
Abstract
Delayed cerebral ischemia (DCI) after aneurysmal subarachnoid hemorrhage (SAH) has been associated with numerous pathophysiological sequelae, including large artery vasospasm and microvascular thrombosis. The focus of this review is to provide an overview of experimental animal model studies and human autopsy studies that explore the temporal-spatial characterization and mechanism of microvascular platelet aggregation and thrombosis following SAH, as well as to critically assess experimental studies and clinical trials highlighting preventative therapeutic options against this highly morbid pathophysiological process. Upon review of the literature, we discovered that microvascular platelet aggregation and thrombosis occur after experimental SAH across multiple species and SAH induction techniques in a similar time frame to other components of DCI, occurring in the cerebral cortex and hippocampus across both hemispheres. We discuss the relationship of these findings to human autopsy studies. In the final section of this review, we highlight the important therapeutic options for targeting microvascular platelet aggregation and thrombosis, and emphasize why therapeutic targeting of this neurovascular pathology may improve patient care. We encourage ongoing research into the pathophysiology of SAH and DCI, especially in regard to microvascular platelet aggregation and thrombosis and the translation to randomized clinical trials.
Keywords
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a highly morbid form of stroke. Though advances in the pathophysiological understanding and management of SAH have occurred, both morbidity and mortality remain high; 30% of patients die 1 and 50% of survivors have long-term cognitive deficits.2,3 Delayed cerebral ischemia (DCI), also known as delayed ischemic neurological deficit, is the largest treatable cause of poor SAH outcome. 4 Historically, large artery cerebral vasospasm was widely considered as the primary mechanism underlying SAH-induced DCI leading to tissue ischemia, cerebral infarction, and ultimately poor neurological outcome.5–7 In recent years, however, this canonical view of DCI pathophysiology has been challenged for several reasons. First, the incidence of angiographic vasospasm following SAH is approximately 70%, while the incidence of DCI is only around 30%.8,9 Second, the temporal relationship between large artery vasospasm and cerebral infarction is often inconsistent.10–12 Third, evidence shows that some SAH patients develop DCI without any angiographic evidence of large artery vasospasm.13–15 Fourth, disappointing results were observed in two randomized, double-blind, placebo-controlled, phase III clinical trials using the endothelin A receptor antagonist, clazosentan – a powerful anti-vasospasm agent. In these trials, clazosentan significantly reduced large artery vasospasm, but failed to improve functional outcome or reduce mortality in SAH patients.10,16–19
Based on these observations regarding the varying relationship between large artery vasospasm and DCI, numerous preclinical and clinical studies have examined whether other mechanisms beyond large artery vasospasm are contributing or even primarily driving the onset of DCI after SAH. To date, microvascular thrombi, cerebral autoregulatory dysfunction, and cortical spreading depression have all, to varying degrees, been implicated as additional contributors to SAH-induced DCI.14,20–22 Of these, microvascular thrombosis – a phenomenon by which microthrombi develop in small arterioles, venules, and capillaries throughout the brain after SAH – has been the most extensively described and studied.23–28 Though definitive experimental data establishing causation and necessary cross-validation studies in patients are currently lacking, a growing body of evidence strongly suggests microvascular thrombi play a key role in DCI and neurological deficits following SAH. Here, we provide a brief review of the platelet aggregation and coagulation cascades that are likely involved with microvascular thrombus formation. We also provide a comprehensive review and synthesis of the preclinical and clinical literature surrounding this important contributor to secondary brain injury after SAH.
Review of platelet aggregation and coagulation cascade
When vascular injuries occur, a chain reaction of pro-inflammatory, wound healing, blood clotting, and vascular repair responses are rapidly triggered, known as hemostasis. 29 This hemostatic process has two main phases, known as primary and secondary hemostasis, wherein platelet aggregation forms the initial platelet plug and subsequent activation of the coagulation system forms the stabilizing fibrin clot.
