
Abstract
Stroke leads to energy failure and subsequent neuronal cell loss. Creatine and phosphocreatine constitute a cellular energy buffering and transport system, and dietary creatine supplementation was shown to protect neurons in several models of neurodegeneration. Although creatine has recently been found to reduce infarct size after cerebral ischemia in mice, the mechanisms of neuroprotection remained unclear. We provide evidence for augmented cerebral blood flow (CBF) after stroke in creatine-treated mice using a magnetic resonance imaging (MRI)-based technique of CBF measurement (flow-sensitive alternating inversion recovery-MRI). Moreover, improved vasodilatory responses were detected in isolated middle cerebral arteries obtained from creatine-treated animals. After 3 weeks of dietary creatine supplementation, minor changes in brain creatine, phosphocreatine, adenosine triphosphate, adenosine diphosphate and adenosine monophosphate levels were detected, which did not reach statistical significance. However, we found a 40% reduction in infarct volume after transient focal cerebral ischemia. Our data suggest that creatine-mediated neuroprotection can occur independent of changes in the bioenergetic status of brain tissue, but may involve improved cerebrovascular function.
Keywords
Introduction
Ischemic stroke results from an acute perfusion deficit leading to the disruption of metabolism, cellular energy supplies, ionic homeostasis and finally loss of cellular integrity in the infarct core. In the surrounding penumbral tissue, cerebral blood flow (CBF) is moderately reduced and a complex pathophysiologic cascade determines neuronal and glial cell fate (Dirnagl et al, 1999). Creatine and phosphocreatine may bolster cell survival, and together with creatine kinase (CK) constitute a temporal and spatial buffering system for cellular energy stores. Creatine kinase can generate adenosine triphosphate (ATP) from phosphocreatine and adenosine diphosphate (ADP) at the sites of high-energy demand and restore phosphocreatine for energy storage (Wallimann et al, 1998). Several recent studies have shown dramatic neuroprotective effects of oral creatine administration in animal models of neurodegeneration, such as Huntington's disease (Matthews et al, 1998; Ferrante et al, 2000), Parkinson's disease (Matthews et al, 1999) and amyotrophic lateral sclerosis (Klivenyi et al, 1999). Moreover, creatine was shown to be protective in models of acute neuronal damage, such as traumatic brain injury (Sullivan et al, 2000) and stroke (Zhu et al, 2004). Creatine protected neurons from anoxia in vitro (Carter et al, 1995; Parodi et al, 2003) and from hypoxia—ischemia in neonatal rat brain (Adcock et al, 2002). In addition, creatine reduced beta-amyloid toxicity, as well as glutamate excitoxicity, in cultured hippocampal neurons (Brewer and Wallimann, 2000). In addition, creatine improved suvival and differentiation of GABA-ergic and dopaminergic neurons from human embryonic brain (Andres et al, 2005a, 2005b). As a nutritional supplement, creatine is used by athletes and in patients afflicted with neuromuscular disorders (Tarnopolsky and Martin, 1999). Clinical trials attempting to assess the neuroprotective potential of creatine in neurodegenerative diseases are ongoing.
In this study, we sought to elucidate the primary target of creatine-mediated neuroprotection in an experimental model of adult stroke. While it has been suggested that creatine reduces tissue injury in the ischemic penumbra by buffering of ATP levels and inhibition of activation of caspase cell-death pathways (Zhu et al, 2004), these effects could be secondary and result as a consequence of improved tissue preservation in creatine-treated animals. Since it is well known that creatine crosses the blood—brain barrier with difficulty (Perasso et al, 2003), we hypothesized that the prophylactic effect of creatine administration in stroke may be mediated in part by improved cerebral vascular function.
Materials and methods
Male 129S6/SvEv mice 7 weeks or older were fed either an unsupplemented control diet or a diet supplemented with 0%, 5%, 1% or 2% creatine (Altromin, Germany) for the times indicated. Anhydrous creatine (purity 98%; Sigma, Germany) was titurated into the chow and pellets were generated at 55°C for <1 min. High-performance liquid chromatography analysis of the chow confirmed the concentrations of creatine and excluded conversion to creatinine. All surgical procedures were approved by the local authorities. Investigators were masked to the treatment groups.
Experimental Stroke
Middle cerebral artery occlusion (MCAO) was performed as described (Prass et al,. 2003). Briefly, a monofilament was introduced into the left common carotid artery under halothane anesthesia, advanced to the origin of the middle cerebral artery (MCA) and left for 45 mins until reperfusion (n=8 to 10/group). During surgery, the rectal temperature was maintained at 37.0°C. Mice were killed after 4 days. The brains were snap-frozen. Infarct areas were quantified on hematoxylin/eosin-stained cryostat sections as described previously (Prass et al, 2003).
