
Abstract
Ischemic-type injury to developing white matter is associated with the significant clinical condition cerebral palsy and with the cognitive deficits associated with premature birth. Premyelinated axons are the major cellular component of fetal white matter and loss of axon function underlies the disability, but the cellular mechanisms producing ischemic injury to premyelinated axons have not previously been described. Injury was found to require longer periods of modelled ischemia than at latter developmental points. Ischemia produced initial hyperexcitability in axons followed by loss of function after Na+ and Ca2+ influx. N-methyl-D-aspartate- (NMDA) type glutamate receptor (GluR) agonists potentiated axon injury while antagonists were protective. The NMDA GluR obligatory Nr1 subunit colocalized with markers of small premyelinated axons and expression was found at focal regions of axon injury. Ischemic injury of glial cells present in early developing white matter was NMDA GluR independent. Axons in human postconception week 18 to 23 white matter had a uniform prediameter expansion phenotype and postembedded immuno-gold labelling showed Nr1 subunit expression on the membrane of these axons, demonstrating a shared key neuropathologic feature with the rodent model. Premyelinated central axons therefore express high levels of functional NMDA GluRs that confer sensitivity to ischemic injury.
Introduction
Glutamate is the major excitatory neurotransmitter of the central nervous system, acting upon a variety of metabotropic and ionotropic receptors. Among the ionotropic receptor class, N-methyl-D-aspartate-type glutamate receptors (GluRs) have unique properties that allow them to act as coincidence detectors, requiring both the presence of agonist and a depolarizing stimulus to evoke a signal in the postsynaptic cell. This is the case not only at synapses but also at the axon growth cone where receptor activation is a positive modulator of extension. 1 N-methyl-D-aspartate GluRs are also present in the axon cylinder neighboring the growth cone where their function is not known.2, 3, 4 Axonal expression is highest during early white-matter development, before the onset of myelination.5, 6 Although axons that have yet to myelinate (premyelinated axons) are generally resistant to ischemic conditions, 7 they are the major cellular component of the fetal white matter that is subject to focal and diffuse ischemic-type injuries giving rise to cerebral palsy and cognitive deficits in premature infants; two large and untreatable patients cohorts.8, 9, 10 A recent report highlighted the loss of unmyelinated axons in a fetal rabbit model of cerebral palsy, with a reduced number of myelinated axons at latter ages suggesting that the lost unmyelinated axons correspond to premyelinating axons. 11
Expression of NMDA-type GluRs on oligodendrocyte processes results in high ischemia sensitivity in these structures,12, 13 but the significance of expression on the premyelinated axon cylinder is not known. Excitotoxicity has been reported in immature central axons that have initiated oligodendrocyte ensheathment,14, 15 but susceptibility to excitotoxic injury in the period in axon development before the consolidation of synapses and loss of axon growth cones has not previously been examined. Indeed, axon injury mechanisms in the period before the consolidation of synapses and loss of axon growth cones have not previously been investigated. We have examined the injury mechanism operating in these clinically significant structures and have confirmed expression of NMDA GluR on human mid-gestation periventricular white-matter axons.
Materials and methods
UK home office regulations were followed for all experimental work, which was conducted in accordance with the relevant guidelines and regulations. The animal welfare and ethics committee of the University of Leicester approved all the experimental protocols. Rat optic nerves (RONs) were dissected from Lister-hooded rats between postnatal day 0 and 4 (referred to as ‘P2’), P8 or adult. Nerves were perfused with artificial cerebrospinal fluid (aCSF), composition (in mmol/L): NaCl, 126; KCl, 3; NaH2PO4, 2; MgSO4, 2; CaCl2, 2; NaHCO3, 26; glucose, 10; pH, 7.45, bubbled with 5% CO2/95% O2 and maintained at 37°C. Zero-Ca2+aCSF: CaCl2 was omitted and 50 μmol/L EGTA was added. Zero-Na+ aCSF: Na+ was substituted with choline. For oxygen—glucose deprivation (OGD), aCSF was replaced by glucose-free aCSF prebubbled with 95% N2/5% CO2 for at least 60 minutes. The chamber atmosphere was switched to 5% CO2/95% N2 during OGD. Osmolarity of solutions was measured and adjusted as required. Data are mean ±s.e.m., significance determined by t-test or ANOVA as appropriate. Glutamate receptor antagonists were from Tocris (Bristol, UK), all other reagents were from Sigma (Gillingham, UK). Drug treatments were initiated 10 minutes before the onset of OGD and were washed out 10 minutes after OGD, followed by a further 50 minutes recovery period.
Electrophysiology
The electrophysiologic recording techniques have been described recently. 16 In brief, compound action potentials (CAPs) were evoked and recorded with glass electrodes and peak-to-peak amplitude was used to assess changes in axonal integrity (see Fern et al 7 ). Compound action potentials were evoked via square-wave constant current pulses (Isostim A320, WPI, Sarasota, FL, USA), amplified (Cyber Amp 320, Axon Instruments, Sunnyvale, CA, USA), subtracted from a parallel differential electrode, filtered (low pass: 800 Hz), digitized (1401 mini, Cambridge Electronic Design, Cambridge, UK) and displayed on a PC running Signal software (Cambridge Electronic Design). Nonrecoverable CAP loss from the optic nerve indicates irreversible failure of axon function. 14
Immunohistochemistry
Rat optic nerves were fixed in 4% paraformaldehyde/0.1 mol/L PBS for 30 minutes before incubation in 0.1 mol/L PBS plus 20% sucrose w/v for 5 minutes and freeze sectioning. In all, 20 μm sections were subsequently blocked for 60 minutes in 0.1 mol/L PBS, 10% normal goat serum plus 0.5% Triton X-100. Sections were then incubated in this solution plus primary antibody overnight at 4°C. Antibodies were raised against: neurofilament-200 (NF-H) and neurofilament-70 (NF-L) (Chemicon, Merck, Temecula, CA, USA; 1:200 and 1:100, respectively), GFAP (1:100, Sigma), Thy-1 (1:100, Santa Cruz, Dallas, TX, USA), pan-NaV (1:100, Sigma), NG-2 (1:100, Upstate, Merck, Temecula, CA, USA) and Nr1 sub-unit of the NMDA GluR (1:100, Santa Cruz). Staining was detected using appropriate Alexa-conjugated secondary antibodies (1:1,000, Cambridge Bioscience, Cambridge, UK). See Salter and Fern 12 for further details of the antibodies used. Primary antibody omission controls were blank. Images were collected using an Olympus confocal microscope (Tokyo, Japan).