Primary hemostasis is the process by which platelets, adhesive proteins, and the vessel wall interact to form a platelet plug. 30 While platelets do not normally adhere to the intact vascular endothelium, vascular injury exposes collagen and von Willebrand Factor (vWF) in the subendothelial tissue. This allows platelets to undergo a morphological change to increase surface area and triggers the formation of “platelet plugs.” Platelets initially adhere to both vWF and exposed collagen (Figure 1). Following adhesion, platelets degranulate and release several factors that aid in plug formation, such as arachidonic acid, which is converted to thromboxane A2. Thromboxane A2 produced by activated platelets triggers further platelet aggregation as it binds to ADP. 30 This ADP binding leads to a conformational change in GpIIb/IIIa receptors on the platelet surface, promoting fibrinogen deposition. The final product of primary hemostasis, the “platelet plug,” is an aggregate of platelets and fibrinogen.
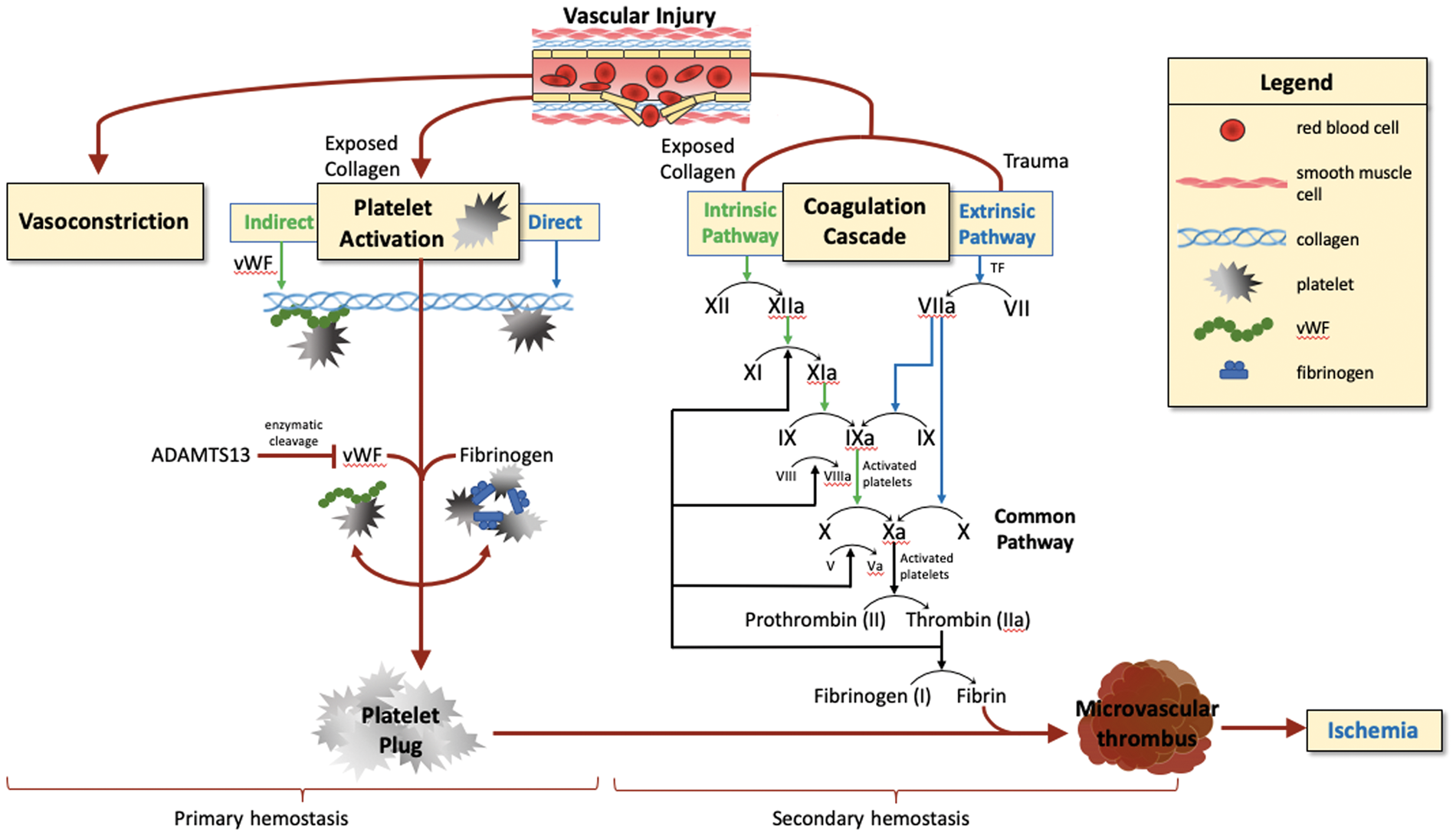
Primary and secondary hemostasis after vascular injury.