Biochemical Analysis
Measurements of creatine, phosphocreatine, ATP, ADP and adenosine monophosphate (AMP) were performed by two independent laboratories (D.M. and S.S.-I.; n=10 and 12). Mice were decapitated and the heads were immediately snap frozen in liquid nitrogen. The brains were removed from the skull at −20°C within 2 mins and homogenized with ice-cold 0.5 mol/L perchloric acid containing 0.5 mmol/L ethylene glycol-bis tetraacetic acid, the acid extracts were treated as described (Ipsiroglu et al, 2001). High-pressure liquid chromatography was performed as described (Furst and Hallstrom, 1992).
Cell Culture
Immortalized rat brain microvascular endothelial cells (rBEC6; Blasig et al, 2001) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (Biochrom, Germany) and 0.8% ECGF/Heparin (Promocell, Germany). At 3 days before hypoxia, cells were plated at a density of 25,000/well in the presence or absence of 2 mmol/L creatine (Sigma, Germany). Oxygen glucose deprivation (OGD) was perfomed for 180 mins as described (Ruscher et al, 2002). Creatine (2 mmol/L) was added to creatine-pretreated wells at the onset of OGD. After 1 h of reoxygenation, 3-[4,5]dimethylthiazol-2,5-diphenyltetrazolium bromide (MTT) assay was performed as described (Mosmann, 1983). Briefly, MTT (Sigma) was added to the cultures at a final concentration of 0.5 mmol/L. After 35 mins of incubation, 0.01
Isolated Middle Cerebral Artery
Middle cerebral arteries were prepared as described (Lindauer et al,. 2003). Mice (n=17 to 18/group) were anesthetized with isoflurane and decapitated. The main trunk of the MCA was dissected from the brain, cannulated on glass micropipettes in an organ chamber, and perfused continuously at a transmural pressure of 80 mm Hg. Luminal diameters were analyzed online using an inverted microscope. Vessels were allowed to equilibrate for 1 h while spontaneous tone developed, reducing the resting diameter to ∼80% of the initial value. Reactivity to increased extraluminal potassium or extraluminal acidosis was examined.
Magnetic Resonance Imaging
Mice were anesthetized with 1.5% isoflurane in an oxygen/air mixture and MCAO was performed as described above. Immediately after MCAO, mice were fixed using a stereotactic frame and positioned in the magnet bore. Respiratory rate and body temperature were monitored with an MR-compatible physiology monitoring unit and temperature was maintained within physiologic limits using a heated water jacket.
MRI was performed on a Bruker 7T PharmaScan® 70/16 with a Bruker 98/38 mm RF Coil, operating on Paravision software platform (Bruker, Germany). Perfusion weighted imaging (PWI) was performed with a spin echo echo planar imaging sequence with a 180° Gauss RF inversion pulse (imaging parameters: echo time=16.23 ms; imaging slice thickness=2 mm; image matrix=64 × 64; field of view=20.141 × 20 mm; inversion parameters: inversion slice thickness=6 mm; pulse length=1ms) modified from published protocols of flow-sensitive alternating inversion recovery (FAIR) imaging (Detre et al, 1992; Kim, 1995). Five neighboring slices at 2 mm distance between olfactory bulb and cerebellum were selected. For each slice, 2 × 11 images were acquired for one CBF image: the first series with a slice selective inversion, the second with a nonselective inversion (Figure 4A). The first image was performed with an inversion time (TI) of 114 ms, in each of the succeeding 10 images the TI was increased by 800 ms (TI=114…8114 ms). From the resulting relaxation curves, the longitudinal relaxation time constant T1 was obtained for the slice selective and slice nonselective inversion by fitting the data points according to S(TI) = A+∣S0(1 − 2eminus;TI/T1)∣ with S(TI) being the signal intensity after TI, A the absolute bias, S0 the signal intensity at TI=0 s (Figure 4B). For each timepoint, T1 was determined for slice selective inversion (T1sel) and for slice nonselective inversion (T1nonsel) (Figure 4C). Cerebral blood flow was quantified using by CBF = λ((1/T1sel) − (1/T1nonsel272)) with ë being the blood—tissue water partition coefficient (assumed to be 0.9 ml/g; Herscovitch and Raichle, 1985) (Figure 4D). After each acquisition of PWI, diffusion weighted imaging (DWI) was performed in the same sample volume with a Stejskal-Tanner-like multislice spin echo echo planar imaging sequence (imaging parameters: echo time=66.5 ms, Δ=25 ms, diffusion gradient duration=5 ms, 10 slices with slice thickness 1 mm, image matrix=128 × 128, field of view=20 × 20 mm, 15 averages). In each sequence, one image without diffusion weighting (b=0 s/mm2) and one with diffusion weighting (b=1500 s/mm2) was acquired to allow for a quantitative analysis of the apparent diffusion coefficient (ADC). The ADC was calculated using ADC = − 1/bln(S(b)/S0) with b being the b-value, S(b) the signal intensity with diffusion gradient, S0 the signal intensity without diffusion gradient (b=0). The sequence was repeated three times, each time with a different direction of the diffusion gradient (orthogonal in read, slice and phase direction), resulting in a total scan time of 20 mins. To obtain an isotropic diffusion estimate, the mean ADC of these three directions was calculated. During MCAO, one PWI (15 to 30 mins post-MCAO) and one DWI acquisition (30 to 55 mins post-MCAO) was performed. After 60 mins of MCAO, reperfusion was allowed and two more sets of PWI and DWI were obtained.
