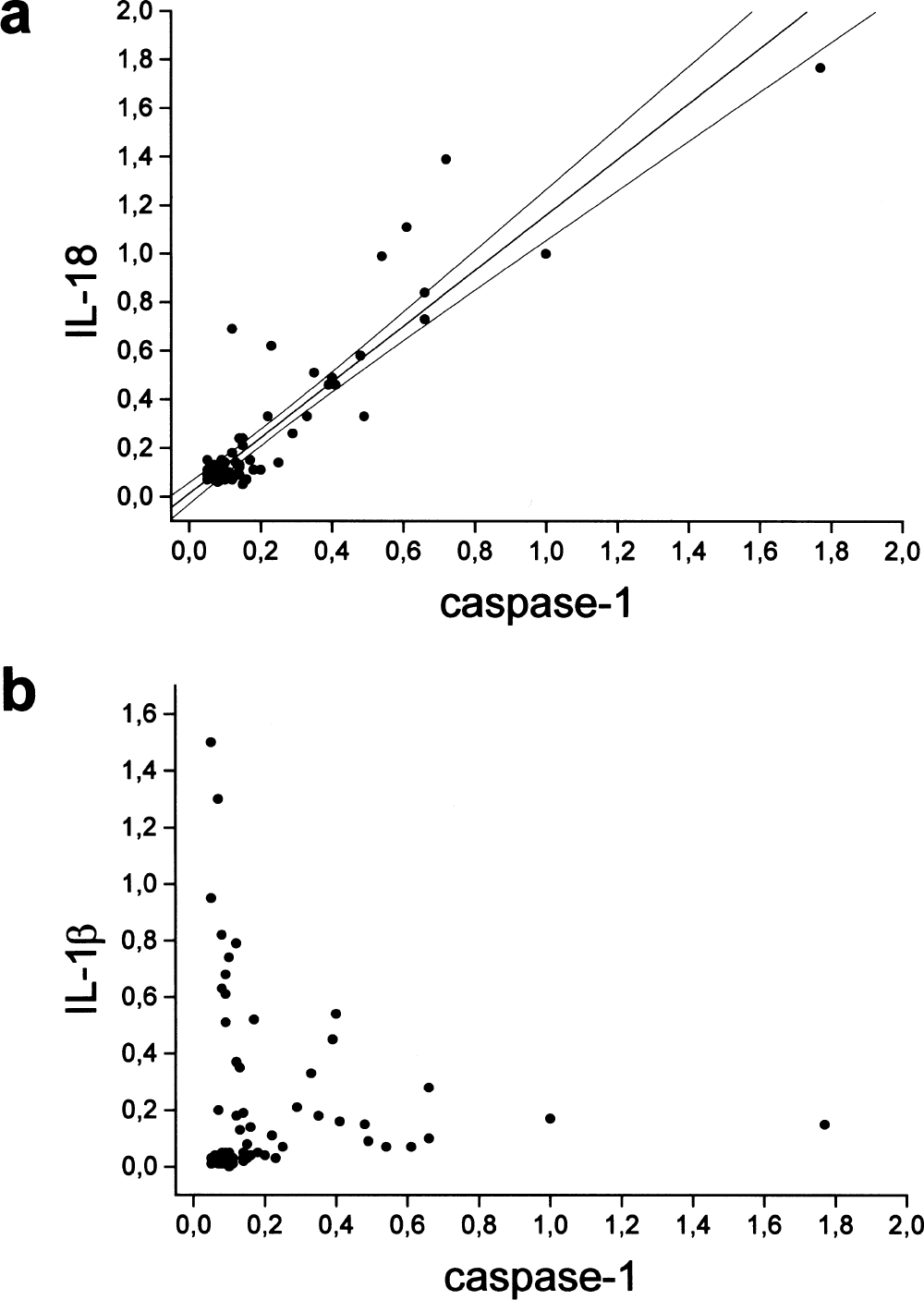
Relation between caspase-1 mRNA and interleukin-18
Interleukin-18 mRNA induction in focal ischemia is not accompanied by interferon γ expression
Because interleukin-18 was originally discovered as an interferon γ-inducing factor (Okamura et al., 1995), we asked if the increase in interleukin-18 mRNA after focal ischemia was accompanied by the induction of interferon γ mRNA. As positive control for interferon γ mRNA expression, we used the EAE model in which interleukin-18 plays a key role in the activation of pathogenic T helper cells producing high levels interferon γ (Jander and Stoll, 1998; Wildbaum et al., 1998). In previous studies, we have shown that the expression of both interleukin-18 and interferon γ in the spinal cord is maximal at the stage of active disease progression between 11 and 13 days after immunization (Jander and Stoll, 1998; Jander et al., 1998a). Accordingly, in our present study we could easily detect significant levels of interferon γ mRNA in the spinal cord of EAE rats 11 days after immunization with a moderate number (29) of amplification cycles (Fig. 4). Under the same conditions, no interferon γ mRNA was detectable in ischemic lesions 3, 6, and 14 days after photothrombosis (Fig. 4); even increasing the cycle number to 35 yielded no reproducible interferon γ signal from these samples (data not shown). Identical findings were obtained in the MCAO model.