Secondary hemostasis is characterized by activation of the coagulation cascade, which involves two distinct pathways (extrinsic and intrinsic) that activate various clotting factors with the goal of activating thrombin. 30 The extrinsic and intrinsic coagulation pathways are parallel processes by which factor X is converted to activated factor Xa, which promotes thrombin activation and conversion of fibrinogen to fibrin (Figure 1). The extrinsic pathway is initiated by vascular injury-induced exposure to tissue factor (TF) expressed in the subendothelium. TF binding to factor VIIa and calcium promotes conversion of factor X to Xa.31,32 The intrinsic pathway is initiated when factor XII, along with co-factors prekallikrein and HMW kininogen, assembles on a suitable surface or polymer, leading to conversion of factor XII into activated factor XII and setting off a cascade of events, including conversion of Factor XI to Factor XIa and Factor IX to Factor IXa, that ultimately leads to conversion of factor X into activated factor Xa.33,34 Factor Xa then interacts with factor V, tissue and platelet phospholipids, and calcium to form a prothrombinase complex and convert prothrombin into thrombin. 30 Thrombin, once active, catalyzes the conversion of fibrinogen to fibrin, which forms crosslinks in the platelet plug and stabilizes it into a true thrombus. Understanding the time course and mechanisms of both primary and secondary hemostasis will be crucial to our understanding of the underlying pathophysiology driving SAH-induced microvascular thrombi.
Microvascular thrombi in autopsy studies of subarachnoid hemorrhage
Autopsy studies show evidence of microvascular thrombi in the brains of patients who have died of aneurysmal SAH. Early studies, such as Kim et al. 35 in 1979, recognized that there was a potential link between vasospasm and microvascular platelet aggregation and thrombosis. Soon after, studies by Hasegawa et al. and Suzuki et al. also discuss the hypothesis that microvascular thrombus formation and vasospasm are linked, discovering that vasospasm can induce a hypercoagulable state that accelerates cerebral ischemia through high platelet aggregability and microvascular thrombosis.36,37 Suzuki et al. 38 identified and characterized the presence of this phenomenon in a SAH patient in 1983. They identified microvascular thrombi within multiple cerebral arteries and arterioles near the site of large artery vasospasm in a SAH patient who died due to complications of DCI. Microvascular thrombi were felt to be too small and too multiple to solely be the result of the patient’s original ruptured aneurysm. The presence of microvascular thrombi following aneurysmal SAH was later confirmed by Suzuki et al. 39 who examined six patients with SAH who died during their hospital course – four of whom died due to complications of DCI, one of whom died due to aneurysm re-rupture, and one of whom died due to acute hydrocephalus. They noted that microvascular thrombi were only present in the four patients who developed severe large artery vasospasm and suffered DCI. They also found that the distribution of microvascular thrombi was greater in the ischemic or infarcted regions of the brains of these four patients. Interestingly, a study by Neil-Dwyer et al. 40 examined the presence of cortical ischemic lesions in SAH patients, and noted that while they were present in 77% of patients, there was no significant association between the presence of microvascular thrombi and angiographic vasospasm.
More recently, Stein et al. 22 analyzed 29 patients who died from aneurysmal SAH. They performed a systematic histological evaluation of the presence and nature of microvascular thrombi in autopsy brain sections from the insula, cingulate gyrus, and hippocampus of these patients. Several important observations were made. First, the development of microvascular thrombi was bimodal, with one peak in patients who died within 3 days of ictus and another peak in patients who died 7 to 14 days after ictus (a peak time window for onset of DCI). Second, they identified a strong correlation between the extent of microvascular thrombi and the presence of ischemic infarctions. Third, they found that microvascular thrombi were most numerous in brain areas where maximal neuronal necrosis was found. These correlative data strongly suggest that microvascular thrombi may play an important role in SAH-induced DCI and suggest that the coagulation cascade producing microvascular thrombi may represent a novel therapeutic target.