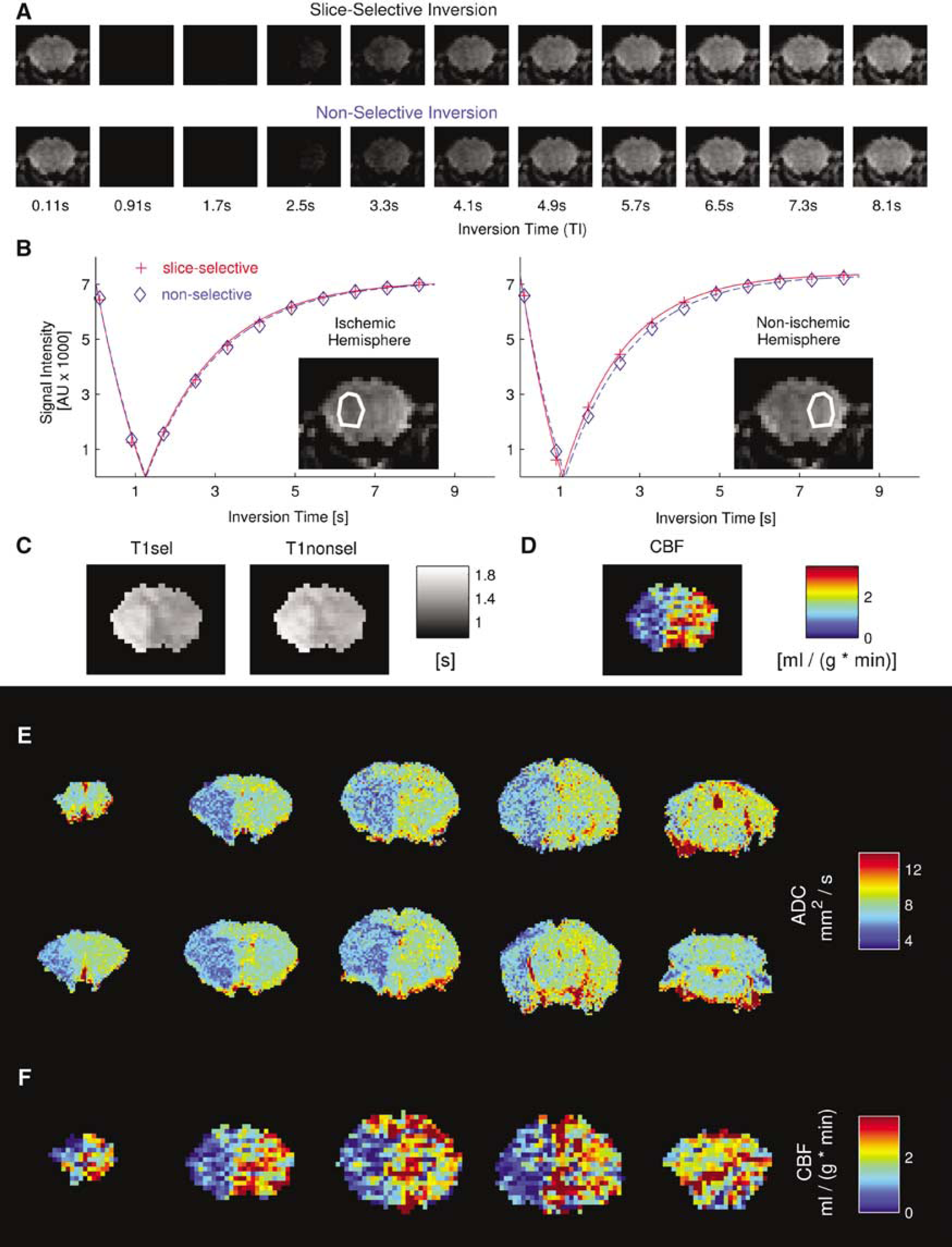
In vivo MRI measurement of perfusion and diffusion during and after cerebral ischemia. Representative PWI/FAIR-MRI analysis (
For quantitative analysis, each slice was divided into two horizontal symmetrical halves. Omitting the two most posterior 1-mm-slices in DWI and the most posterior 2-mm-slice in PWI, hemispheric ADC and CBF values of the remaining slices were averaged for each time point.
