
Abstract
Activation of platelets induces interactions with platelets, endothelial cells, and leukocytes. In vivo observation of these interactions in the cerebral microcirculation is rare. The purpose of the present study was to develop a model enabling the in vivo observation of platelet kinetics in the cerebral microcirculation. Intravital fluorescence microscopy was performed in the Mongolian gerbil. Platelets of a donor were labeled ex vivo with carboxyfluorescein diacetat-succinimidylester (CFDA-SE), providing long-term fluorescence. Platelet function was tested ex vivo by flow cytometric analysis and in vivo by analyzing platelet-endothelium interactions. Labeled platelets stimulated with adenosine diphosphate ADP (200 μmol/L) or thrombin (1000 U/L) showed aggregation in flow cytrometric analysis, whereas unstimulated platelets were not aggregated. Irradiation of the brain surface after intravenous injection of the photosensitizing dye Photosan first induced rolling and firm adherence of platelets on arteriolar and venular endothelium, followed by the formation of a thrombus obstructing the vessel. Quantitative analysis (nx100 μm−1 min−1) before and after 6 mins of irradiation showed 2.6±3.2 versus 29.0±28.9 rolling, and 0.0±0.0 versus 1.7±2.3 firm adherent platelets in arterioles, and 3.9±3.3 versus 36.6±20.9 rolling and 0.0±0.0 versus 13.6±8.9 firm adherent platelets in venules. Thus, we conclude that ex vivo labeling of platelets with CFDA-SE does not activate platelets. Platelet aggregation and adhesion was achieved by platelet-specific stimulation such as ADP, thrombin or irradiation. In vivo assessment of physiologic and pathophysiologic mechanisms of platelets in the cerebral microcirculation can be achieved in this model.
Introduction
By the preservation of vascular integrity and the control of hemorrhage after injury, platelets contribute to the maintenance of the normal circulation of blood (Ruggeri, 2000).
Furthermore, growing evidence supports a role for platelets in inflammation. Activation of endothelial cells through an inflammatory reaction results in interactions of platelets with endothelial cells. Platelets roll on endothelial cells via endothelial P-selectin, followed by platelet activation and firm adherence of platelets to the vessel wall (Frenette et al, 1995; Katayama et al, 2000; Lou et al, 1997).
On activation, platelets generate reactive oxygen species, eicosanoids, chemokines, and growth factors, thus providing mechanisms for tissue injury and the recruitment of leukocytes (Seno et al, 2001; Finazzi-Agro et al, 1982; Ogletree, 1987; Weyrich et al, 1996; Deuel et al, 1981). Interaction of platelet CD40L (CD154) with CD40 on endothelial cells leads to further aggravation of the inflammation reaction (Henn et al, 1998).
Besides their interactions with endothelial cells, leukocytes roll on P-selectin expressed by adherent platelets, followed by firm adherence of leukocytes to adherent platelets via leukocyte Mac-1 (Evangelista et al, 1999; Salter et al, 2001; Diacovo et al, 1996).
The importance of these mechanisms has been shown in variety of different organs and in response to different pathophysiologic stimuli.
Concerning the cerebral microcirculation, only few data exist, suggesting a role for platelets in the inflammation of cerebral vessels. Histologic studies after focal cerebral ischemia show platelet deposiin cerebral microvessels (Jafar et al, 1989; Okada et al, 1994). Acute ischemic stroke has been found to enhance platelet activation (van Kooten et al, 1994; Zeller et al, 1999). These data, however, provide no information on dynamics of platelet activation, platelet-endothelium interactions, or platelet-leukocyte interactions in the cerebral microcirculation and their contribution to tissue injury.
The purpose of the present study was to develop a model enabling the visualization and quantitative assessment of platelet kinetics, and their interaction with leukocytes and endothelial cells. We decided to use a model of intravital fluorescence microscopy, which has been used in different studies to analyze leukocyte-endothelium interactions, vessel diameters, Blood–brain barrier function, and cerebral blood flow (Uhl et al, 2000; Lehmberg et al, 2003). Modifying a previously described protocol (Manegold et al, 2003) platelets were labeled ex vivo with the fluorescent dye carboxyfluorescein diacetat-succinimidylester (CFDA-SE). Their functionality was tested ex vivo by flow cytometric analysis and in vivo by intravital fluorescence microscopy. Isolated and labeled platelets showed marked reactivity to specific stimulation ex vivo and in vivo. Accordingly, this study provides a model enabling the observation of platelet behavior in the cerebral microcirculation.