Microvascular thrombi in experimental models of subarachnoid hemorrhage
Although microvascular thrombi were first described in SAH patients in 1983, their identification in the setting of experimental SAH did not occur until the mid-2000s when Sehba et al. 41 documented microvascular platelet aggregation in a rat model of experimental SAH in 2005 and Jeon et al. 42 documented microvascular thrombi formation in a rat model of experimental SAH in 2010. Since that time, this pathophysiological phenomenon has been reported in numerous animal species (mouse, rat, and rabbit)23,25,43–48 using a variety of SAH induction techniques (endovascular perforation, prechiasmatic injection, and cisterna magna injection).23,25,43–48 Importantly, two, likely interrelated, microvascular thrombotic pathologies have been described in animals models of SAH: (1) Microvascular platelet aggregation, defined as histological demonstration of platelet plugs within small arterioles and capillaries; and (2) Microvascular thrombus formation, defined as platelet plugs stabilized with fibrin deposition within small arterioles and capillaries. It is important to note that the studies below did not differentiate between thrombus and embolus; it cannot be ruled out that some of what is being defined as microvascular thrombus is in fact embolus.
In this subsection, we provide a synthesis of available preclinical evidence for the presence, timing, and character of microvascular platelet aggregation and microvascular thrombi formation following experimental SAH. We also review the underlying molecular cascades that have thus far been implicated in these two microvascular pathologies.
Temporal and spatial characterization of SAH-induced microvascular platelet aggregation and thrombi formation
Microvascular platelet aggregation
Using an endovascular perforation rat model of SAH to study microvascular platelet aggregation as assessed by GPIIb/IIIA immunostaining, Sehba et al. 41 found that experimental SAH induced a biphasic time course of microvascular platelet aggregation within the basal cortex, frontal cortex, and striatum. The first phase of platelet aggregation began 10 min after SAH, which was lost by 6 h after SAH. The second phase of platelet aggregation occurred 24 h after SAH, which then dissipated by 48 hours after SAH. The discovery of microvascular platelet aggregation following SAH, along with the capability of platelets to induce vascular injury, led the authors to suggest that administration of antiplatelet agents after SAH might be beneficial if timed appropriately (i.e. after securing the ruptured brain aneurysm). 41
Microvascular thrombi formation
Jeon et al. 42 were the first described microvascular thrombi formation following experimental SAH. Using a prechiasmatic injection rat model of SAH, they examined the presence and localization of microvascular thrombi via fibrinogen immunostaining. They found a significant increase in microvascular thrombi in the cortex and cerebellum eight days after SAH (the only time point tested). Since this report, several independent laboratories using a variety of species and complementary induction techniques have similarly documented and characterized the development of widespread microvascular thrombi after experimental SAH.
Using a prechiasmatic injection mouse model of SAH, Sabri et al. 25 identified microvascular thrombi via fibrinogen immunostaining in the cortex and hippocampus 48 h after SAH (the only time point tested). They also noted microvascular thrombi in the subcortical/deep brain tissues via Western blot. Importantly, an association between the presence of arteriole constriction and greater burden of microvascular thrombi was noted, suggesting a potential mechanistic link between SAH-induced vasoreactivity and the formation of microvascular thrombi. Similarly, Vergouwen et al. 48 showed that microvascular thrombi develop in the prechiasmatic injection mouse model, showing this pathology in both the cortex and hippocampus 48 h after SAH (the only time point tested).
Using an endovascular perforation mouse model, Pisapia et al. 47 and Muroi et al. 45 rigorously examined the time course of microvascular thrombi development after experimental SAH. Pisapia et al. used anti-thrombin immunostaining to quantify microvascular thrombi 24, 48, 72, and 96 h after induction of SAH. They found that microvascular thrombi were present throughout both ipsilateral and contralateral hemispheres at all-time points, but that the severity of microvascular thrombi peaked at 48 h. Similarly, Muroi et al. showed that microvascular thrombi (as assessed by fibrinogen immunostaining) develop in the hippocampus and cerebral cortex in delayed fashion, peaking on post-SAH days 2 and 3. Importantly, these delayed time courses closely correlate with the timing of large artery vasospasm in these animal models of SAH,46,49 which suggests both pathophysiological events likely contribute to DCI. A similar correlation between the timing of microvascular thrombi and larger artery vasospasm was noted in the endovascular perforation mouse model of SAH by Milner et al. 46
Using a cisterna magna injection rat model of SAH, Wang et al. 50 studied the time course of microvascular thrombi development after experimental SAH, as assessed by fibrin and GLUT1 immunostaining. They discovered that diffuse thrombosis of the cortical capillaries was significantly elevated 24 h post-SAH in comparison to sham mice (the only time point tested). Finally, Andereggen et al. used a cisterna magna injection rabbit model of SAH to study the development of microvascular thrombi as assessed by fibrinogen immunostaining. They found a significant increase in microvascular thrombi in the hippocampus and a trend towards an increase in microvascular thrombi in the cerebral cortex 24 h after SAH (the only time point tested).