Statistics
A two-tailed Student's t-test (two groups) or one-way ANOVA with subsequent Bonferroni correction (>2 groups) were used. Values for P<0.05 were considered statistically significant.
Results
Infarct Volumes
We examined the effects of 3 weeks of dietary creatine supplementation on infarct volumes in mice (Figure 1). Oral administration of 1% and 2% creatine resulted in a significant reduction in infarct volumes compared with control diet. We found a dose-dependent neuroprotective effect of creatine, leading to the reduction of lesion sizes by 8%, 37% and 42% when 0.5%, 1% and 2% oral creatine were administered, respectively. To determine whether creatine could also protect mice from stroke when given after the onset of ischemia, we injected 30 mg/g body weight of creatine intraperitoneally at the end of surgery. The altered route of administration was required since we found that food ingestion varied in mice after stroke. Based on previously published results (Perasso et al, 2003), a more than 1000-fold increase in serum creatine concentration will result from this treatment. However, this high dose of creatine did not reduce infarct sizes when administered after stroke (Figure 1).
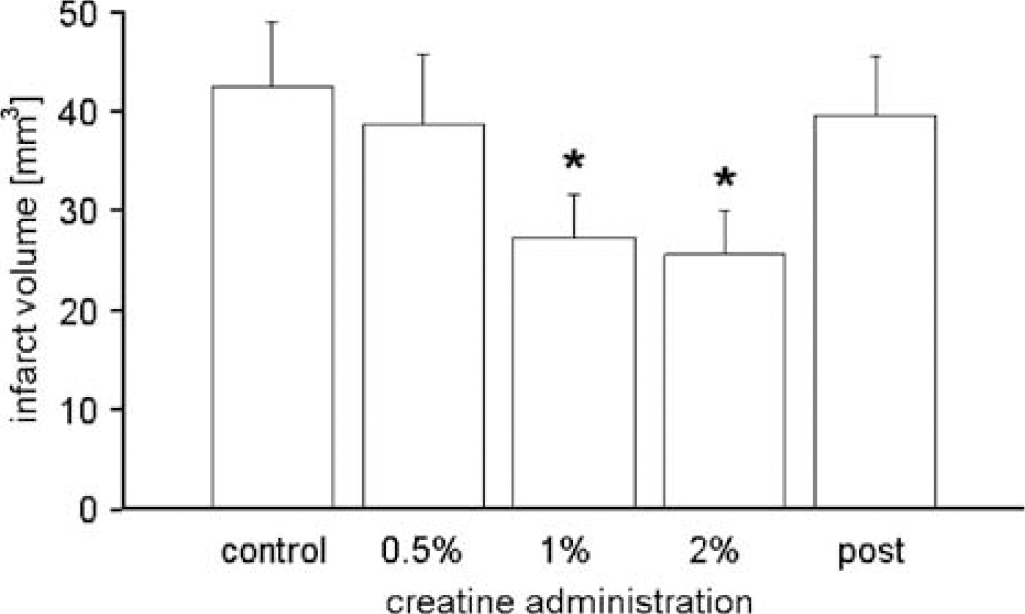
Reduced infarct size after oral creatine administration. Infarct volumes after 45 mins of MCA occlusion in mice fed for 3 weeks with control diet (control) or diet supplemented with 0.5%, 1% and 2% creatine. Post-treatment (post) of mice with a single dose of creatine i.p. (30 mg/g body weight) after MCAO. Data are presented as means+s.d. (n=8 to 10/group). ∗ indicates statistical significance between control and creatine-administered groups at P<0.05 (ANOVA with Bonferroni's correction).
Since the occurrence of ischemic events in patients at risk for stroke cannot be anticipated, long-term oral creatine supplementation would be required. We therefore administered 2% oral creatine or control diet to mice for 12 months and subjected them to 45 mins of MCAO. Surprisingly, the protective effect of creatine was abolished and no differences in infarct volumes were observed (control: 60.9 mm3±37 versus creatine: 58.4 mm3±35).
Brain Energy Metabolites
We presumed that the neuroprotective effects of creatine in stroke would be a function of the concentration of energy metabolites in the brain. In studies performed in two independent laboratories (DM and SS-I), total creatine, creatine, phosphocreatine and ATP concentrations in brain were found to be similar, irrespective of whether the animals had received control or 2% creatine-supplemented diet for 3 weeks (Figure 2). The creatine/phosphocreatine ratio approximated 3:1. Absolute ATP concentrations and the ratios of ATP/ADP and ATP/AMP were not significantly different between both groups, and provided no evidence for run down of high-energy phosphates. HPLC-analysis of the chow confirmed the concentrations of creatine and excluded conversion to creatinine (data not shown).