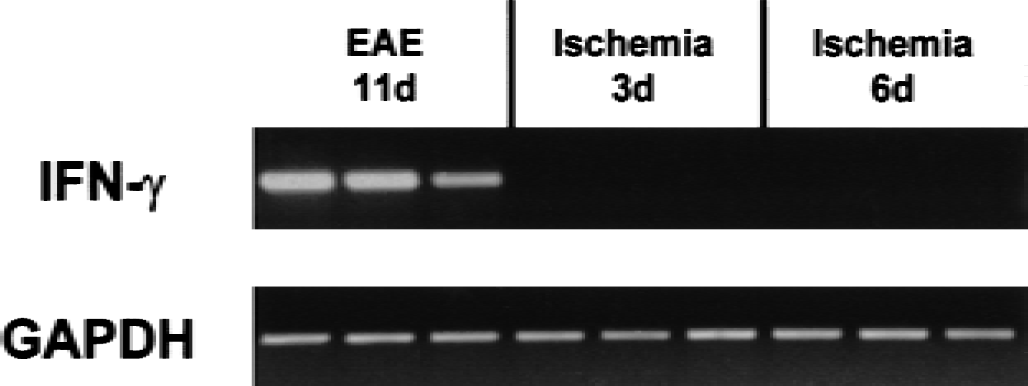
Lack of interferon γ mRNA induction after focal brain ischemia. Total RNA from either spinal cord of animals in the progressive phase of experimental autoimmune encephalomyelitis (11 days after immunization, lane 1–3) or ischemic lesions 3 (lane 4–6) or 6 days (lane 7–9) after photothrombosis were analyzed by reverse-transcriptase polymerase chain reaction using rat interferon γ-specific primers. Interferon γ mRNA is easily detectable in the experimental autoimmune encephalomyelitis spinal cord, but not in the ischemic brain lesions. Representative original findings are from three animals per group.
Localization of interleukin-18 immunoreactivity in focal ischemia
To extend the mRNA data to the protein level, we performed immunocytochemical studies using an affinity-purified goat polyclonal antibody against rat interleukin-18 on free-floating sections of paraformaldehyde-fixed tissue (Fig. 5). In the brains of untreated control rats, we found weak constitutive expression of interleukin-18 immunoreactivity that was localized to ramified microglia-like cells. There was no significant increase of interleukin-18 immunoreactivity in the lesions up to 24 hours after ischemia. At day 3 and more strongly at day 6, increased interleukin-18 staining was found in the lesions and was most pronounced in round phagocyte-like cells in the inner border zone and necrotic core of the infarcts (Figs. 5a, 5b). Additionally, ramified microglia in the outer border zone of the lesions expressed enhanced interleukin-18 immunoreactivity (Figs. 5a, 5c). At day 14, interleukin-18 immunoreactivity was decreasing. Thus, immunocytochemistry confirmed the delayed time course of interleukin-18 induction with a peak approximately 6 days after ischemia. Specificity of interleukin-18 immunostaining was proven by control experiments in which the primary interleukin-18 antibody was omitted or preadsorbed with excess recombinant rat interleukin-18 before staining. These procedures led to a complete disappearance of immunostaining in the ischemic lesions (Fig. 5d).