Cell Imaging
Live astrocyte imaging was performed on P2 RONs loaded for 60 minutes at room temperature in aCSF containing 10 μmol/L FURA-2-AM. After washing, nerve ends were fixed to a glass coverslip and sealed onto a Plexiglas perfusion chamber (atmosphere chamber; Warner Instruments, Hamden, CT, USA) with silicone grease. Artificial cerebrospinal fluid was run through the chamber (2 to 3 mL/min) and 95% O2/5% CO2 was blown over the aCSF. Cells were imaged on a Nikon Eclipse TE200 inverted epifluorescence microscope (Melville, NY, USA). Chamber temperature was maintained closely at 37°C with a combination of flow-through feedback tubing heater (Warner Instruments) and a feedback objective heater (Bioptechs, Butler, PA, USA). Cells were illuminated at 340, 360, and 380 nm (Optoscan monochromator; Cairn Research, Faversham, Kent, UK), and images were collected at 520 nm using an appropriate filter set (Chroma Technology, Bellows Falls, VT, USA). Images were taken with a cooled CCD camera (CoolSNAP HQ; Roper Scientific, Martinsried, Germany) every 60 seconds. Changes in 340:380 ratio were taken to indicate changes in [Ca2+]i. The 360 intensity (isosbestic point) was monitored to assess the capacity of cells to retain dye. The sudden loss of 360 signal correlated with loss of cell membrane integrity and the release of dye into the extracellular space (ECS). See Fern 17 for further details.
Electron Microscopy
P2 RONs were washed in Sorenson's buffer and postfixed in 3% gluteraldehyde/Sorenson's. Nerves were fixed (2% osmium tetroxide) and dehydrated before epoxy infiltration. Ultrathin sections were counter-stained with uranyl acetate and lead citrate and examined with a Jeol 100CX electron microscope (Welwyn Garden City, UK). Since effectively all axons in the adult RON are myelinated, nonmyelinated axons of the P2 nerve can be considered to be in an early stage of preparation for myelination. For morphometric analysis and viability scoring, axons within a minimum of three grid sections were outlined by hand (Image-J software, NIH) and axon area and perimeter measured. Axon diameter was taken as the mean of the longest and shortest widths. Grids were randomly selected and all identifiable axons within the area were included. Axon viability scores were assigned blind using the following scoring system. Axons were given one point for each of three well-established indicators of viability: 16 (1) the presence of an intact axolemma, (2) the presence of microtubules, and (3) the presence of a debris-free axoplasm. Axons that showed all three attributes were therefore given a viability score of ‘3’, and axons with none were given a score of ‘0’. Postembedded immuno gold labelling for Nr1 was performed as previously described for P10 RON. 16
Human Tissue
Tissue from five human fetuses ranging in age between 18 and 23 postconception weeks (PCWs) was studied. Collection of the tissue has been described previously 18 and involved a postmortem delay of 2 to 5 hours after medically induced abortion. Tissue was obtained with signed consent and followed institutional guidelines. In all, 1-mm3 subcortical blocks were cut from temporal cortex before fixation (1% paraformaldehyde/1.25% glutaraldehyde in PBS) overnight at 4°C. Tissue was then treated as for rat nerve described above, with large ultrathin sections mounted to allow identification of cortical layers and underlying white matter. Immuno-staining for Nr1 protein expression followed previously described protocol. 14
Results