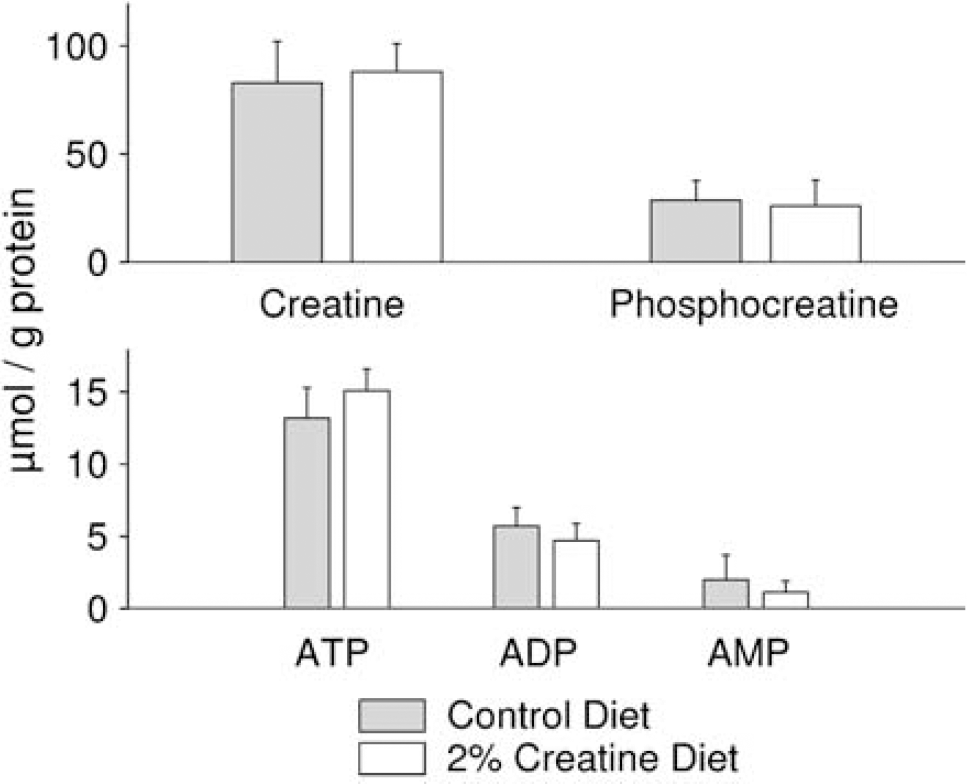
Bioenergetic status of brain tissue is not altered by creatine administration. Concentrations of creatine, phosphocreatine and energy-rich phosphates in the brains of mice fed for 3 weeks with control diet (n=10, gray bars) and diet supplemented with 2% creatine (n=10, white bars). The data are presented in μmol/g protein as means+s.d. No significant differences in metabolite levels were detected between the groups (unpaired two-tailed Student's t-test).
In the setting of cerebral ischemia, phosphocreatine can help restore depleted ATP levels through the activity of CK. Thus, the neuroprotective effects of oral creatine administration could also be mediated by the modulation of CK activity in the brain. We measured CK activities 24 and 72 h after cerebral ischemia in the ischemic and nonischemic hemispheres of mice fed control or 2% creatine-enriched diet. No significant differences in CK activities were found between both groups (data not shown).
Vascular Function
Since creatine did not cross the blood—brain barrier in our model, we hypothesized that its neuroprotective effect could result from improved cerebral vascular function. In immortalized brain microvascular endothelial cells, treatment with 2 mmol/L creatine for 3 days significantly reduced MTT cleavage by viable cells (Gerlier and Thomasset, 1986) after oxygen-glucose deprivation (control: OD 510.5±69.9 creatine-treated: OD 469.4±65.8; P=0.03) without affecting the cell number (data not shown). Vascular reactivity of isolated MCA was examined after 3 weeks of 2% creatine versus control diet (Figure 3). After equilibration, spontaneous tone developed and reduced vessel diameters to 109±10 and 105±9 μm in the arteries from creatine-treated and control mice, respectively (P=0.23). The dilation in response to elevated extraluminal potassium concentrations of 40 and 60 mmol/L was significantly increased in arteries from creatine-treated compared with control animals (P=0.01 and 0.03, respectively). Moreover, the dilatory response of isolated MCA to acidosis was also significantly enhanced (P=0.001). In contrast to non-cerebral vessels or the basilar or posterior cerebral artery, middle cerebral arteries only constrict in response to very high concentrations of extraluminal potassium (hypertonic, >80 mmol/L), and dilate in response to concentrations up to 70 to 80 mmol/L, as shown previously for rats (Golding et al, 2000).
