
Abstract
Accumulation of neutrophils in brain after transient focal stroke remains controversial with some studies showing neutrophils to be deleterious, whereas others suggest neutrophils do not contribute to ischemic injury. Myeloperoxidase (MPO) has been used extensively as a marker for quantifying neutrophil accumulation, but is an indirect method and does not detect neutrophils alone. To elucidate the interaction of macrophages in the neutrophil inflammatory response, we conducted double-label immunofluorescence in brain sections at 0, 1, 2, 3, 7, and 15 days after ischemia. Each of these results was obtained from the same animal to determine correlations between neutrophil infiltration and ischemic damage. It was found that MPO activity increased up to 3 days after cerebral ischemia. Dual-staining revealed that macrophages engulf neutrophils in the brain and that this engulfment of neutrophils increased with time, with 50% of neutrophils in the brain engulfed at 3 days and approximately 85% at 15 days (N=5, P < 0.05). Interestingly, at 7 days the amount of dual-staining was decreased to 20% (N=5, P < 0.05). Neutrophil infiltration was positively correlated with ischemic damage in both the cortex and striatum (r2 = 0.86 and 0.80, respectively, P < 0.01). The results of this study indicate that the MPO from neutrophils phagocytized by macrophages may continue to contribute to the overall MPO activity, and that previous assessments that have utilized this marker to measure neutrophil accumulation may have miscalculated the number of neutrophils within the ischemic territory and hence their contribution to the evolution of the infarct at later time points. Thus any biphasic infiltration of neutrophils may have been masked by the accumulation of macrophages.
Introduction
Transient ischemia of the cerebral vasculature followed by reperfusion leads to a secondary cascade of pathophysiologic events, characterized by a complex inflammatory response (del Zoppo et al, 2001; del Zoppo and Mabuchi, 2003). The hallmark of the inflammatory response is the initial infiltration of polymorphonuclear leukocytes (neutrophils) into the ischemic region of the brain (Feuerstein et al, 1998). The recruitment and extravasation of neutrophils into the brain parenchyma is initiated by both chemoattractants within the brain parenchyma and, by integrins on neutrophils binding to adhesion molecules (such as endothelial intercellular adhesion molecules or vascular cell adhesion molecules) expressed on the surface of endothelial cells, and can be classified into three stages; cell rolling, adhesion, and emigration/diapedesis (Panes et al, 1999). Neutrophil adhesion to the endothelium can also cause reocclusion of the microvasculature and thus exacerbation of the ischemia, in a process known as the ‘focal no-reflow’ phenomenon (Mori et al, 1992; Zhang et al, 1994).
In the brain parenchyma, activated neutrophils can release a variety of proinflammatory mediators such as tumor necrosis factor (TNF)-α, and interleukin (IL)-1β (for reviews see Cassatella, 1995; Witko-Sarsat et al, 2000) and possess the ability to produce reactive oxygen species (ROS) (such as superoxide anion and hypochlorous acid via two enzymes, the membrane-associated nicotinamide adenine dinucleotide phosphate dehydrogenase oxidase and myeloperoxidase (MPO), respectively), which could contribute to oxidative stress and subsequently neuronal damage (Anderson et al, 1991; Babior 2000; Hansen 1995; Witko-Sarsat et al, 2000). In addition, neutrophils can also release chemotactic factors (such as macrophage inflammatory protein (MIP)-1α, MIP-1β, monocyte chemoattractant protein (MCP)-1 and IL-8) that act on other inflammatory cells (Cassatella, 1999), potentially amplifying the cellular inflammatory response (Ainsworth et al, 1996) that may further exacerbate ischemic injury.
The infiltration of neutrophils into the brain parenchyma after ischemia-reperfusion has been shown to be deleterious in numerous studies that were designed to inhibit or deplete neutrophil infiltration and accumulation (for summary see Beray-Berthat et al, 2003a. These studies showed that inhibition of neutrophil infiltration led to a reduction in infarct volumes, resulting in the view that neutrophils contribute to the ischemic damage observed after stroke. Yet the pathogenic role of neutrophils after cerebral ischemia and reperfusion remains uncertain, with some studies failing to show a clear correlation between neutrophil infiltration and infarct formation (Beray-Berthat et al, 2003b; Emerich et al, 2002; Fassbender et al, 2002; Hayward et al, 1996; Soriano et al, 1999; Takeshima et al, 1992). Furthermore, the role of neutrophils after cerebral ischemia is made more complex by recent studies that have reported variability in the extent of ischemic injury associated with neutrophil infiltration, between cerebral regions, including the cortex and striatum (Batteur-Parmentier et al, 2000; Beray-Berthat et al, 2003a, b; Kitagawa et al, 1998; Yenari et al, 1998).