Mechanisms of SAH-induced microvascular platelet aggregation and thrombi formation
Vasoconstriction-induced stasis
Friedrich et al. 23 used an endovascular perforation mouse model of SAH and intravital fluorescence microscopy to assess the development of microvascular thrombi in pial arterioles and venules in live mice over time. They found that microvascular thrombi occurred as early as 3 h after SAH, and that microvessel thrombi virtually only occurred in pial arterioles that were significantly vasoconstricted. Sabri et al. 25 used the prechiasmatic mouse model of SAH and measured nitric oxide (NO) levels via DAF-2DA-based spectrofluorometry. They found that NO decreased significantly with decreased microvascular area. They also found an association between microvascular thrombi and vasoconstriction related to thickening of the endothelial and sub-endothelial layers.
Thromboinflammation
An overlap between inflammation and thrombosis has been reported to play a potential role in a variety of brain diseases including SAH.51–54 SAH causes the release of toxic molecules such as endothelins and oxygen free radicals that potentiate pro-inflammatory pathways and endothelial cell-mediated thrombosis. 55 This mechanism can lead to an increase in thromboinflammation, which in turn causes activation of platelets and eventually may precipitate the formation of microvascular thrombi. 56 In addition, the release of reactive oxygen species can damage endothelial cells, leading to the release of prothrombotic factors including tissue factor and von-Willebrand factor (vWF). Both molecules activate platelets via processes controlled by ADAMTS-13, a vWF-cleaving metalloprotease further discussed below as a therapeutic target against SAH-induced microvascular thrombi. 57 Additional data linking thromboinflammation to DCI including evidence that aneurysmal SAH patients who go on to develop DCI become more hypercoagulable than patients who do not develop DCI.58–60 Finally, a recent study by Frontera et al. 61 showed that platelet activation and inflammation in a single-institution cohort of SAH patients were associated with greater incidence of brain edema and DCI and worse patient outcome.
P-selectin
Ishikawa et al. 62 used two endovascular perforation mouse models (single and double perforation) to study platelet aggregation in the initial hours after SAH induction – likely an important early step in the formation of platelet plugs in microvessels. Intravital fluorescence microscopy was used to observe platelet dynamics in the cerebral microcirculation after experimental SAH. They found progressive platelet rolling and adhesion from 30 min to 8 h after SAH, with platelet recruitment being slightly greater in the double perforation model. Interestingly, most of the platelet-endothelial cell interactions took place in venules, although some platelet recruitment was also observed in arterioles. In a second set of experiments, a subgroup of SAH mice were administered a control antibody or a monoclonal antibody against P-selectin, a cell adhesion molecule expressed on activated vascular endothelium and activated platelets. This antibody treatment reduced rolling and adherent platelets in SAH mice to near-baseline levels – an effect driven by a combination of a reduction in platelet-endothelial cell adhesions as well as decreased platelet-leukocyte interactions. In support of these findings, Sabri et al., 25 examined P-selectin expression levels in a prechiasmatic mouse model of SAH and documented a significant increase in P-selectin expression in SAH mice. Additionally, Frijns et al. 63 analyzed blood samples of 106 patients after aneurysmal SAH and discovered a significant increase in P-selectin in patients who experienced DCI. In total, these results indicate P-selectin likely mediates early microvascular platelet aggregation and thrombus formation after experimental SAH and suggest that early P-selectin targeting in SAH patients has promise as a novel therapeutic approach towards reducing DCI. 62
Therapies targeting SAH-induced microvascular platelet aggregation and thrombi formation
Recombinant human ADAMTS13
Muroi et al. 45 used an endovascular perforation mouse model of SAH to investigate whether ADAMTS13 has therapeutic potential against microvascular thrombi formation, DCI, and neurological deficits following experimental SAH. Wild-type mice were administered intravenous recombinant human ADAMTS13 protein 20 min after SAH surgery. Recombinant human ADAMTS13 was found to significantly reduce microvascular thrombi in both cortex and hippocampus, while also decreasing neuronal degeneration and improving neurological outcome. Recombinant human ADAMTS13 treatment, however, did not reduce severity of cerebral vasospasm, and it did not alter bleeding time. This is an important distinction, as it strengthens the hypothesis that microvascular thrombi contribute to the neurological deficits associated with DCI in a manner that is separate from the known neurological deficits caused by vasospasm. Similarly, Vergouwen et al. 48 used a prechiasmatic injection model of SAH to assess the potential therapeutic benefit of ADAMTS13 treatment on SAH-induced microvascular thrombi formation. Recombinant human ADAMTS13 was intravenously administered in both wild-type mice and in ADAMTS13 knockout mice following SAH induction. A trend towards reduction in microvascular thrombi in wild-type mice and a significant reduction in microvascular thrombi in ADAMTS13 knockout mice was noted.