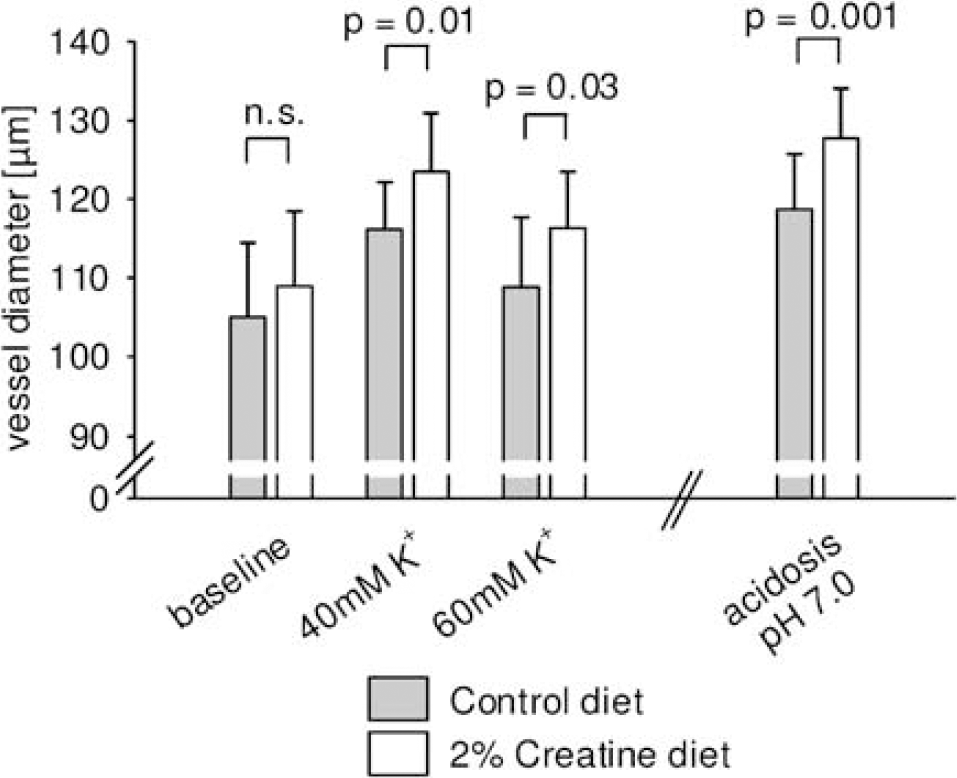
Improved vasodilatory responses in middle cerebral arteries from creatine-treated mice. Absolute diameters of isolated MCA derived from mice fed for 3 weeks with control diet (n=18, gray bars) and diet supplemented with 2% creatine (n=17, white bars) in response to increased extraluminal potassium (40, 60 mmol/L) and acidosis (pH 7.0). The data are presented as means+s.d. Under baseline conditions, mean vessel diameters in creatine-fed mice were not significantly different (NS) compared with controls. All other conditions showed significantly higher mean vessel diameters in creatine-fed mice compared with controls (P-values from unpaired two-tailed Student's t-test with Bonferroni's correction).
In vivo Magnetic Resonance Imaging Measurement of Perfusion and Diffusion
To assess the functional consequences of altered vascular reactivity by creatine, we performed in vivo MRI measurements of CBF and tissue damage during ischemia and reperfusion. Respiratory rate and rectal temperature were kept within physiologic limits in creatine-treated and control mice (Table 1). We developed a novel technique of in vivo CBF measurement using FAIR-MRI in mice as described in detail under Material and methods (Figures 4A to 4D). Comparison of CBF values obtained using FAIR-MRI with autoradiographic analysis of CBF in mice revealed matching results (Leithner et al, unpublished observations). During MCAO, DWI (Figure 4E) and PWI (Figure 4F) showed a pronounced reduction in the ischemic MCA territory. For quantitative analysis, DW and PW images were averaged across each hemisphere. During acute ischemia, ADC and CBF decreased significantly (P<0.000001) in the ischemic compared with the nonischemic hemispheres of both creatine-treated and control animals. The reductions in ADC or CBF did not differ between treatment and control groups, suggesting that the severity of ischemia was comparable at the time of analysis (data not shown). However, creatine-treated mice recovered faster from ischemia (Figure 5): 15 to 35 mins after reperfusion, mean CBF was significantly higher in creatine-treated mice compared with controls (P=0.01). After 61 to 81 mins of reperfusion, the difference in CBF values between the groups was not significant (P=0.18). Interestingly, ADC recovery in creatine-treated mice lagged behind CBF recovery: 35 to 55 mins after reperfusion, only a trend towards higher ADC was observed in the treatment group (P=0.21), whereas ADC was significantly higher in the ischemic hemispheres of creatine-treated mice compared with controls after 75 to 95 mins of reperfusion (P=0.01).