Ischemic Injury of Premyelinated Central White-Matter Axons
The P2 RON CAP was biphasic and stable under control conditions (Supplementary Figure 1A). Consistent with prior reports, 7 initial experiments showed that a 60-minute period of OGD produced injury in adult RON but failed to damage P2 RON (Supplementary Figure 1B). Longer periods of OGD were tested on P2 RON and a significant CAP loss was produced by a 90-minute challenge. The initial response to OGD at this age was frequently a rise in amplitude that could be as large as 188.3% (e.g., Figure 1A, filled circles), with a mean of 122.0±10.3% after 15.5 minutes of OGD (Figure 1B, n=13). Mean CAP amplitude then decreased progressively, reaching 40.3±8.9% of baseline. Unlike white matter from older animals subject to 60 minutes OGD (e.g., Supplementary Figure 1A), P2 RONs exposed to 90 minutes of OGD failed to recover significantly after reinstitution of normoxic conditions (37.5±5.0% after 60 minutes aCSF, P>0.05; Figures 1A and 1B).The absence of recovery after 90 minutes of OGD and the tolerance to 60 minutes OGD indicate progressive and nonreversible conduction failure between these two time points.
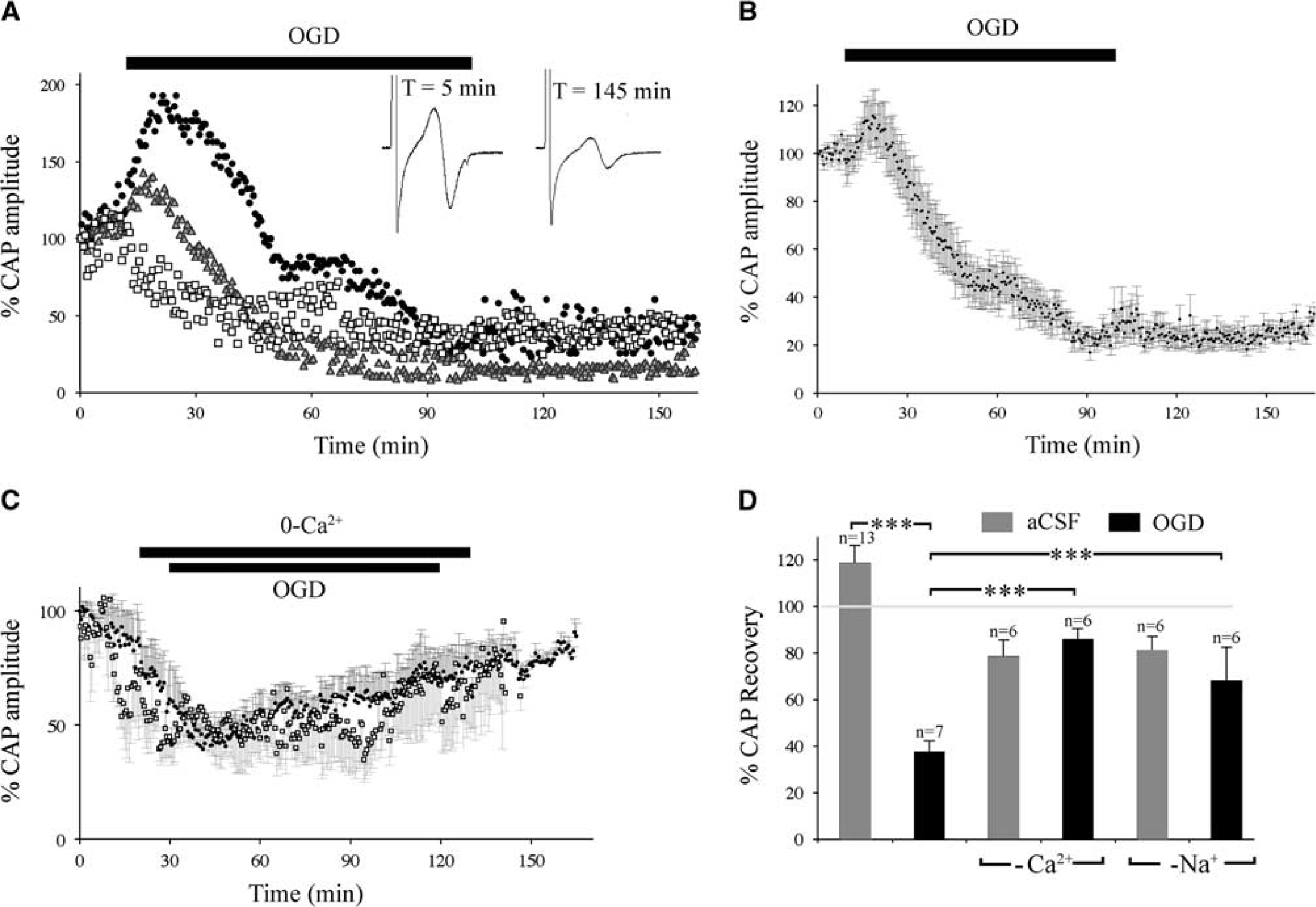
Ischemic injury in premyelinated central white matter. (
Ischemic-type injury of adult RON is both Ca2+ and Na+ dependent, 19 while at P10 RON injury is Ca2+ dependent/Na+ independent. 14 This difference is accounted for by the clustered expression of voltage-gated calcium channels on axons in the P10 RON that are yet to initiate myelination. 16 In the P2 RON, CAP declined under zero-Ca2+ conditions in otherwise normal aCSF (48.1±12.6% 90 minutes, n=6; Figure 1C), consistent with a large contribution to action potential conduction from voltage-gated calcium channels at this age. 16 This effect was largely reversed when normal aCSF was reinstituted (78.5±7.1%, n=6; Figure 1C). Surprisingly, exposure to 90 minutes OGD 10 minutes after initiating zero-Ca2+ conditions, with reintroduction of Ca2+ 10 minutes after OGD, resulted in a similar profile of CAP amplitude loss and recovery (Figure 1C, open symbols) to that seen in zero-Ca2+ alone (Figure 1C, filled symbols), suggesting that Ca2+-influx, progressive conduction failure and permanent injury occur over a similar time course under ischemic conditions. Consistent with this, there was no significant permanent loss of the CAP after OGD in zero-Ca2+ compared with zero-Ca2+ control (85.8±4.7%; Figure 1D). A similar protocol performed under zero-Na+ conditions had a similar protective effect (Figure 1D).
The Role of Ionotropic Glutamate Receptors
Application of the excitotoxin kainic acid (30 μmol/L: Supplementary Figure 2A) or the NMDA GluR coagonists NMDA (1 mmol/L)+glycine (10 μmol/L: Figure 2A) had no permanent significant effect on the CAP recorded from P2 RON (103.4±7.6%, n=6 and 88.4±11.1%, n=7 of initial CAP area, respectively). Similar findings have been reported in P10 14 and adult 20 RON and these current and prior observations suggest that GluR-mediated injury does not proceed rapidly in the presence of O2 and glucose. However, we found significant potentiation of OGD-induced injury by superfusion with NMDA+glycine (Figure 2D) and perfusion with the NMDA GluR antagonists MK-801 (1 μmol/L) or memantine (1 μmol/L) resulted in significant protection against OGD (Figures 2B and D). The less specific glutamate antagonist kynurenic acid had no effect either upon control CAPs or the response to OGD (Supplementary Figure 2B).
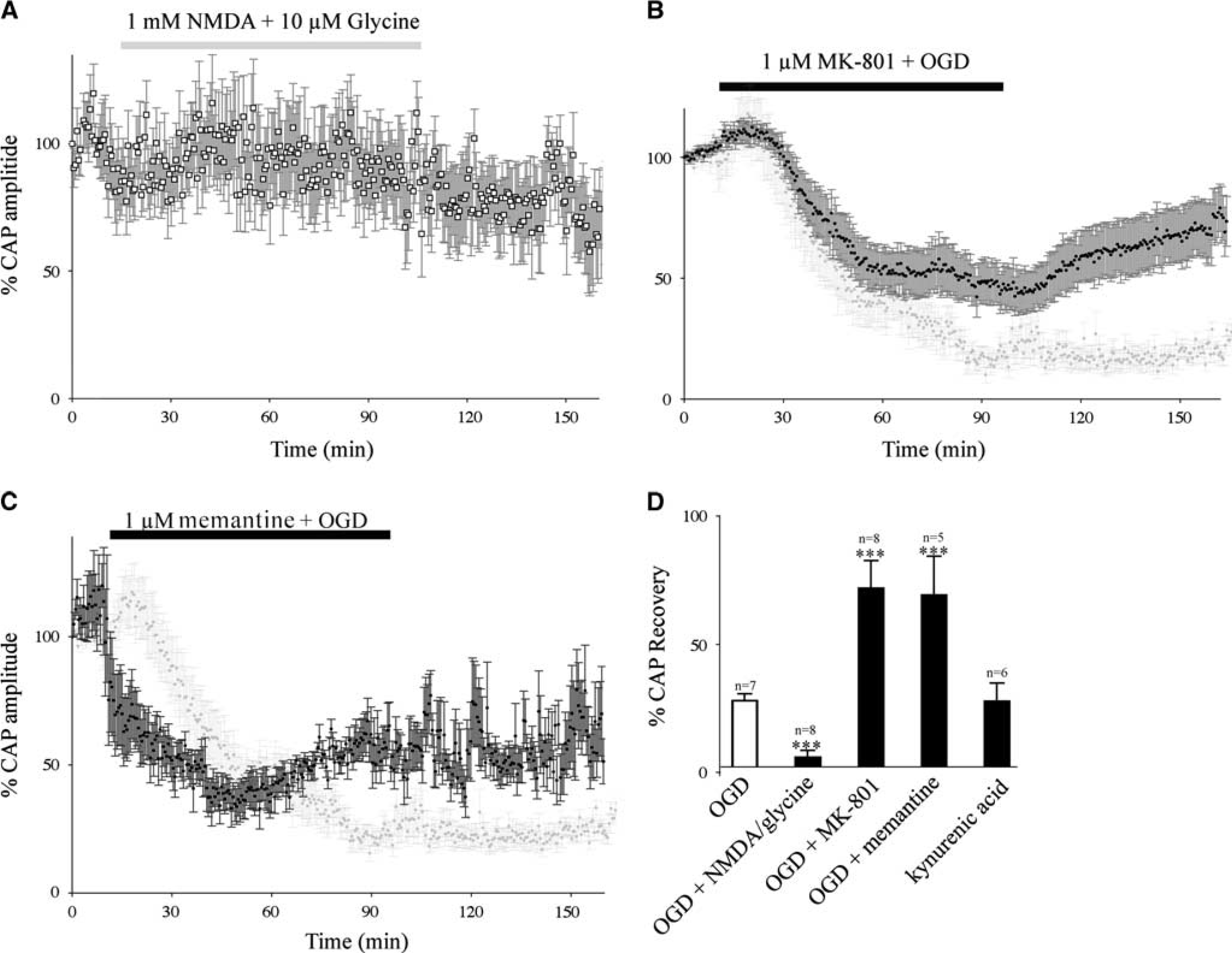