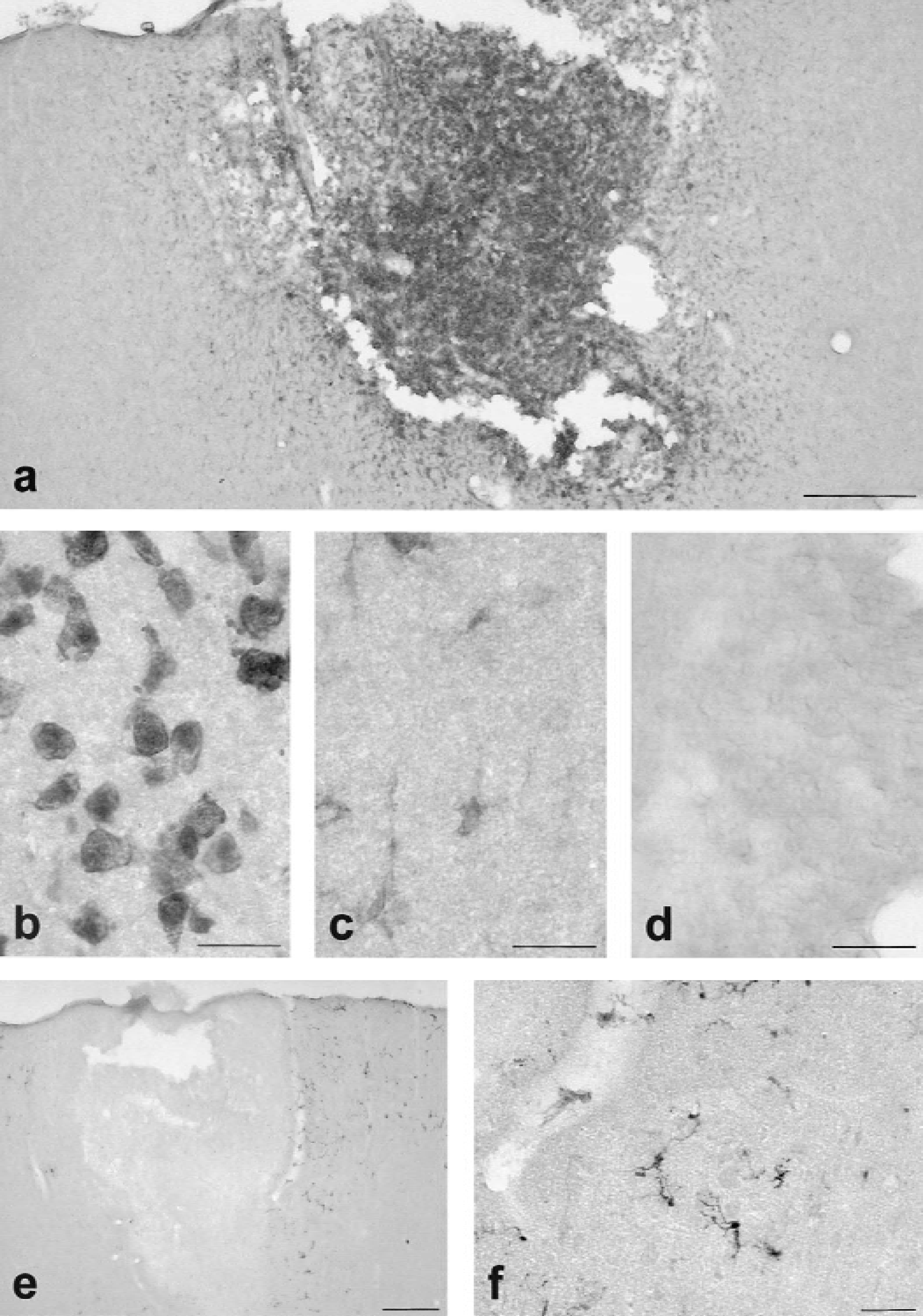
In contrast to interleukin-18, upregulation of interleukin-1β immunoreactivity was evident 4 hours after ischemia, peaked between 8 and 16 hours (Figs. 5e, 5f), and decreased thereafter. After 6 days, no significant interleukin-1β staining was visible. Morphologically, interleukin-1β immunoreactive cells had the typical appearance of ramified microglia and were essentially restricted to the outer border zone of the lesion with considerable extension into the neighboring nonischemic cortex (Figs. 5E, 5F). In line with our previous findings (Jander et al., 2000; Jander et al., 2001), significant numbers of interleukin-1β expressing ramified microglia were found in the ipsilateral cortex far remote from the lesion (data not shown). In contrast, the remote nonischemic cortex was always devoid of significant interleukin-18 induction.
To clarify the cellular source of interleukin-18 in cerebral ischemia, we additionally performed immunofluorescent double-staining combining the polyclonal interleukin-18 antibody with the monoclonal antibody ED1 against phagocytic macrophages and microglia. Analysis by confocal microscopy revealed extensive colocalization of interleukin-18 with ED1-positive microglia/macrophages in the ischemic lesions (Figs. 6a–6c).