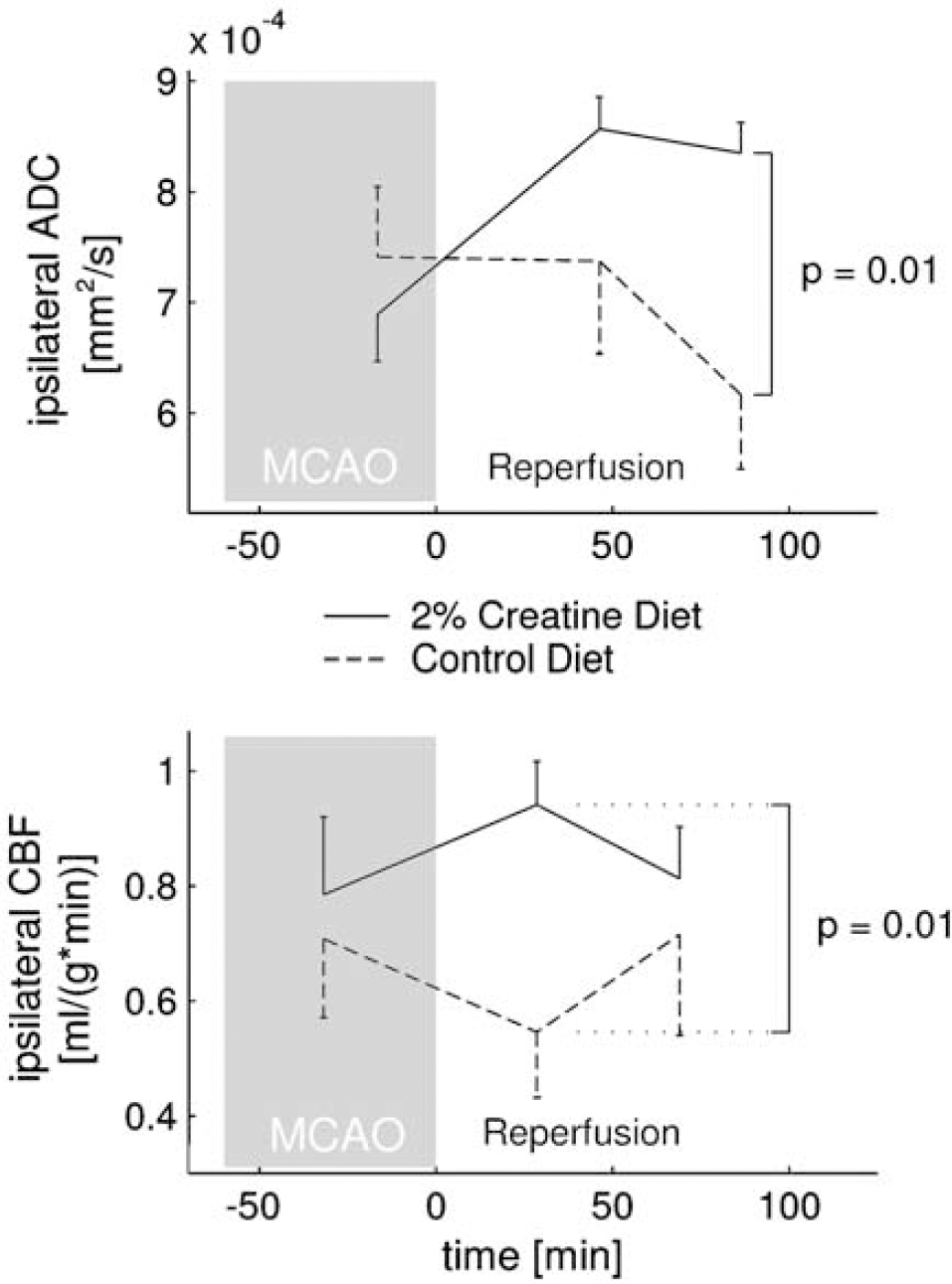
Reduced stroke size as a result of accelerated restoration of cerebral blood flow in creatine-fed mice. Diffusion weighted imaging of ADC (upper panel) and PWI/FAIR-MRI analysis of CBF (lower panel) during 60 mins of MCAO and up to 90 mins of reperfusion in mice fed for 3 weeks with control diet (n=6, dashed line) and diet supplemented with 2% creatine (n=6, solid line). Time courses of ADC and CBF in the ischemic hemispheres expressed as means±s.e.m. Where differences between groups are significant, P-values from unpaired two-tailed Student's t-test with Bonferroni's correction are shown.
Physiological parameters during MRI
Respiratory rate and body temperature of mice treated with creatine (n=6) and controls (n=6) were monitored during MRI. Alternating measurements of perfusion weighted imaging (PWI) and diffusion weighted imaging (DWI) were performed. One set of PWI/DWI was obtained during ischemia (15–55 mins after MCAO), two sets were obtained during reperfusion (15–95 mins of reperfusion). Data are presented as means±s.d.