The significance of excitotoxicity. (
N-methyl-D-aspartate Glutamate Receptor Expression and Axon Injury
Expression of the obligatory NMDA GluR Nr1 subunit was probed in P2 RON. At the light level, costaining for the astrocyte marker GFAP revealed a degree of colocalization (Supplementary Figure 3A, e.g., arrows in middle panel), but the majority of Nr1 reactivity existed in the spaces between astrocytes (Figure 3A, ‘∗’ in middle panel). Note that the large majority of cells in P2 RON are GFAP(+) astrocytes. 21 Omitting Nr1 antibody from the staining protocol resulted in no staining using identical image collection and processing settings (Supplementary Figure 3, right panels). Costaining for Nr1 and the axonal markers NF-L (Supplementary Figure 3B), Thy-1 (Supplementary Figure 3F), and NF-H (Supplementary Figure 3G), all showed high levels of colocalization. In the P2 RON, NG-2 staining revealed a population of pericytes aligned along blood vessels that did not express Nr1 (Supplementary Figure 3C). Note, NG-2(+) oligodendrocyte precursors invade the nerve from >=P222, 23 and NG-2(+) cells with the morphology of oligodendrocyte precursors extended processes that were Nr1(+) in P10 RON (Supplementary Figure 3D, arrows), consistent with prior reports.12, 13
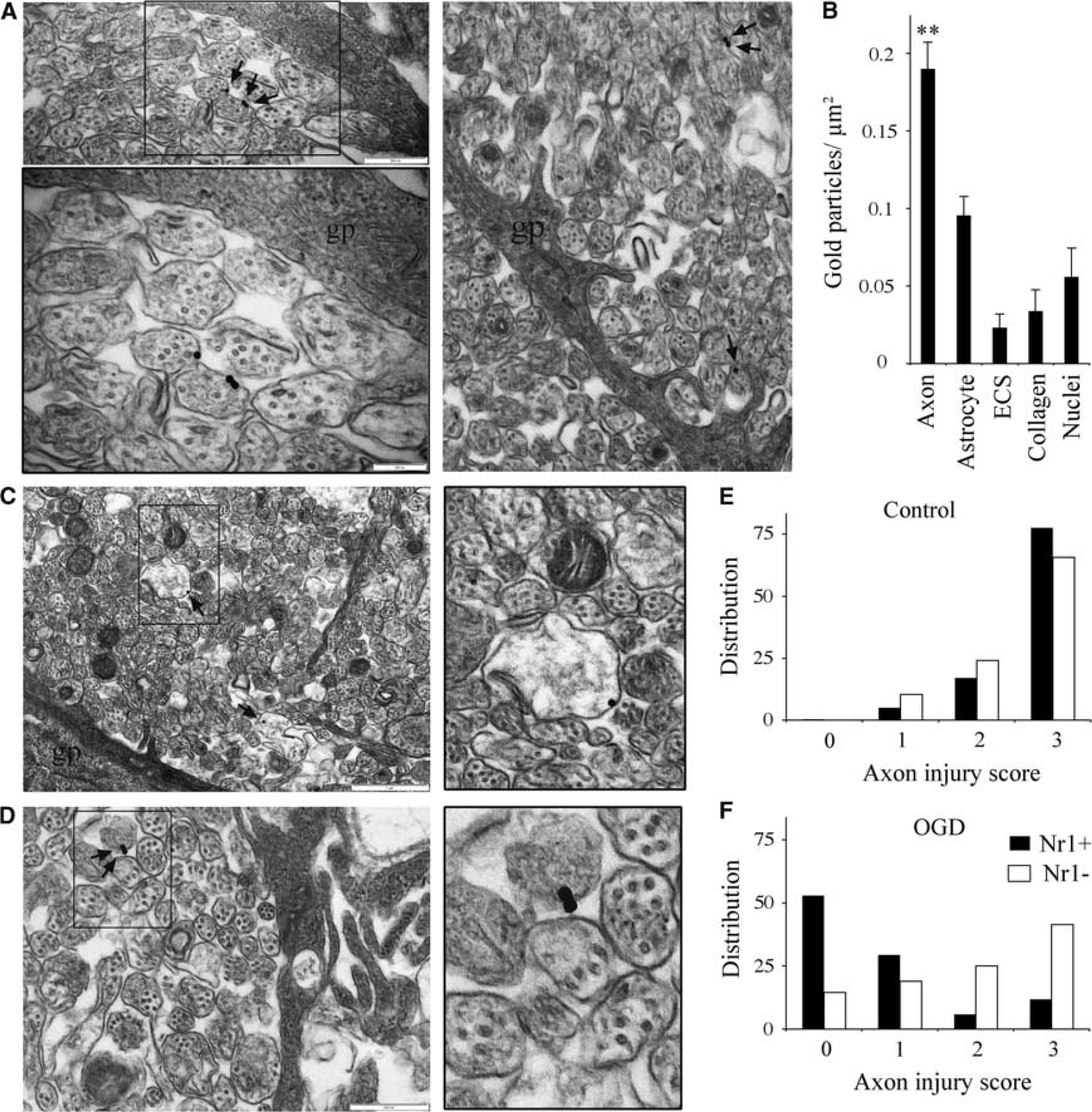
Ultrastructural colocalization of Nr1 on P2 axons correlates with injury. (
Colocalization of markers at the light level is difficult in a structure like the neonatal RON, where numerous and heterogeneous small structural elements are closely apposed. We therefore used postembedded immuno-gold localization of Nr1 expression at the ultrastructural level to confirm regions of reactivity on axolemma at sites proximal and distal to astrocyte somata and processes (Figure 3A). To assess the selectivity of this staining, gold-particle distribution was assessed throughout six whole sections of P2 RON. Gold particles had a significantly higher density in axons compared with astrocytes and the ECS, and lower densities were found in structures such as collagen fibers of the perineurium and cell nuclei which will represent background levels of staining (Figure 3B).
In P2 RONs subject to 90 minutes of OGD, pathologic changes in the ultrastructure of axons included swelling, loss of microtubules, accumulation of debris in the axoplasm and frank axolemma dissolution (Figures 3C, 3D, and 6). Immuno-gold localization of Nr1 expression was successful in areas of the nerve where axon injury was less extensive (Figures 3C and 3D), revealing frequent reactivity on damaged axons. Staining was also present in more severely damaged regions of the nerve, but it was not possible to ascribe such staining to any specific cell structure and these area are not therefore included in this analysis. The extent of axonal injury in control perfused and postOGD RONs was assessed blind using an established viability scoring system. In control nerves, the mean viability score was 2.72±0.03 (where a score of 3 is no pathology) for Nr1(−) axons (n=240), and 2.55±0.10 for Nr1(+) axons (n=58; P>0.05). The distribution of viability scores was similar for these two groups of axons (Figure 5E). In postOGD nerves, the mean viability score was 1.94±0.18 for Nr1(−) axons (n=364), and 0.77±0.06 for Nr1(+) axons (n=34; P<0.001 versus, Nr1(−) axons). The distribution shows viability scores skewed to low values in the Nr1(+) axons (Figure 3F). The data, therefore, show a strong correlation between sites of high Nr1 reactivity and axon injury.
