
Abstract
While the detrimental role of non-regulatory T cells in ischemic stroke is meanwhile unequivocally recognized, there are controversies about the properties of regulatory T cells (Treg). The aim of this study was to elucidate the role of Treg by applying superagonistic anti-CD28 antibody expansion of Treg. Stroke outcome, thrombus formation, and brain-infiltrating cells were determined on day 1 after transient middle cerebral artery occlusion. Antibody-mediated expansion of Treg enhanced stroke size and worsened functional outcome. Mechanistically, Treg increased thrombus formation in the cerebral microvasculature. These findings confirm that Treg promote thrombo-inflammatory lesion growth during the acute stage of ischemic stroke.
Introduction
It has been observed for many years that immune cells transmigrate over the blood—brain barrier into infarcted brain tissue. This process follows a defined time course, with neutrophils and macrophages beginning to accumulate in the brain some hours after stroke, and lymphocytes reaching maximum infiltration by approximately day 3. 1 While the detrimental role of non-regulatory T-cell subsets in the acute phase of ischemic stroke is widely accepted,2, 3, 4 there is an ongoing debate about the contribution of regulatory T cells (Treg) to this pathology.4, 5, 6, 7
We were able to show that Treg cause microvascular dysfunction and consequently thrombus formation and secondary infarct growth in the acute phase after experimental ischemic stroke by interaction with endothelial cells and platelets.4,8 This interplay between inflammatory processes and thrombus formation has recently been referred to as ‘thrombo-inflammation’. 8
Other groups reported secondary deterioration of stroke volumes and functional outcome in animals 7 days after blocking of Treg. 5 Therefore, the aim of this study was to clarify the impact of Treg in the acute phase of ischemic stroke by the means of a superagonistic anti-CD28 antibody (CD28 SA) that leads to an expansion of pre-existing Treg in the lymphoid organs and the dissemination of increased Treg numbers in the peripheral blood.9,10
Materials and Methods
Details of the experimental procedure are provided in the Supplementary Information. The animal experiments were conducted in accordance with the recommendations of the European Convention for the Protection of Vertebrate Animals used for Experimentation and the current ARRIVE guidelines (http://www.nc3rs.org/ARRIVE). Experiments were approved by legal state authorities (Government of Lower Franconia).
Ischemia Model
Focal cerebral ischemia was induced in 6 to 8-week-old male C57BL/6, DEREG or
Expansion of Treg by CD28 SA Treatment in vivo
CD28 SA (clone D665, Exbio, Praha, Czech Republic, 50 μg/mouse) was applied 3 days before tMCAO (prophylactic application) or immediately after tMCAO (therapeutic application) by an intraperitoneal injection. MOPC-21 antibody (BioXCell, West Lebanon, NH, USA) served as control. Regulatory T cells were quantified by flow cytometry analysis (Foxp3 staining). Depletion of Treg in DEREG mice was achieved by the administration of diphtheria toxin (DTX, Merck, Darmstadt, Germany, 1 μg/mouse per day intraperitoneally) on three consecutive days before tMCAO.
Immunohistochemistry
Cryoembedded brain slices were stained with antibodies against CD31 (Abcam, ab9498, Cambridge, UK), CD4 (100506, BioLegend, San Diego, CA, USA), Ly6B.2 (MCA771GA, Serotec, Puchheim, Germany) or AF488-conjugated anti-platelet glycoprotein IX (generated by B Nieswandt, Würzburg, Germany). For quantification of occluded vessels hematoxylin—eosin staining was performed (10 μm slices), and the percentage of occluded vessels in every tenth slice was counted under 40-fold magnification.
Results
First, we confirmed that CD28 SA treatment leads to an expansion of Treg in mice (maximum on day 3), as has previously been described.