Discussion
In this study, we found that short-term dietary (1% to 2%) creatine supplementation for 3 weeks dose-dependently reduced stroke size in mice. These findings are in line with recently published results using a more severe MCAO model in mice (Zhu et al, 2004). All studies found that creatine needed to be administered for weeks before stroke onset to be effective. Based on the fact that creatine crosses the blood—brain barrier poorly (Perasso et al, 2003), it is not surprising that 1 week of dietary creatine administration failed to protect mice from stroke and that even after 4 weeks of creatine-enriched diet, no increase in creatine concentration was observed in the nonischemic brain (Zhu et al, 2004). We also did not detect significant changes in the bioenenergetic status of brain tissue (levels of creatine, phosphocreatine and ATP) after 3 weeks of dietary creatine supplementation in mice. It should be noted that a comprehensive analysis of the pharmacokinetics of oral supplementation of creatine-monohydrate in different species revealed comparatively poor uptake of creatine into mouse brain, albeit a 32% increase in total brain creatine concentration was observed when mice were fed with creatine for 8 weeks (Ipsiroglu et al, 2001).
In addition to most of the previous studies that have linked neuroprotection with increased levels of creatine and/or phosphocreatine in brain (Matthews et al, 1998; Klivenyi et al, 1999; Matthews et al, 1999; Ferrante et al, 2000; Sullivan et al, 2000), we observed beneficial creatine effects in stroke that did not directly relate to the concentrations of creatine and energy-rich phosphates in the brain. These findings do not diminish the role of creatine as a buffering system for cellular energy stores, but suggest that creatine may have additional neuroprotective properties. Thus, creatine was recently reported to afford cytoprotection via direct antioxidant activity (Sestili et al, 2006). Moreover, creatine was found to act in concert with mitochondrial CK in inhibiting permeability transition pore opening, an early trigger of apoptosis (Dolder et al, 2003). While it has been suggested that creatine buffers ATP levels and inhibits the activation of caspase cell-death pathways in the striatal ischemic core (Zhu et al, 2004), these effects could result from improved tissue preservation in creatine-treated animals. In fact, ATP loss can be used to differentiate infarct core from energetically compromised penumbral tissue after stroke (Hata et al, 2000). It is therefore tempting to speculate that the prophylactic effect of creatine administration in stroke is not mediated primarily by buffering of ATP levels.
Instead, we propose that creatine may be taken up by vascular endothelial and/or smooth muscle cells without measurable contribution to total brain creatine concentration at 3 weeks of dietary supplementation. Both vascular endothelial cells (Decking et al, 2001) and smooth muscle cells (Ishida et al, 1991) contain creatine, as well as cytosolic and mitochondrial isoforms of CK. In support of our hypothesis, we found that creatine prevents the activation of brain endothelial cells in reponse to OGD. Moreover, ex vivo vascular reactivity to vasodilatory stimuli, which are pathophysiologically relevant in stroke, such as acidosis and increased extracellular potassium concentrations, is improved in response to creatine. This could be mediated by Ca2+-activated and/or ATP-sensitive potassium channels, which are known to be involved in vasodilatory responses to hypoxia/ischemia, acidosis and 40 mmol/L potassium (Reid and Paterson, 1996; Lindauer et al, 2003). Interestingly, sodium/potassium ATPase and ATP-sensitive potassium channels were recently found to be functionally coupled to CK (Guerrero et al, 1997; Selivanov et al, 2004).
To assess whether oral creatine administration also improves vascular function in vivo, we wanted to monitor CBF and ADC in the same animal during cerebral ischemia and reperfusion. To this end, we used MRI and adapted a FAIR-based technique of quantitative CBF measurement to mice. In the past, FAIR-MRI was used in rats (Detre et al, 1992; Tsekos et al, 1998) and gerbils (Pell et al, 1999). Recently, FAIR-MRI has also been used to investigate global CBF changes in a model of sickle cell disease (Kennan et al, 2004). To our knowledge, this is the first study that employs FAIR as a tool to monitor acute CBF impairment in a mouse model of stroke, opening the possibility of using this technique in future studies involving knockout mice. Interestingly, we found that creatine-treated animals recovered significantly faster from cerebral ischemia compared with controls. Notably, CBF was increased during reperfusion and tissue diffusion recovered faster as a result of creatine administration, which is more likely to result from direct effects of creatine on the vascular system as suggested by our ex vivo and in vitro data than increased demand by surving neurons as a result of neuroprotection.
Our findings may provide a novel additional mechanism of creatine-induced neuroprotection and suggest that creatine improves CBF after stroke by preserving vascular cell function. Interestingly, we found that life-long creatine administration failed to protect adult mice from stroke, suggesting that adaptive mechanisms could compromise the beneficial roles of creatine. This needs to be taken into consideration when creatine is to be employed long-term as a neuroprotective agent in patients afflicted with neuromuscular or neurodegenerative disorders. It would be interesting to determine whether the CBF changes we observed with short-term creatine administration persist with prolonged treatment.