In support of these pre-clinical findings, Kumar et al. 64 reported from a small autopsy case series that ADAMTS13 activity levels were lower in SAH patients vs. non-SAH patients, and that the ratio of ADAMTS13 to vWF antigen was significantly lower in SAH patients vs. non-SAH patients. Additionally, Li et al. 65 found that SAH patients suffering from DCI, as compared to SAH patients without DCI, had decreased plasma ADAMTS13 levels on post-SAH day 1 and increased plasma vWF levels on post-SAH days 1 and 4. Similar results were noted in patients with angiographic evidence of large artery vasospasm vs. those without. Collectively, results from these autopsy and clinical cohort studies are consistent with the notion that SAH promotes microvascular thrombi formation by lowering ADAMTS13 and subsequently increasing vWF. 66 They also suggest: (1) Treatment directed at microvascular thrombi may produce improvement in neurological outcome after SAH; and (2) ADAMTS13-directed therapy has promise as a novel therapeutic approach against SAH-induced microvascular thrombi, DCI, and neurological deficits.
Plasminogen activation
Pisapia et al. 47 examined the impact of plasminogen activation on SAH-induced microvascular thrombi formation. First, they intravenously administered mutant thrombin-activated urokinase-type plasminogen activator (scFv/uPA-T) to wild-type mice subjected to endovascular perforation SAH and found that the plasminogen activator significantly reduced extent of microvascular thrombi. Second, they intravenously administered scFv/uPA-T along with the anti-vasospasm endothelin antagonist, clazosentan, to wild-type mice subjected to endovascular perforation SAH and found that the plasminogen activator attenuated the increased microvascular thrombi formation associated with clazosentan treatment alone. In total, these results suggest: (1) plasminogen activation carries therapeutic promise against SAH-induced microvascular thrombi formation and DCI; and (2) a potential reason for the reported lack of functional improvement in SAH patients treated with clazosentan in Phase 3 clinical trials16–19 may have been promotion of microvascular thrombi formation despite reduction in cerebral vasospasm.
Conditioning-based therapy
Conditioning-based therapy harnesses the brain’s inherent resistance to injury by exposure to a sub-lethal injurious stimulus, capitalizing on powerful endogenous protective cascades. Milner et al. 46 examined the effect of isoflurane conditioning treatment on microvascular thrombi as well as cerebral vasospasm, autoregulatory dysfunction and neurological deficits using an endovascular perforation mouse model of SAH. Isoflurane conditioning involved exposing wild-type mice to 2% isoflurane for 1 h beginning 1 h after SAH or sham surgery. They noted a marked reduction in microvascular thrombi in isoflurane-treated animals as well as significant reductions in cerebral vasospasm, autoregulatory dysfunction, and neurological deficits. These results suggest that isoflurane conditioning likely has a pluripotent protective effect against SAH-induced neurovascular dysfunction including a positive impact on microvascular thrombi.