Materials and Methods
Adult male Mongolian gerbils weighting 65 to 75 g were used in this study (age 12 to 16 weeks; in vitro: four platelet donors; in vivo: four experimental animals and four platelet donors). Animals had free access to tap water and pellet food and were kept in a 12-h light/dark cycle at room temperature of 22°C. The State Government of Upper Bavaria had approved the experiments, which were conducted according to institutional guidelines.
The surgical preparation has been described in detail previously (Uhl et al, 2000). In brief, anaesthesia was induced with halothane (0.6% to 1.5%). Oxygen content of the inspired gas was 27%. Body temperature was maintained at 37°C by using a rectal probe connected to a feedback controlled heating pad (Effenberger, Pfaffing, Germany). A polyethylene catheter (Portex Ltd, Hythe, England) was inserted into the tail artery for continuous blood pressure monitoring and the femoral vein for application of fluorescent dyes and labeled platelets. Animals did not receive heparin at the time of vessel catheterization. Catheters were not heparinized either.
The skull was then fixed in a stereotactic frame (Model 51600; Stoelting Co., Wood Dale, IL, USA). After a midsagittal skin incision from the forehead to the neck, the calvaria was exposed and a rectangular 4 × 4 mm2 window prepared by trephination over the left parietal hemisphere, with the dura mater left intact. The dura mater was rinsed continuously with physiologic saline at 37°C.
Platelets were separated and labeled with fluorescence ex vivo similar to a protocol previously described (Manegold et al, 2003). The original protocol had to be modified, since gerbil platelets were activated during the isolation process using this protocol (Figure 1B). For the isolation of gerbil platelets, 1.5 mL of whole blood were withdrawn from a syngenic donor gerbil by cardiac puncture and distributed to polypropylene tubes containing 0.4 mL volume of 23 mmol/L citric acid, 53 mmol/L trisodium citrate, and 74 mmol/L glucose, 20 μL prostaglandin E1 (PGE1) (560 nmol/L final concentration) (Serva, Heidelberg, Germany) and 0.5 mL Dulbecco phosphate-buffered saline (D-PBS) (Pan Systems, Aidenbach, Germany). The blood was centrifuged at 130g for 10 mins. Platelet-rich plasma was transferred to a fresh tube containing 0.6 mL volume of 23 mmol/L citric acid, 53 mmol/L trisodium citrate and 74 mmol/L glucose, 40 μL PGE1 (747 nmol/L final concentration) (Serva) and 1 mL D-PBS (Pan Systems). After adding the fluorescent marker aminoreactive CFDA-SE (Molecular Probes, Eugene, OR, USA), platelet-rich plasma was centrifuged at 1430g for 10 mins. The pellet of platelets was resuspended in 0.5 mL of D-PBS (Pan Systems). Platelet concentration and purity of the sample were examined using a Coulter AcT Counter (Coulter, Miami, FL, USA). A total of 200 × 106 labeled platelets were injected intravenously before intravital microscopy was performed.
Functionality of labeled platelets was examined by flow cytometric analysis (FACSort flow cytometer; Becton Dickinson, Heidelberg, Germany) before and after in vitro stimulation by adenosine diphosphate (ADP) (200 mmol/L) and human thrombin (1 U/mL) (Sigma, Deisenhofen, Germany) for 10 mins.