To analyze the extent of glial and axonal injury after OGD, random high-power fields were collected blind from cross-section ultra-micrographs. Typical fields of axons in control perfused, postOGD and postOGD+MK-801 are shown in Figures 4A and 4C. The density of identifiable axons fell from 12.1±1.8 axons/μm2 to 2.3 axons/μm2 afte 90 minutes of OGD+60 minutes recovery (n=6 sections each; P<0.01); with a density of 5.75±1.5 axons/μm2 after OGD+MK-801 (n=6 sections; P<0.01 versus, control; Figure 4D). The mean viability score of those axons was 2.69±0.03 in control (n=298), 1.12±0.07 after OGD (n=398; P<0.001 versus, control) and 2.20±0.05 after OGD+MK-801 (n=327; P>0.05 versus, OGD alone and <0.05 versus, control; Figure 4E). This degree of axonal protection against OGD-induced injury is similar to the electrophysiologic protection found in Figure 2D. When the distribution of injury scores was examined, OGD+MK-801 was similar to control (Figure 4F).
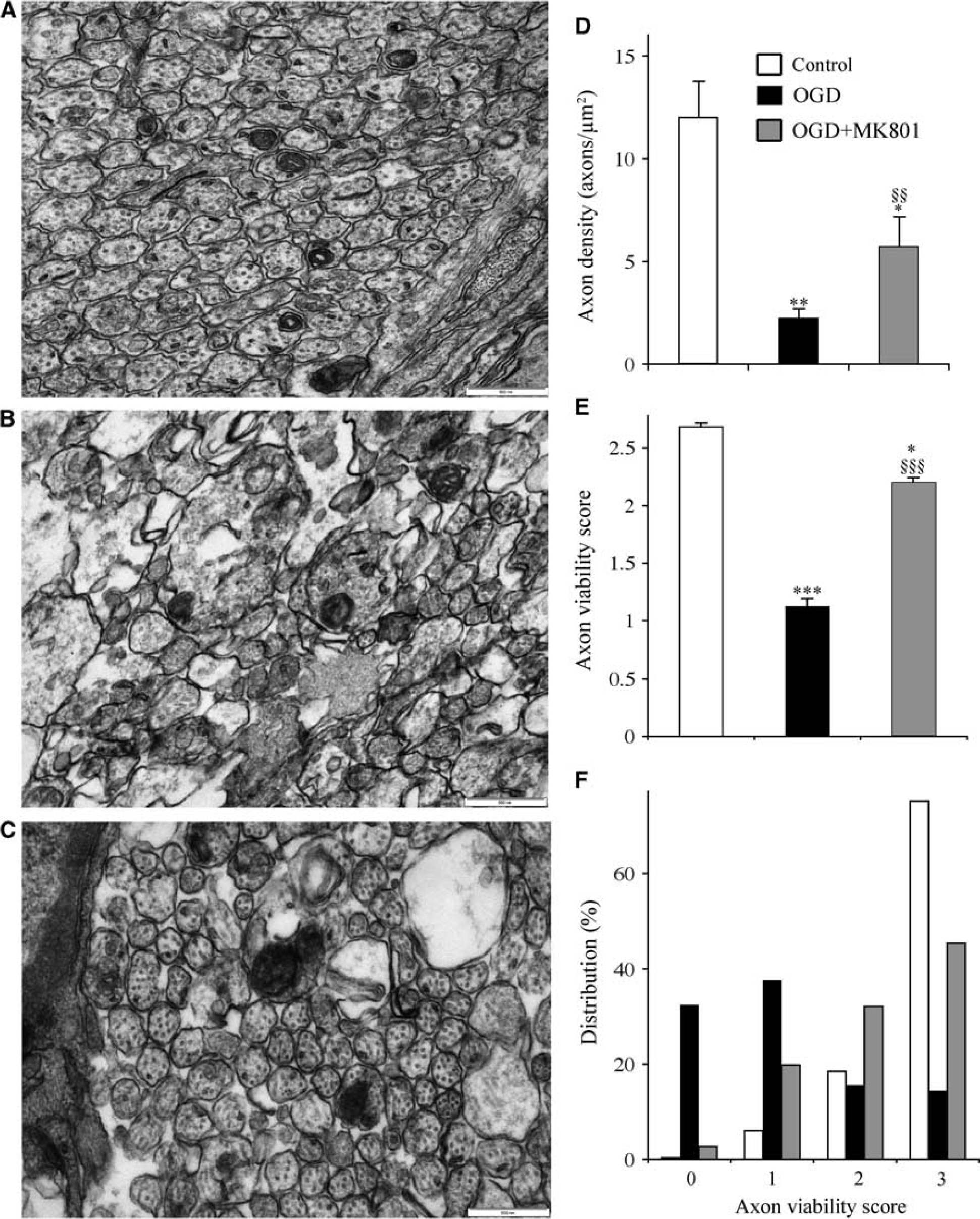
N-methyl-D-aspartate (NMDA) receptor block is protective against oxygen—glucose deprivation (OGD)-induced disruption of axon structure. (
Since NMDA GluR Nr1 reactivity was found in some astrocytes at the light level, intracellular [Ca2+] ([Ca2+]i) and cell viability in astrocytes was assessed via FURA-2 cell imaging. As previously reported, 17 FURA-2-AM loading produced dye filling of numerous cells in RON at this age (Figures 5A and 5E). The onset of a 90-minute period of OGD resulted in elevation in the Ca2+-dependent 340/380 ratio in the presence of MK-801 (Figure 5B). Under these conditions, Ca2+ rises were frequently followed by breakdown of cell membrane and release of FURA-2 into the extracellular space, apparent as loss