Antiplatelet therapy
Though not specifically examining the issue of microvascular platelet aggregation and thrombosis, there are a series of randomized controlled trials (RCTs) that have tested the use of a variety of antiplatelet agents as a novel therapy for aneurysmal SAH patients.67–77 A cochrane review by Dorhout Mees et al. 67 titled “Antiplatelet Therapy for Subarachnoid Hemorrhage” discussed 7 of these RCTs totaling 1385 SAH patients. Treatments used in these RCTs included aspirin, OKY-046 (a selective thromboxane synthetase inhibitor), dipyridamole, and ticlopidine. Though all included RCTs showed a trend towards reducing DCI and a trend towards increasing intracranial hemorrhagic complications in SAH patients, only one reported a statistically significant reduction in DCI – a study that was complicated by multiple concerns such as small sample size, no power calculations, and trial stoppage at 133 patients without a given reason. The overall conclusion from this systematic review was that no definitive evidence for a beneficial effect of single antiplatelet therapy on DCI or neurological outcome in SAH patients exists. 67
Moving away from single antiplatelet agents, Nagahama et al. 70 conducted a single-institution retrospective study of aneurysmal SAH patients who were managed with dual antiplatelet therapy (DAPT; aspirin and clopidogrel). Of 161 patients, 85 were managed with DAPT – a group of patients compared to a historical control group of 76 patients who did not receive antiplatelet therapy. They reported that SAH patients managed with DAPT had a significantly decreased incidence of DCI without a significant increase in intracranial hemorrhagic complications. 70 This is the first study to examine the use of DAPT in SAH patients, with findings that suggest additional investigation into this novel therapeutic approach is warranted.
Senboyuka et al. 68 examined the effect of cilostazol, a selective PDE3 inhibitor having dual therapeutic properties including an antiplatelet effect and a vasodilatory effect, in aneurysmal SAH patients in a multicenter prospective open-label RCT. They reported that cilostazol treatment led to a lower incidence of DCI in SAH patients. Matsuda et al. 78 reported a similar finding with cilostazol in a more recent RCT examining 148 SAH patients. Three additional RCTs have examined the efficacy of cilostazol in the prevention of DCI in SAH patients,79–81 and a meta-analysis by Saber et al. summarizing all five RCTs has shown positive results for cilostazol against DCI, 82 though no definitive evidence for improve in long-term neurological outcome currently exists. While these RCTs do not specifically show benefit against microvascular thrombi, the mechanism of action of cilostazol suggests this may be one of its therapeutic properties. Cilostazol inhibits platelet aggregation and relaxes intact smooth muscle cells, and has proven protective effects on endothelial damage,83,84 which itself leads to platelet aggregation, coagulation, and thrombosis.
Anticoagulant therapy
Though not specifically examining the issue of microvascular platelet aggregation and thrombosis, there are a series of retrospective studies and RCTs that have investigated the potential therapeutic benefit of anticoagulant therapies such as enoxaparin and low-dose intravenous heparin (LDIVH) for patients with aneurysmal SAH.85–88 In the early 2000s, two RCTs examining the effects of enoxaparin were published with conflicting results. Siironen et al. 87 enrolled 170 SAH patients in their RCT and found no difference in clinical outcome when comparing patients who received subcutaneous enoxaparin to those who received placebo. One year later, however, Wurm et al. 88 enrolled 120 SAH patients in their RCT and reported a significant reduction in both angiographic vasospasm and DCI in patients who received subcutaneous enoxaparin in comparison to those who received placebo. Neither of these RCTs evaluated long-term cognitive outcome.
Another anticoagulant therapy that has been applied to SAH patients is unfractionated heparin. Simard et al. 89 first examined the effect of this anticoagulant approach in a rat model of SAH, in which autologous blood was injected into the subarachnoid space of the entorhinal cortex under stereotactic guidance. Rats were administered low-dose intravenous unfractionated heparin (LDIVH) or vehicle and sacrificed 48 h after SAH. Rats in the LDIVH treatment group had a significant decrease in neuroinflammation and neuronal apoptosis in comparison to the vehicle-treated group. Simard et al. 85 followed up this preclinical study with a retrospective analysis of 86 aneurysmal SAH patients who were managed postoperatively with either conventional subcutaneous heparin twice daily for DVT prophylaxis, or with the Maryland LDIVH infusion protocol (8 U/kg/h progressing over 36 h to 10 U/kg/h, beginning 12 h after surgery and continuing until day 14 post-SAH). They reported a significant decrease in the incidence of DCI as well as a reduction in the use of rescue therapy in SAH patients who received LDIVH. The same LDIVH protocol was examined in an additional retrospective analysis by James et al. 86 who found that not only was DCI reduced, but that cognitive outcome as measured by the Montreal Cognitive Assessment (MoCA) was improved in SAH patients who received the Maryland LVIDH treatment. These promising preclinical and early phase RCT results have led to initiation of a multi-center prospective, randomized phase II trial called ASTROH (Aneurysmal Subarachnoid hemorrhage Trial RandOmizing Heparin) that will examine the efficacy of LDIVH treatment in aneurysmal SAH patients (NCT02501434; estimated completion date of December 2020).