For intravital microscopic observations, the animals were placed on a computer-controlled microscope stage. The intravital fluorescence microscope (Leitz, Wetzlar, Germany) was equipped with a 75 W xenon bulb, L3 and TX2 Ploemopak filter blocks for epiillumination, and a salt-water immersion objective x 25. Enhancement of microvessels was achieved by intravenous injection of Texas Red-labeled dextran (molecular weight: 70,000, bolus of 0.1 mL of a 0.5% solution; Molecular Probes), and photothrombosis was enabled by a intravenous injection of Photosan (hematoporphyrin-equivalent, bolus of 2 mG/kG; Seehof Laboratorium, Wesselburenerkoog, Germany) before the first measurement. Recordings lasted 10 secs with the TX2 filter block for assessment of vessel diameters and a total of 8 mins with the L3 filter block for irradiation of each vessel and concomitant observation of platelet-endothelium interactions. The images were recorded by a SIT-video camera (Hamamatsu Photonics, Herrsching, Germany) and stored on an S-VHS videorecorder. Arteriolar and venular diameters (μm) and the number of rolling and adherent platelets (n x 100 μm−1 min−1) were quantified using a computer assisted microcirculation analysis system (CapImage; Ingenieurburo Dr Zeintl, Heidelberg, Germany). Platelet-endothelium interactions were classified similar to rolling and adherent leucocytes (Uhl et al, 2000). Rolling platelets were defined by their intermittent contacts with the endothelium, thereby advancing definitively slower than the freely moving thrombocytes in the center of flow axis of a microvessel. Firm adherent thrombocytes were defined by their attachment to the vessel wall for more than 20 secs. Vessel segments of 100 μm in length were studied.
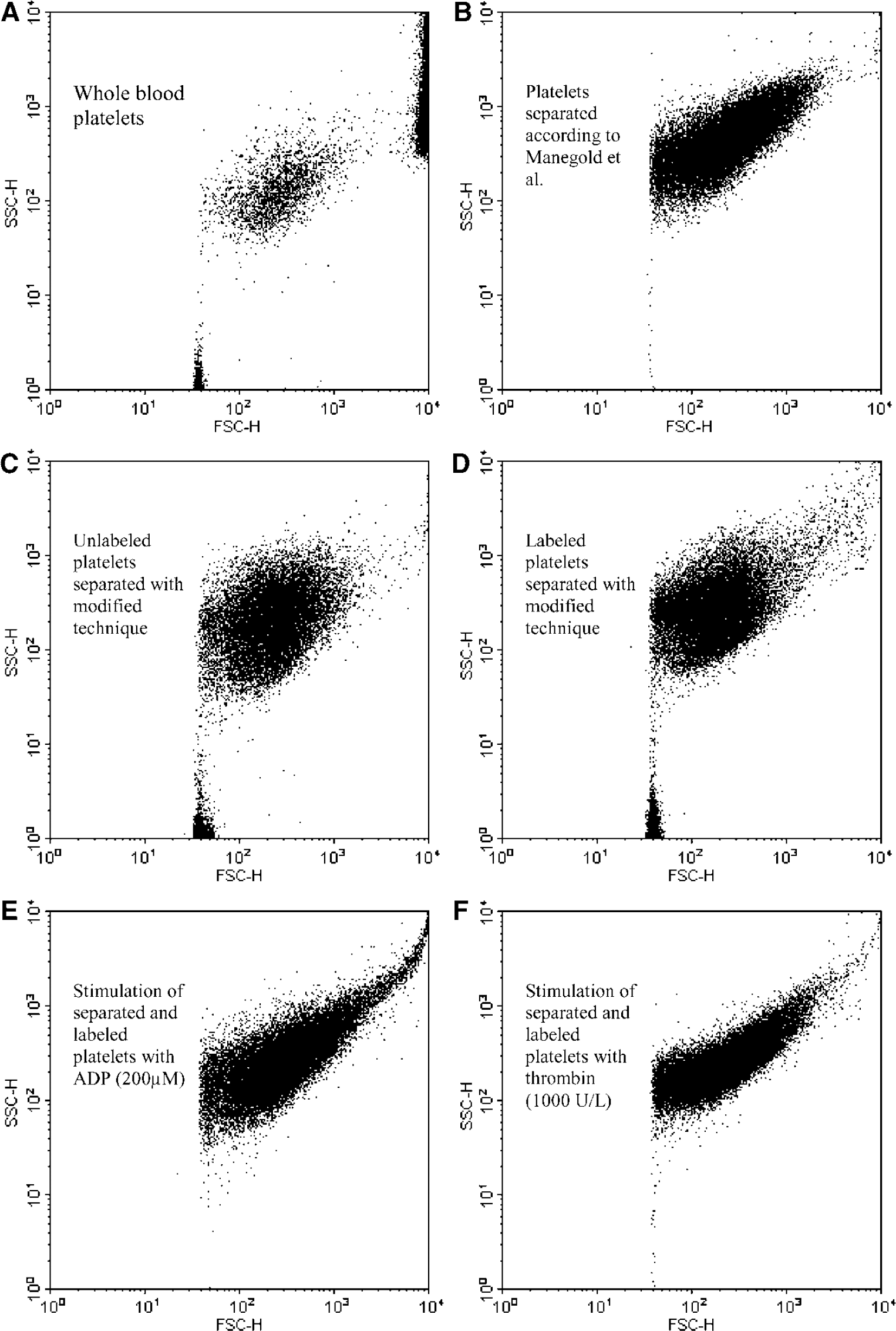
Flow cytometric assessment of platelet functionality. Log forward/log side light scatter FACS analysis demonstrates a distinct population of whole-blood platelets (
After surgical preparation, intravital microscopy was performed. A total of 16 venules (diameter: 38.3 ± 2.3 μm) and 14 arterioles (diameter: 37.3 ± 3.7 μm) were occluded photochemically in four experimental animals. At the beginning of each vessel occlusion, the diameter of the vessel was recorded. Rolling and adherent platelets were analyzed before and at 2, 4, and 6 mins after the beginning of irradiation for 30 secs. Ensuing analyses were not performed as thrombosis prevented differentiation of individual platelets.