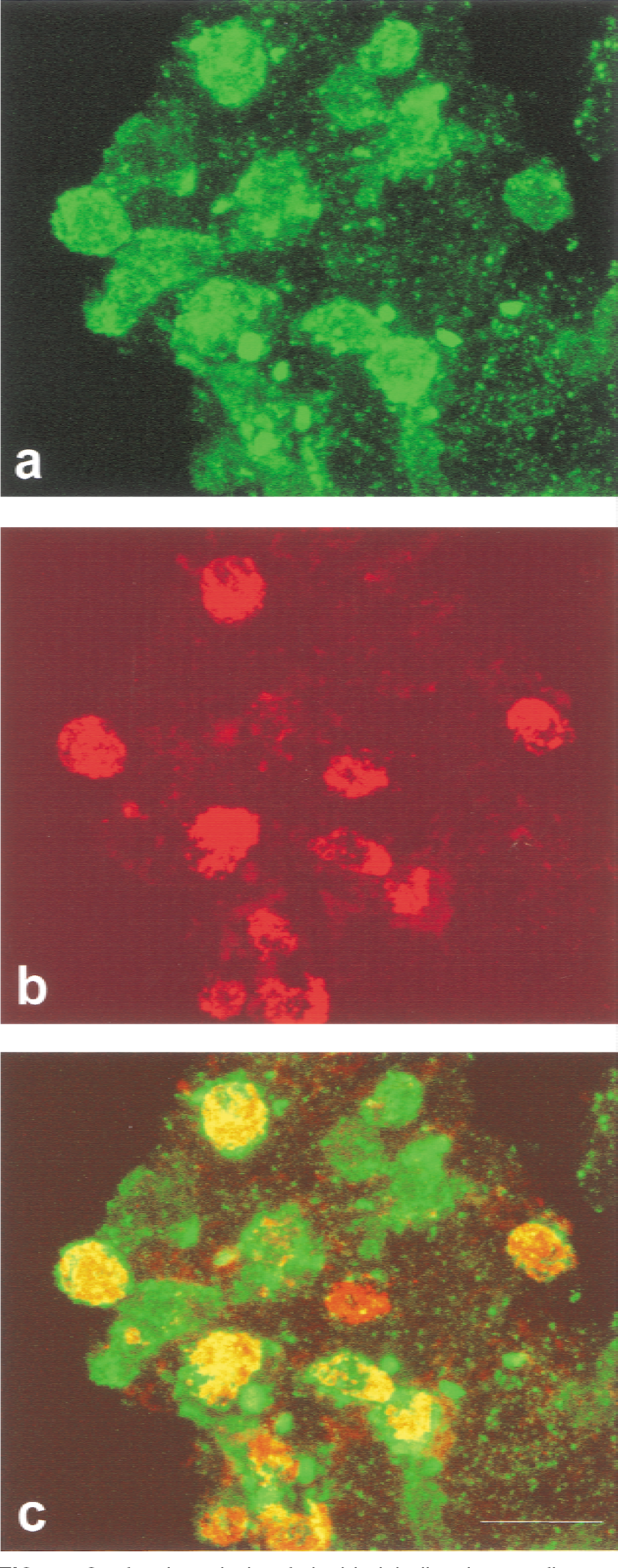
Confocal analysis of double-labeling immunofluorescence for interleukin-18 (green in
DISCUSSION
Interleukin-18 is a newly discovered cytokine that exhibits structural homology to the prototypic proinflammatory cytokine interleukin-1β. Therefore, interleukin-18 is considered a member of the growing family of interleukin-1–like cytokines (Okamura et al., 1995; Bazan et al., 1996; Kohno and Kurimoto, 1998; Touzani et al., 1999). Our present study is the first to show that focal ischemia of the rat brain leads to local induction of interleukin-18 in infarcted cortex and its immediate surroundings. Interleukin-18 mRNA and protein expression exhibited a delayed time course of induction, commencing at 24 to 48 hours and peaking 6 days after ischemia. Thereby, interleukin-18 differs from interleukin-1β, which is induced rapidly during the first few hours after ischemia and is dramatically downregulated after 2 days (Liu et al., 1993; Davies et al., 1999).