Limitations of the microvascular thrombosis hypothesis
There is considerable evidence indicating microvascular thrombosis is a significant contributor to SAH-induced DCI. Widespread histological evidence documenting microvascular thrombi across multiple experimental models of SAH exists,23,25,42,43,45–48,50 and at least one example of the contribution of microvascular thrombi to the neurological deficits caused by SAH-induced DCI (separate from large artery vasospasm) has been reported. 45 However, consistent direct evidence of the independent impact of microvascular thrombosis on SAH-induced neurological deficits is currently lacking. Microvascular thrombi are widespread and present at multiple time points after SAH, but how long these thrombi persist after formation and how they contribute to ischemic injury and neurological outcome has not been fully elucidated. Future studies linking microvascular thrombi to important endpoints including reduction of regional and/or global cerebral blood flow would help establish a causal relationship. Experiments correlating regional density of microvascular thrombi with histological evidence of microinfarction and functional endpoints related to clinical outcome would also strengthen the microvascular thrombosis hypothesis. Finally, more innovative and systematic approaches to studying this phenomenon in humans are desperately needed including more rigorous autopsy studies focused on elucidating the association and role of microvessel thrombi in ischemic brain injury and neurological outcome as well as development of novel imaging strategies to permit dynamic evaluation of the timing, extent, and impact of microvessel thrombi in live patients following aneurysmal SAH.
Conclusion
DCI is the largest modifiable risk factor for patients suffering aneurysmal SAH 4 and remains an attractive therapeutic target for improving long-term patient outcome. SAH-induced DCI is a multifactorial pathophysiological process, rather than a singular pathological event linked exclusively to large artery vasospasm. One of the most important and well-studied non-vasospasm contributors to SAH-induced DCI is microvascular platelet aggregation and thrombi formation. In the present review, we show that platelet aggregation and thrombi develop in cerebral microvessels in a similar time frame to other components of DCI including large artery vasospasm. We also show that microvascular platelet aggregation and thrombi formation occur after experimental SAH across species and induction techniques, and that this neurovascular pathology occurs after experimental SAH in cortex and hippocampus and across both hemispheres. Importantly, these experimental observations have been confirmed in autopsy studies in SAH patients where microvascular thrombi have been documented in cortical and hippocampal arterioles as well as other regions, usually near infarcted areas with maximal neuronal cell death, and in line with the timing of DCI in humans. Excitingly, the underlying pathophysiological and molecular mechanisms responsible for microvascular platelet aggregation and thrombi formation following SAH have begun to be elucidated, and several therapeutic approaches to prevent or reduce this neurovascular deficit have shown promise in experimental studies. Despite this progress, additional investigation is required. First, the molecular pathways responsible for SAH-induced microvascular platelet aggregation and thrombi formation need further elucidation. Second, therapeutic approaches targeting microvascular platelet aggregation and thrombi formation require more rigorous testing including cross-validation studies with differing SAH induction techniques, multiple animal species, and long-term endpoints such as neurobehavioral outcome. Third, novel techniques to measure and quantify platelet aggregation and thrombi formation in cerebral microvessels in patients with aneurysmal SAH need development so that the full impact of this pathology can be determined and a biomarker to track this pathology can be established permitting pharmacodynamic dose optimization studies for promising anti-thrombotic treatments. The considerable body of experimental and clinical evidence implicating the importance and impact of microvascular platelet aggregation and thrombi formation in SAH-induced brain injury and neurological outcome argues strongly for further investigation.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20921974 - Supplemental material for Microvascular platelet aggregation and thrombosis after subarachnoid hemorrhage: A review and synthesis
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20921974 for Microvascular platelet aggregation and thrombosis after subarachnoid hemorrhage: A review and synthesis by Julian V Clarke, Julia M Suggs, Deepti Diwan, Jin V Lee, Kim Lipsey, Ananth K Vellimana and Gregory J Zipfel in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke (grant number R01NS091603 [to G.J.Z.] and R25NS090978 [to G.J.Z.]).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.