of cell fluorescence (e.g., the cell indicated by a white arrow in Figure 5A, which disappears from the image after 40 minutes of OGD; these data are plotted in Figure 5B where cell death is apparent as a sudden decline in the 360 signal indicated by the black arrow). MK-801 did not significantly reduce the extent of cell death produced by OGD (Figure 5C), and did not greatly change the time distribution of cell death (Figure 5D). To ensure that FURA-2 loaded cells were astrocytes, the dye was fixed in whole mount nerves that were subsequently antibody labelled for GFAP (see Fern 17 ). This analysis showed that the great majority of FURA-2 loaded cells were GFAP(+) astrocytes (Figure 5E), consistent with the previous observations. 17
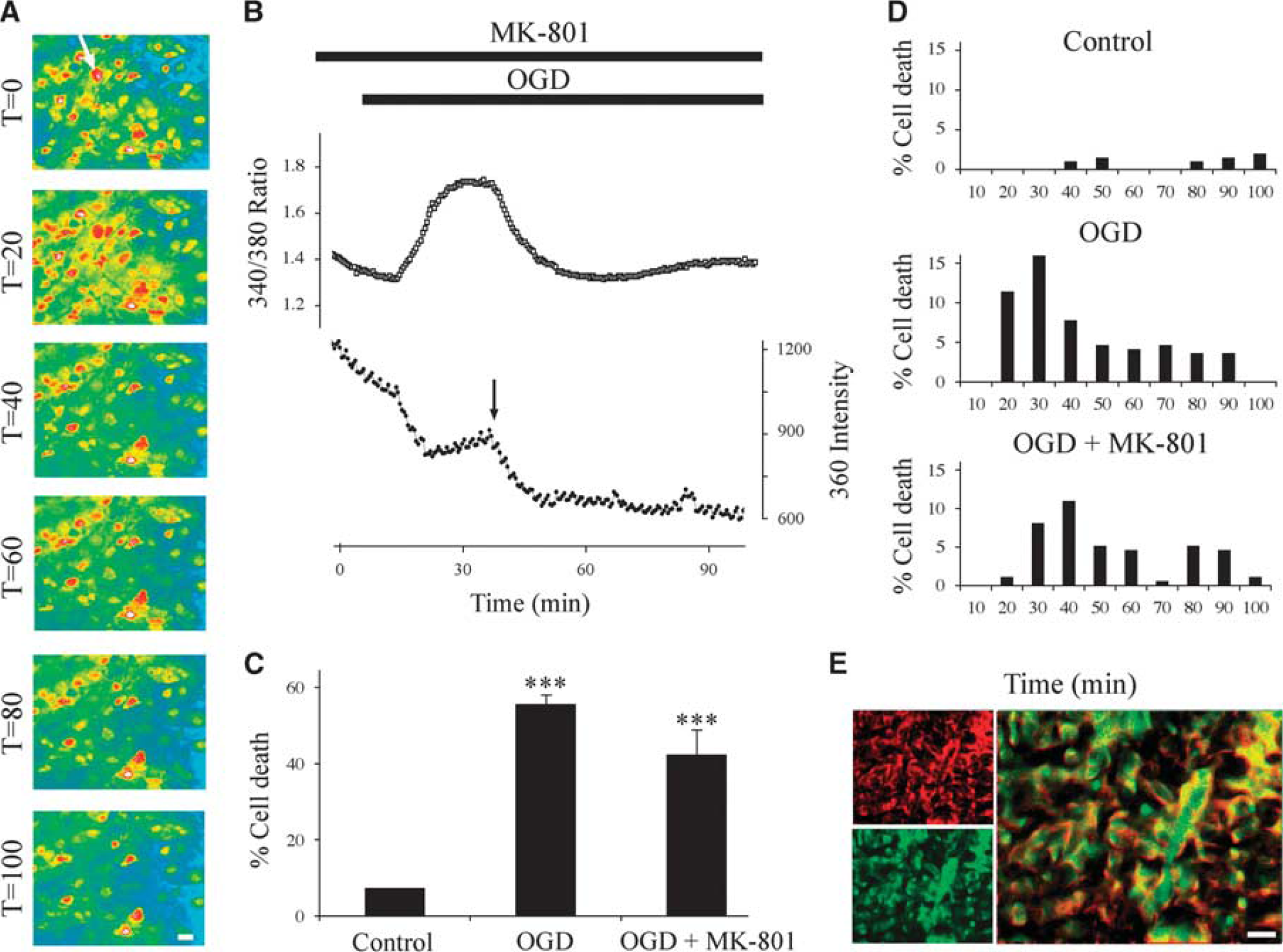
N-methyl-D-aspartate (NMDA) receptors do not mediate Ca2+ influx or cell death in P2 rat optic nerve (RON) astrocytes. (
Ultrastructural analysis revealed wide scale swelling of astrocyte organelles (Figure 6A), cell necrosis (Figure 6B) and gross swelling and dissolution of astrocyte processes (Figure 6C) in postOGD+MK-801 P2 RONs. Disruption of astrocyte processes was particularly evident at the nerve perimeter, where the astrocytic elements of the glial limitans neighboring the basement membrane were frequently grossly enlarged with a fragmented cell membrane (e.g., Figure 6C, ‘∗’). In contrast, axons with a normal structure were generally preserved even when surrounded by severely damaged astrocyte processes (Figure 6C, inset). Blinded assessment of astrocyte injury (Figure 6D) and the distribution of injury scores (Figure 6E) confirmed the absence of protection from OGD-induced injury in ultrastructurally identified astrocytes by MK-801.