The initial inflammatory response to focal ischemia is mainly exerted by resident microglia, whereas hematogenous macrophages are recruited with a delay of at least 2 days (Schroeter et al., 1997). Accordingly, interleukin-1β-immunoreactive cells around the ischemic lesion had the morphology of resident ramified microglia, whereas interleukin-18 was mainly localized to round phagocytes infiltrating the infarct at the end of the first week after ischemia. Of note, by using mouse monoclonal antibody ED1 against a phagolysosomal antigen, activated resident microglia cannot be formally differentiated from hematogenous macrophages. Therefore, a contribution of both resident and blood-derived phagocytes to interleukin-18 synthesis during late stages after ischemia cannot be excluded. Similar to interleukin-1β, other proinflammatory cytokines, such as tumor necrosis factor α and interleukin-6, are upregulated within a narrow time window of the first 12 to 24 hours after ischemia (Liu et al., 1994; Wang et al., 1995; Jander et al., 2000). Thus, interleukin-18 represents the first proinflammatory cytokine associated with the delayed inflammatory response to focal brain ischemia.
In previous studies, we have shown that induction of interleukin-1β and tumor necrosis factor α after focal ischemia is not restricted to the infarcts, but is likewise observed to a significant extent in noninfarcted cortex of the entire ipsilateral hemisphere (Jander et al., 2000). Using pharmacologic blocking experiments with the N-methyl- d -aspartate receptor antagonist MK-801, we could show that this remote cytokine response in nonlesional cortex is elicited by cortical spreading depression (Jander et al., 2000; Jander et al., 2001). Spreading depression can be triggered by the local application of high-molar potassium chloride to the intact cortex, and is furthermore commonly observed in the surrounds of focal ischemic lesions (Hossmann, 1994). In our present study, interleukin-18 expression was restricted to macrophages and microglia in the ischemic lesion and the immediate surroundings, whereas the nonischemic cortex remote from the lesion was devoid of interleukin-18 immunoreactivity. Therefore, our data show highly differential spatiotemporal regulation of proinflammatory cytokines of the interleukin-1 family after focal brain ischemia.
Both interleukin-18 and −1β are primarily synthesized as inactive precursor molecules that exhibit considerable structural homology (Bazan et al., 1996) and necessitate cleavage by caspase-1 (previously designated interleukin-1β–converting enzyme) for conversion to the mature active cytokines (Ghayur et al., 1997; Gu et al., 1997; Akita et al., 1997). Thus, apart from a direct effect on early apoptotic cell death (Friedlander et al., 1997), blockade of caspase-1 may represent an intriguing way to downregulate inflammation as a potential mechanism of delayed lesion growth (Yang et al., 1999; Rabuffetti et al., 2000). In our present study, expression of caspase-1 mRNA was tightly correlated with the delayed upregulation of interleukin-18, but not with the early induction of interleukin-1β mRNA. In fact, in both the MCAO and photothrombosis model, caspase-1 mRNA levels did not increase significantly until 2 days after ischemia. This finding is consistent with a previous Northern blot study by Asahi et al. (1997) showing constitutive expression of caspase-1 mRNA that did not change within the first 24 hours after ischemia. Therefore, the early increase of caspase-1 bioactivity described by Rabuffetti et al. (2000) may be explained by proteolytic cleavage of caspase-1 precursor molecules rather than by de novo synthesis of caspase-1 mRNA. Activation of caspase-1 early after ischemia is also suggested by the beneficial therapeutic effect of interleukin-1β antagonists in experimental stroke models (Relton and Rothwell, 1992).
Interleukin-18 was discovered based on its ability to stimulate secretion of interferon γ by T helper cells and was subsequently characterized as an important regulator of T helper cell differentiation, which overall promotes delayed type hypersensitivity reactions mediated by T helper type-1 cells (Kohno and Kurimoto, 1998). However, at least at the time points examined in our study, we did not find significant expression of interferon γ mRNA in the ischemic lesions under conditions that easily allow for its detection in the model of EAE as a classical Th1-mediated central nervous system autoimmune disease. This finding is consistent with previous findings in traumatic brain injury (Rostworowski et al., 1997) and cutaneous wound healing (Kampfer et al., 2000). However, independent from its interferon γ–inducing properties, interleukin-18 induces expression of the adhesion receptor ICAM-1 on endothelial and monocytic cells, which might provide a basis for interferon γ–independent proinflammatory actions of interleukin-18 (Kohka et al., 1998; Kitching et al., 2000). It remains to be studied whether interference with the expression of interleukin-18 or its activation by caspase-1 provides a means to alleviate late-stage inflammatory responses to focal ischemia.
Footnotes
Acknowledgments:
The authors thank B. Blomenkamp and A. Tries for excellent technical assistance, and Dr. R. Kubitz for help with confocal microscopy.