10
We found a significant increase in the relative numbers of Treg in the blood and lymph nodes of wild-type mice (Supplementary Figure 1,
Next, we assessed if the increase in Treg numbers before tMCAO influences stroke development in wild-type mice. Stroke volumes on day 1 were significantly larger (82.0±35.2 mm3) compared with wild-type animals that had received isotype control antibodies (46.5±36.4 mm3,
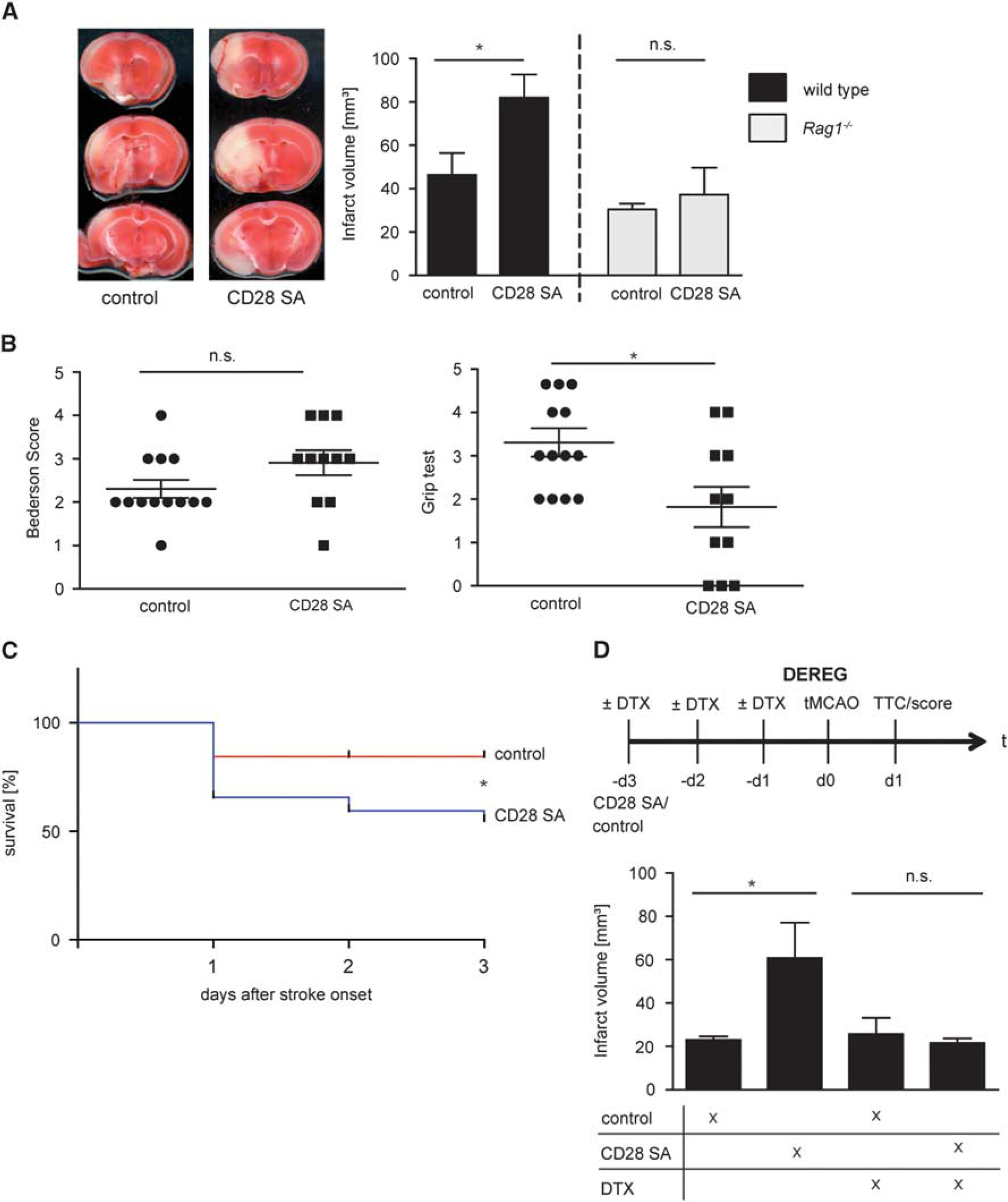
Increase in superagonistic anti-CD28 antibody (CD28 SA)-induced Treg worsens stroke outcome. (
In the next step, we analyzed if the increase in peripheral Treg alters the composition of the cellular infiltrate within the ischemic brain. Indeed, we found more ipsilesional CD4+ cells (Figure 2A,
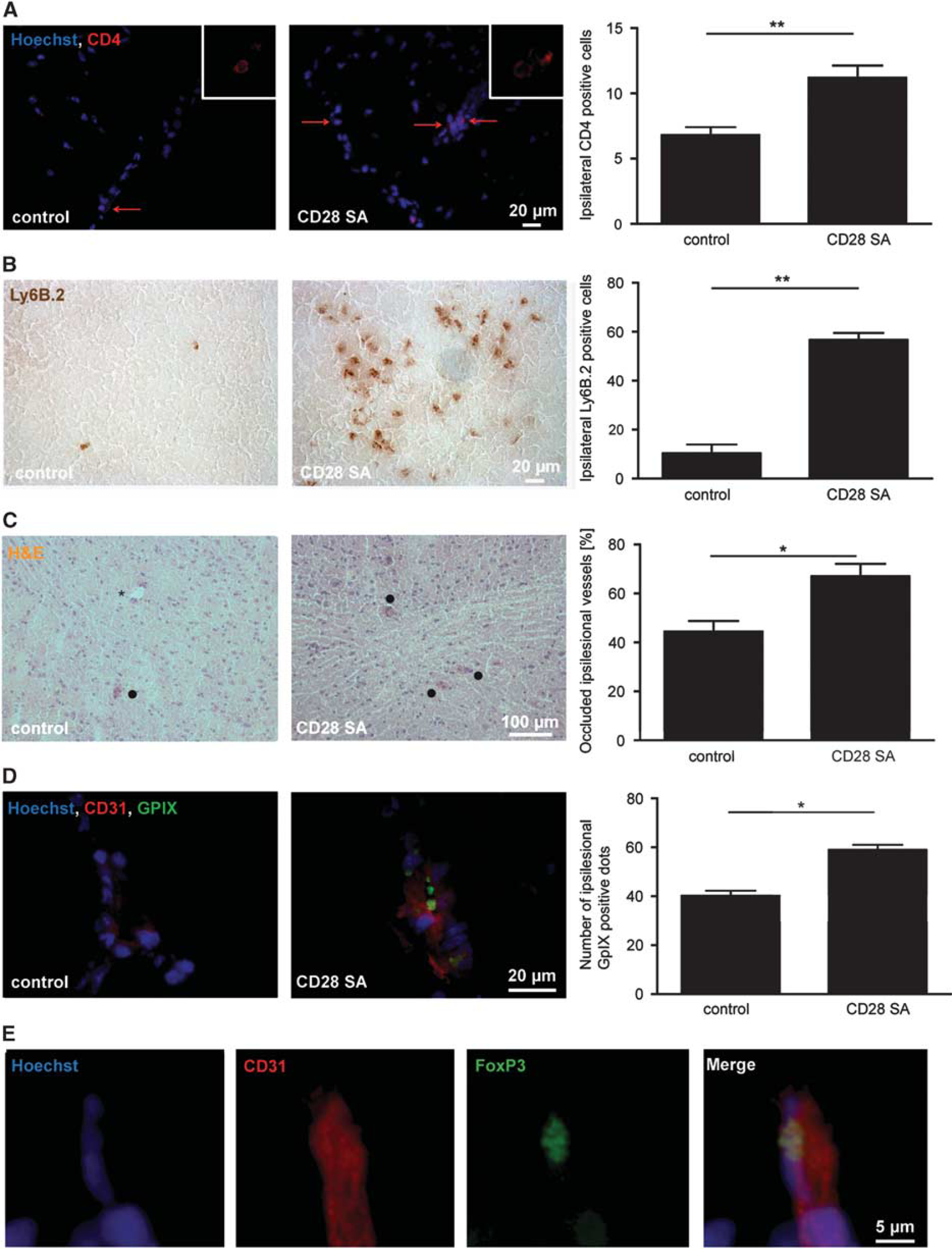
CD28 SA treatment increases intracerebral immune cell accumulation and thrombosis after transient middle cerebral artery occlusion (tMCAO). (
To confirm that Treg contribute to ischemic brain injury by boosting ‘thrombo-inflammation’,
8
we demonstrated a higher number of occluded brain vessels (Figure 2C,
Discussion
In the present study, with the use of a pharmacologic expansion of Treg, we independently confirm that Treg are strong potentiators of acute ischemic stroke. 4 Superagonistic anti-CD28 antibody-induced expansion of Treg positively correlated with increased stroke size 24 hours after ischemia. In analogy to our previous study, Treg interact with endothelial cells and platelets to induce microvascular dysfunction and thrombosis. 4 In contrast to our ancestor study 4 and the results of this study—both strongly arguing for a detrimental role of Treg during the acute phase of brain ischemia/reperfusion injury—it has been reported that Treg are key modulators of cerebroprotection in brain ischemia in mice in the late phase. 5 Therefore, it would be worthwhile to study whether CD28 SA application at later time points influences stroke outcome although Treg expansion with CD28 SA after tMCAO had no impact on the neurological status on day 3 in our hands (Supplementary Figure 4).