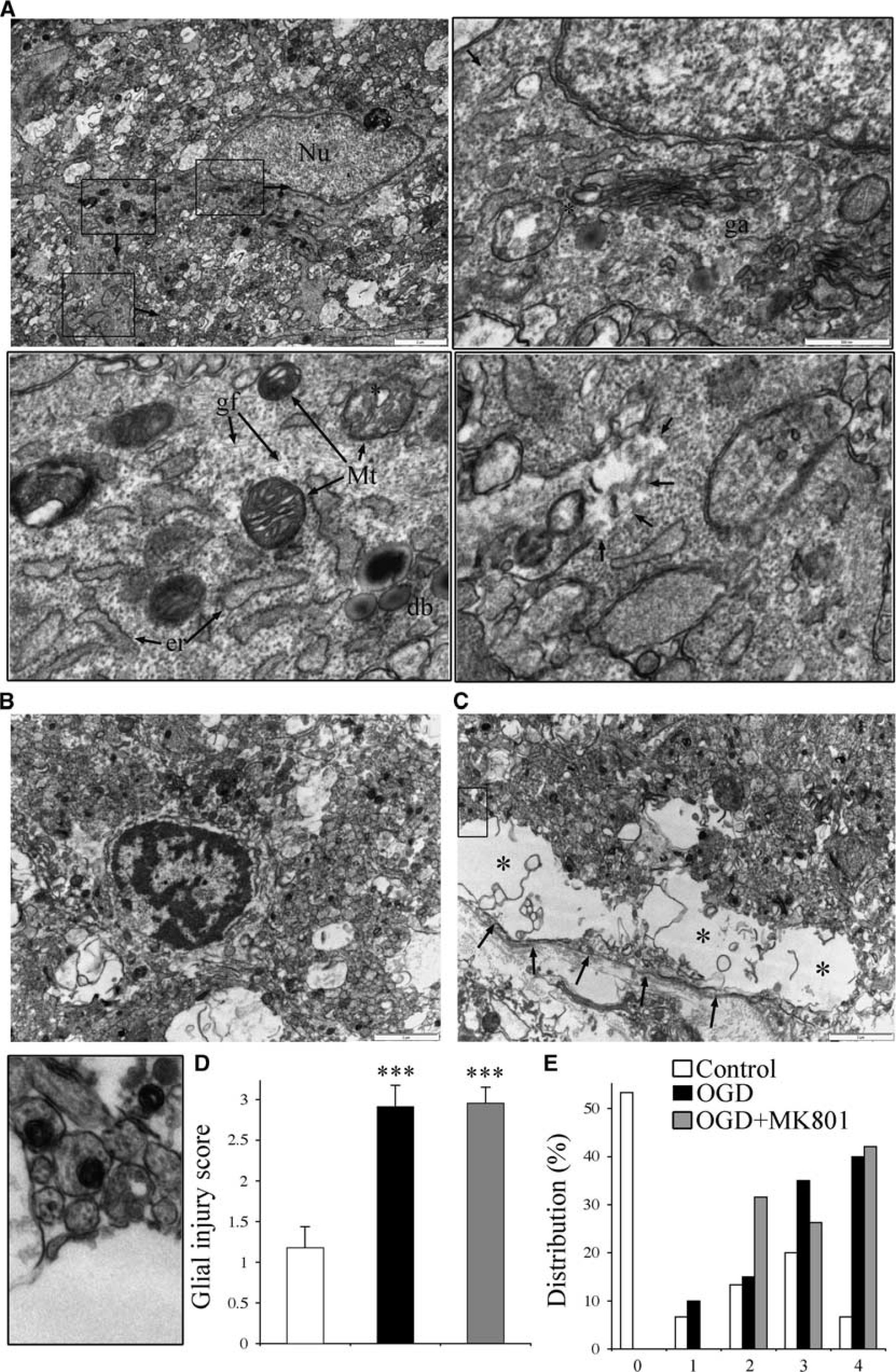
Glial injury is non-NMDA glutamate receptor (GluR)-R mediated. (
The incidence of periventricular leukomalacia (PVL) in the human fetus peaks between PCW 23 and 32.8, 24 It is unclear whether axon maturation has initiated in periventricular white matter at the beginning of this period since the major phase of oligodendroglial progression from the O4(−) precursor to O4(+)/O1(−) pre-oligodendrocyte stage does not accelerate until PCW 28.25, 26 In animal models such as the RON, axon diameter expansion initiates several days before the maturation of O4(+) oligodendroglia, 27 and is accompanied by loss of Nr1 reactivity (Figure 3). Formative white matter in fetal intermediate zone at PCW 18 to 23 was therefore analyzed and axons were compared with those of the overlying cortical layers. Axons were readily identified in both regions by their cylindrical structure and the presence of microtubules (Figure 7, e.g., ‘∗’). Axons were generally found in clusters embedded within fields of larger, less electron dense and less cylindrical processes that contain either a few tubular or filamentous elements (Figure 7, ‘P;) that may be growth cones, or contained glial filaments identifying them as astrocyte processes (Figure 7G, ‘AP’). In formative gray-matter layers of the cortex, neurons (Figure 7A, ‘N’) were often densely packed around small fields of processes. Gray-matter axons at this stage were rarely larger than 1 μm in diameter and most were in the 100- to 200-nm range (Figure 7A, e.g., ‘∗’). Astrocytes (Figure 7F, ‘A’) and occasional glioblasts (Figure 7H, ‘G’) populated the underlying intermediate zone were axons were observed in both cross-section and long-section with axon diameters under 400 nm (Figures 7B and 7H).
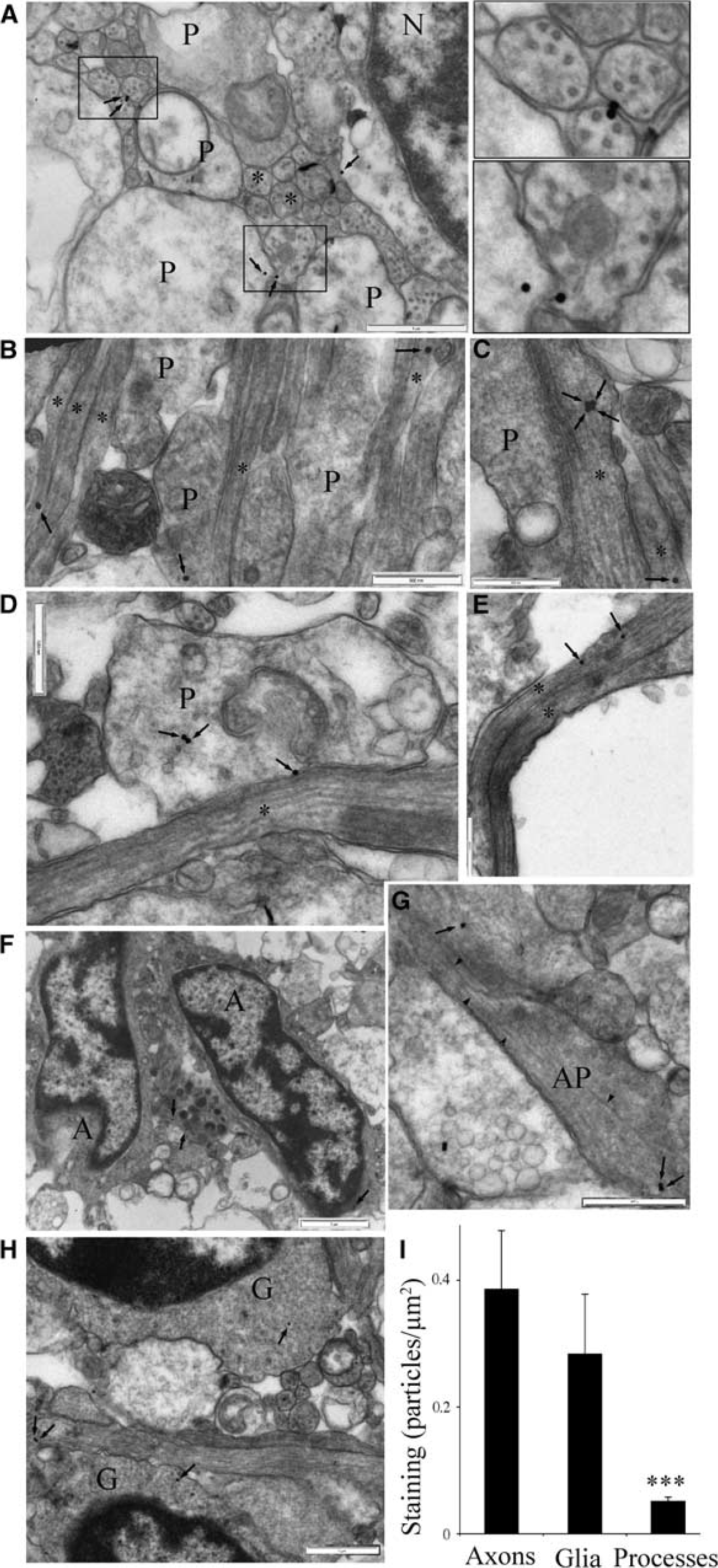
Nr1 expression in mid-gestation human cortex. (
Postembedded immuno-gold labelling for Nr1 revealed reactivity in both putative gray-matter and white-matter axons at this developmental stage (Figure 7, e.g., arrows). Axonal staining was generally located within the axolemma in both regions, but axoplasmic staining was also observed (Figures 7B, 7C, and 7E). Staining was observed on occasion at sites where axons extended small hillock-like structures (Figure 7C, four arrows), and at sites where cell processes came into close apposition with axons (Figure 7D). Reactivity was also seen in astrocyte somata (Figure 7F), unidentified processes (Figure 7D), identifiable astrocyte processes (Figure 7G), and glioblasts (Figure 7H). Immuno-gold staining density for each structure type was calculated for all gold particles in whole grid sections (n=8 sections), revealing significantly higher density in axons and glial cell somata compared with processes (Figure 7I).