Statistical evaluation of data was performed on a personal computer using Sigma Stat 2.0 (SPSS Science, Chicago, IL, USA). A Friedman-test followed by the Dunnet's method was used for analyzing differences between baseline levels at the beginning of irradiation and consecutive measurements. A statistically significant difference was assumed at P < 0.05. All data are presented as mean ± s.d.
Results
Fluorescence-actuvated cell sorter (FACS) analysis of whole-blood thrombocytes showed a distinct population in log forward/log side light scatter. The circular shape of the cloud of single events expresses the uniform granularity and size of the thrombocytes (Figure 1A). Separation of Gerbil platelets after the protocol of Manegold et al (2003) reveals a shift toward higher size and granularity (Figure 1B). Platelets separated with the modified protocol for Gerbils show a distinct population in log forward/log side light scatter, expressing their uniform size and granularity (Figure 1C). Labeling with CFDA-SE had no influence on granularity and size (Figure 1D). After application of the weak stimulus ADP (200 mmol/L), the shape shifted towards an ellipse expressing the alteration in platelet size and platelet aggregation (Figure 1E). After application of the strong stimulus human thrombin (1000 U/L), this phenomenon was pronounced (Figure 1F). FACS analysis was performed with the blood of three donors. Figure 1 shows the plot of one representative experiment.
In vivo, at the onset of irradiation, rolling platelets could rarely be observed in pial venules or arterioles and adherent platelets were completely absent. The number of rolling platelets (n x 100 μm−1min−1) in venules increased from 3.9 ± 3.3 at the beginning to 36.6 ± 20.9 after 6 mins of irradiation and the number of firm adherent platelets (n x 100 mm−1min−1) from 0.0+ 0.0 to 13.6 + 8.9 (Figure 2A). The number of rolling platelets at the arteriolar endothelium was 2.6 ± 3.2 at the beginning and 29.0 ± 28.9 after 6 mins of irradiation (Figure 2B). The number of firm adherent platelets at the arteriolar endothelium was 0.0 ± 0.0 at the beginning and 1.7 ± 2.3 after 6 mins of irradiation (Figure 2B). After 6 mins of irradiation, a solid thrombus was formed, which finally occluded the irradiated vessels, venules as well as arterioles (Figure 3). Vessels segments without continuous irradiation showed no changes in rolling and adherent platelets over up to 210 mins (data not shown).