Treg promote stroke progression within a few hours after cessation of cerebral blood flow. At this early stage, they are mainly found within cerebral blood vessels and interact with endothelial cells and platelets. This interplay between thrombotic and inflammatory processes has recently been described as ‘thrombo-inflammation’. 8 Nevertheless, we also found an increased number of CD4+ and Ly6B.2+ cells in the brain parenchyma as early as day 1 after tMCAO. While it is well-known that neutrophils start to transmigrate into the brain within hours after stroke, the pathophysiologic role of these cells in acute ischemic stroke is still under debate.1,4,14 Moreover, the mechanisms through which peripheral Treg expansion by the CD28 SA enhances neutrophil recruitment to the ischemic brain need to be further established. One potential explanation could include the formation of a pro-inflammatory milieu triggered by activated T cells present in high numbers within the brain vasculature and subsequent upregulation of cell adhesion molecules. However, based on the observation that Treg (and other T cells) trigger ischemic brain damage already after a few hours after vessel occlusion, it can be assumed that mechanisms operating mainly within the brain-perfusing vessels rather than within the brain parenchyma contribute to the detrimental T-cell effect to a large extent.2, 3, 4
In summary, the present study independently confirms that Treg promote lesion growth during the acute stage of ischemic stroke. Short-term inhibition of Treg might become a promising therapeutic approach to combat this devastating neurologic condition.
Footnotes
ACKNOWLEDGMENTS
The authors thank Daniela Urlaub, Andrea Sauer, and Melanie Glaser for excellent technical assistance.
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.