Discussion
The formative white matter of the early mid-gestation fetus is composed of radial glial elements, immature axons, navigating growth cones, glioblasts, and astrocytes.25, 28, 29, 30, 31, 32, 33, 34 This mix of cellular elements is subjected to ischemic-type lesions such as PVL and diffuse periventricular white-matter injury. The incidence of PVL is elevated between PCW 23 and 32 while diffuse injuries are a common sequelae of very premature birth.8, 24, 35 Loss of axon function is the ultimate white-matter deficit in these patients, whether after direct axonal injury 9 or hypomyelination,36, 37 and may result in the common birth disorder cerebral palsy and a range of cognitive deficits. 35 Oligodendroglial lineage progression to the preoligodendrocyte stage accelerates from PCW 28,25, 26 and it has been assumed that premyelinated axon diameter expansion initiates immediately before this key pathophysiologic event, as it does in other mammals. 27 Recent findings suggest that axon diameter expansion above 400 nm is sufficient to initiate axon wrapping, 38 a threshold we show has not been reached by PCW 18 to 23. Occasional myelination has been observed from ∼23 to 30 PCW25, 26 and proceeds for years postpartum. 39 A large population of small premyelinated axons therefore populate periventricular white matter until axon ensheathment peaks; a period spanning the windows of heightened susceptibility to all forms of neonatal white-matter injury from PVL to neonatal stroke.
Direct Injury of Small Premyelinated Axons
Axon destruction is a defining feature of focal necrosis in cystic PVL, where severed axon elements are widely described. The incidence of cystic PVL has declined in the West and noncystic PVL has become the predominate form, which may incorporate wide-spread gliosis punctuated by microinfarcts. 9 Axonal injury assessed via fractin or amyloid precursor protein staining together with imaging studies suggests extensive axonal damage in areas of diffuse gliosis, in addition to within microinfarcts.9, 40, 41, 42, 43 The significance of axon loss in noncystic cases may have declined since 2000, 37 and recent studies using in vivo models vary significantly in the degree to which they report axon damage.11, 44 Drobyshevsky et al 11 in particular point to selective loss of premyelinated central axons in the development of hypertonia in cerebral palsy.
In addition to direct axon injury in focal and possibly diffuse white-matter pathology, the injury pathway described in the Results may be significant for the longer-term viability of surviving axons and their neighboring elements. For example, immature axons contain glutamate in both vesicular and axoplasmic pools,16, 45 and can act as a primary source for excitotoxic glutamate release. 46 Glutamate release from small premyelinating axons may occur during sublethal activation of axonal NMDA GluRs after axoplasmic rises in Ca2+/Na+ and the associated axon swelling and vesicle docking; initiating injury pathways on adjacent oligodendroglia. Similarly, sublethal injury to axons at this early developmental stage is likely to disrupt their proper maturation, leading to delayed axon pathology and/or dystrophic axon-glial signalling.
In actively myelinating white matter (P10 RON), oligodendrocyte processes and adjacent small premyelinated axons are sensitive to NMDA GluR-mediated injury. 14 Immuno electron microscopic localization of Nr1 subunit protein to these processes indicates that the primary injury is to the glial elements with secondary injury to small axons, consistent with the rapid injury in P10 RON compared with the earlier stage white matter studied here. Very immature white matter lacks axo-glial junctions and has an expanded extracellular space, allowing unambiguous localization of Nr1 subunit to the axolemma; while the limit of resolution (∼15 to 30 nm 47 ), makes discrimination between axolemma and glial cell membrane problematic at latter ages. The current findings suggest that NMDA GluR on the smallest, most immature premyelinating axons may contribute to injury in actively myelinating white matter and argue against the actions of an intermediate released from damaged glial processes onto neighboring axons. 14 Once axons initiate diameter expansion, any sensitivity to NMDA GluR-mediated injury is masked by a transient heightened sensitivity to ischemic injury mediated by expression of clustered voltage-gated calcium channels,15, 16 a stage insensitive to NMDA GluR blockers. 14
Human Early Mid-Gestation Axons
The ultrastructural features of mid-gestation human formative white-matter axons have not previously been described, and the maturation stage of axons in the tissue has only been inferred in earlier studies. 31 We report numerous identifiable small diameter axons in this clinically important tissue, with no axons with a diameter of larger than 400 nm and no ensheathed axons. The RON model allowed delineation of the injury mechanisms operating in the smallest premyelinated axons. NMDA GluR expression was localized to the axolemma and mediated acute cytotoxic Ca2+/Na+ influx in a manner similar to that found in oligodendroglial processes 12 and mature neurons. Protection was afforded by competitive and noncompetitive NMDA GluR blockers but not by the nonspecific antagonist kynurenic acid, which acts at the glycine site of the NMDA GluR and has limited anti-excitotoxic action (e.g., Fatokun et al 48 ).
Nr1 GluR subunit expression examined by quantitative immuno-gold postembedding EM revealed high levels of focal reactivity in small premyelinated axons of formative white matter in the PCW 18 to 23 fetus. Previous light-level studies have shown reactivity in O4(+) oligodendroglia throughout mid-gestation, in addition to levels of background staining consistent with axonal expression. 49 When studied in a rat PVL model, significant white-matter protection was produced using memantine, 49 and the current findings indicate the potential of a similar approach for human cases after early diagnosis.
Footnotes
ACKNOWLEDGMENTS
The authors thank Ms Natalie Allcock for excellent technical assistance in electron microscopy. The support of the Libyan (TH) and Saudi Arabian (BA) government is gratefully acknowledged as are the comments on the manuscript by James Alix. Professor Nada Zecevic of the University of Connecticut Health Center was generous with provision of fetal tissue and for her comments on the manuscript. Professor Richard Reynolds and Dr Djordje Gveric are thanked for help in supplying MS tissue from the Multiple Sclerosis and Parkinson's Tissue Bank held at Imperial College London.
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.