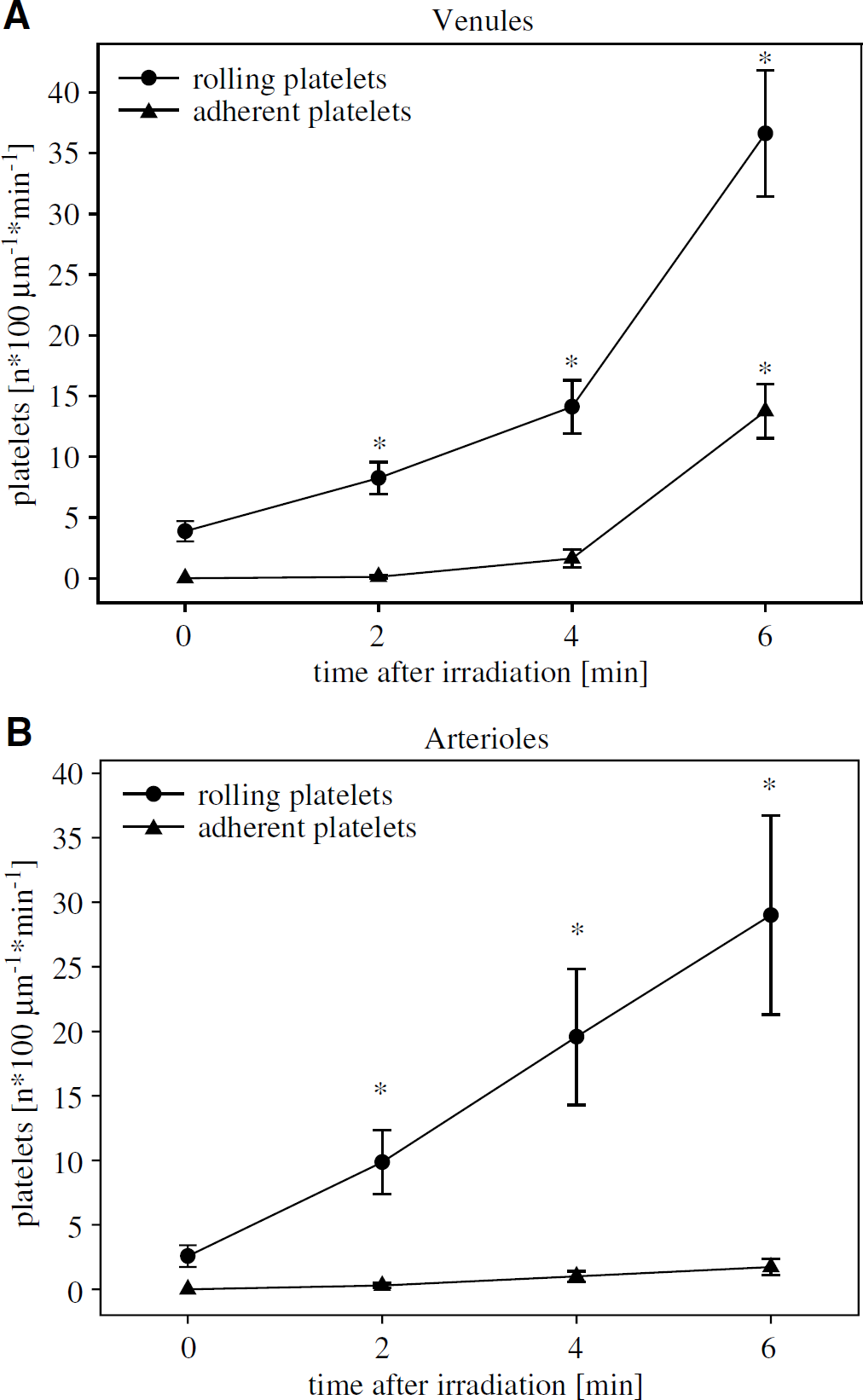
Platelet-endothelium interactions in the cerebral microcirculation induced by photothrombosis. Frequency of rolling and adherent platelets in pial venules (n = 16) (
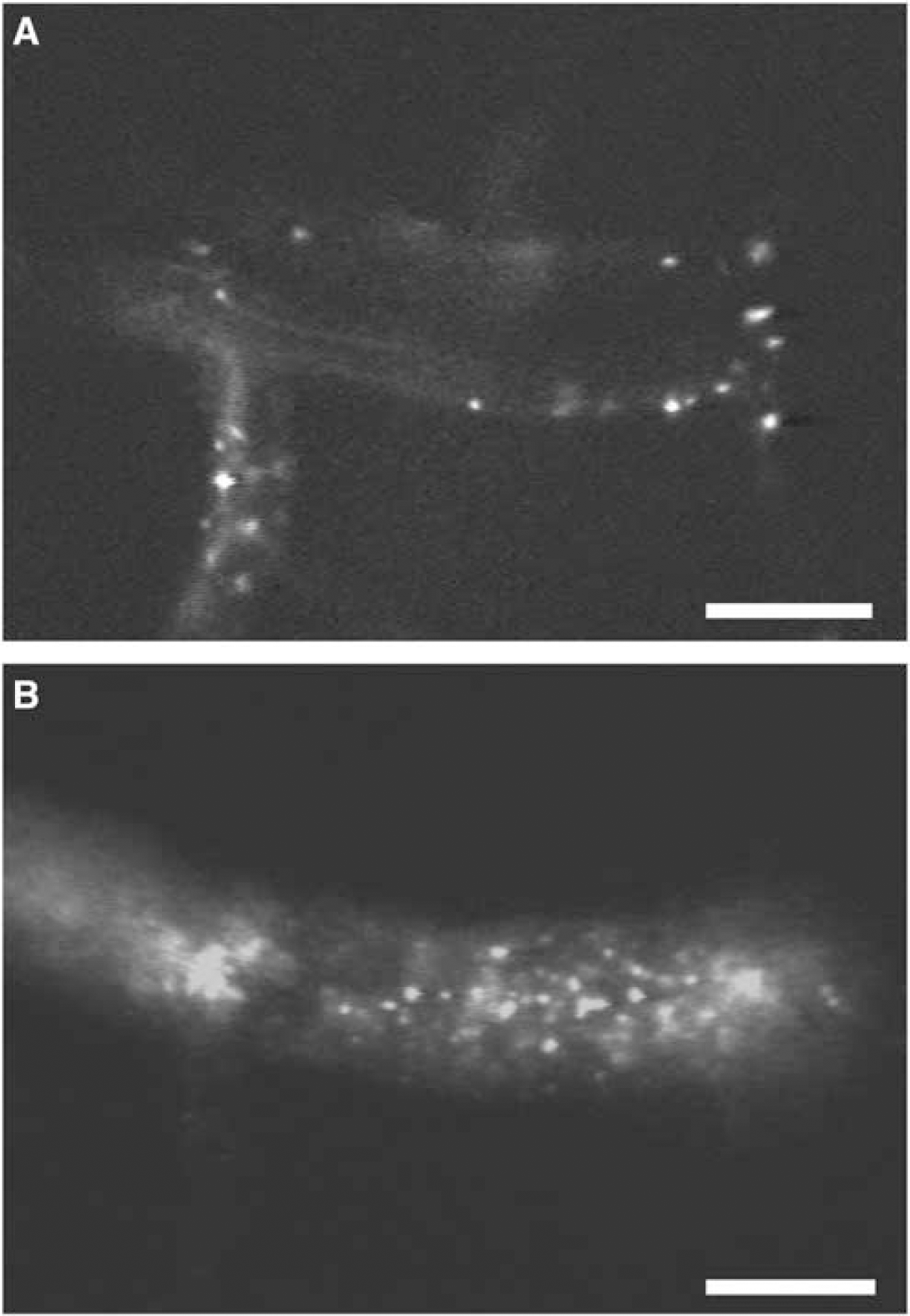
Intravital fluorescence microphotographs of the Mongolian gerbil brain surface after 2 mins (
Discussion
Intravital fluorescence microscopy has been widely used to analyze Blood–brain barrier disturbances, vascular reactivity, blood flow, and leukocyte-endothelium interactions after pathologic insults in the cerebral microcirculation (Wahl et al, 1985; Schuerer et al, 1989; Dirnagl et al, 1994; Uhl et al, 1999; Abels et al, 2000). The present study is the first to present a method enabling quantitative analysis of platelet–endothelium interactions during long-term observation of the cerebral microcirculation.
The model of intravital fluorescence microscopy in the Mongolian gerbil has been first described by Uhl et al (2000), and has been utilized in later studies analyzing leukocyte-endothelium interactions, capillary density, arteriovenous transit-time, and vessel diameters in the cerebral microcirculation simultaneously (Lehmberg et al, 2003). As in this model the dura mater can be left intact for microscopic observations, microcirculatory disturbances due to the surgical preparation can be limited.
In the above-mentioned studies, leukocyte-endothelium interactions have been investigated after intravenous injection of Rhodamine 6G, a commonly used fluorescent dye for in vivo leukocyte labeling. Unfortunately, no fluorescent dye exclusively staining platelets after in vivo injection is available. Ishikawa et al (2002) observed platelet behavior in the cerebral microcirculation in a rat closed cranial window model after in vivo injection of Rhodamine 6G. They could not, however, analyze platelet kinetics quantitatively.
Intravenous injection of Rhodamine 6G in tensively stains leukocytes due to their high content of mitochondria. The intense fluorescence of leukocytes thereby outshines the fluorescence of the smaller platelets, making the detection of individual platelets difficult. The simultaneous labeling by in vivo injection of Rhodamine 6G complicates the discrimination of leukocytes, platelets, platelet–platelet interactions, or platelet–leukocytes interactions.
Khandoga et al (2002) investigated platelet–endothelium interactions after ex vivo labeling of platelets with Rhodamine 6G. Unfortunately, platelets labeled ex vivo with Rhodamine 6G can be observed only for short periods of time, as the fluorescent intensity decreases very quickly.
In contrast, platelets labeled ex vivo by CFDA-SE can be observed for several hours and can be easily discriminated from Rhodamine 6G labeled leukocytes. The ex vivo labeling technique has been validated in a mouse model of tumor microcirculation by Manegold et al (2003). Since gerbil platelets were activated after separation and labeling with the protocol provided by Manegold et al, the protocol was modified and platelet function was tested ex vivo and in vivo after separation and labeling of platelets with CFDA-SE. In response to ADP and thrombin labeled platelets showed aggregation in flow cytometric analysis. In vivo, irradiation induced a rise in platelet-endothelium interactions, showing the intact function of platelets.
The initiation of thrombosis by irradiation of vessels after systemic injection of a photosensitizing dye has been first described by Rosenblum and ElSabban (1977), and is commonly called ‘photothrombosis‘. Various studies have used this model with different photosensitizing dyes and different light sources (Jander et al, 1995; Dietrich et al, 1994; Mosinger and Olney, 1989) to induce thrombosis in cerebral vessels. Although the exact mechanisms of photothrombosis are not clear, a specific activation and aggregation of platelets is produced by this method. Futrell and Riddle (1993) described endothelial damage with exposure of the subendothelium and the formation of nonocclusive thrombus in irradiated vessels after induction of photothrombosis. Thrombus formation after endothelial damage has been observed through the microscope in vivo in earlier studies (Nishimura et al, 1996). Single platelet-endothelium interactions could not be detected with these methods. As mentioned above, growing evidence supports a role for platelets in inflammation reactions, as seen with secondary brain damage after ischemia or traumatic brain injury. However, these mechanisms seem to depend on platelet activation, platelet–endothelium interactions, and the subsequent release of proinflammatory mediators without the need of thrombus formation (Frenette et al, 1995; Katayama et al, 2000). The possibility to analyze platelet–endothelium interactions in the cerebral microcirculation in vivo could help to further describe the contribution of platelets to important cerebral insults.
In conclusion, the method described in this study enables the quantitative analysis of platelet kinetics in the cerebral microcirculation of gerbils, without changing platelet function. Furthermore, platelet–endothelium interactions can be assessed simultaneously with leukocyte–endothelium interactions, vessel diameters, capillary density, cerebral blood flow, blood–brain barrier function and other parameters.