The involvement of macrophages in neutrophil function (Meszaros et al, 1999; Meszaros et al, 2000) adds additional complexity to the role of neutrophils in ischemic injury. Previous research has investigated ways to limit neutrophil infiltration and accumulation (see above), but factors involved in the resolution of the inflammatory response are also important. The major pathway for clearance of neutrophils and their potentially cytotoxic substances from the inflammatory site is programmed cell death (apoptosis) followed by engulfment by macrophages (Meszaros et al, 1999; Savill, 1997). Neutrophils normally only survive for a short period once leaving the blood before undergoing apoptotic cell death (Savill et al, 1989). However, on entering injured tissue, inflammatory mediators, such as adenosine, can activate these cells (Yu et al, 2004). This finding is also supported by in vitro studies that have shown that inflammatory mediators can inhibit neutrophil apoptosis and prolong their function (Lee et al, 1993). Extended survival and activation of neutrophils can exacerbate the inflammatory response and increase tissue damage (Misso et al, 2000). Macrophages can resolve neutrophil activation and therefore reduce neuronal injury by triggering apoptosis in neutrophils (Meszaros et al, 2000) and engulfing them, thereby preventing the release of cytotoxic substances into the surrounding tissue (Meszaros et al, 1999). Meszaros et al (1999) demonstrated that the large accumulation of macrophages and their subsequent phagocytosis of neutrophils leads to the establishment of macrophages as the dominant inflammatory cell in the inflammatory site 3 to 5 days after the initial injury, in peripheral (dorsal peritoneal) tissue, in rats.
Neutrophils are not the only inflammatory cells to possess MPO, both monocytes and macrophages also contain MPO but in a smaller quantity (Zhang and Chopp, 1997). Monocytes can lose their myeloperoxidase activity during conversion to tissue macrophages (Locksley et al, 1987; Zeller et al, 1988); however, macrophages may acquire MPO from their environment by pinocytosis or from ingested neutrophils (Locksley et al, 1987; Neill et al, 1985; Zeller et al, 1988). In this way, macrophages can gain MPO (or other peroxidases), particularly from sites of inflammation with intensive cell destruction. Thus, these enzymes can participate in cytotoxic secretory pathways of macrophages (Locksley et al, 1987; Neill et al, 1985). Myeloperoxidase has also been observed within endothelial cells at sites of inflammation (Eiserich et al, 2002). This has ramifications for studies that use MPO to quantify neutrophil infiltration, as both macrophages and cerebral endothelial cells could also contain MPO and thus MPO may not be the best marker for neutrophil infiltration. Recently, a review of neutrophil function after cerebral ischemia by Emerich et al (2002), commented on the apparent lack of correlation between neutrophil infiltration and infarct formation seen in previous studies, suggesting that the long held hypothesis that neutrophils contribute to ischemic damage may be unfounded. Additional comment was made on the intrinsic design problems associated with previous studies that did not allow correlation between neutrophil infiltration and infarct formation, and that the animal models used may have confounded the results obtained.
Several drugs specifically designed to inhibit neutrophil adhesion and subsequent infiltration have been developed as potential therapies for ischemic stroke. Two such drugs were tested in clinical trials; a monoclonal antibody to intracellular adhesion molecule-1 (ICAM-1) (enlimomab, R6.5) and a humanized antibody to the CD18 integrin (Hu23F2G or LeukArrest) (Becker, 2002). Clinical trials with these drugs were unsuccessful because a lack of neuroprotective efficacy and side effects such as leukopenia, which occurs at the therapeutic doses required to achieve successful inhibition of the infiltration (Becker, 2002; Sherman, 2001). These outcomes further intensify debate over the importance of the neutrophil inflammatory response after cerebral ischemia. Thus, there is a need to elucidate all aspects of the neutrophil inflammatory response, including its resolution not merely neutrophil infiltration.
The events surrounding neutrophil infiltration in cerebral ischemia have been investigated using a number of models including, the electrocoagulation model (Barone et al, 1991), the air embolism model (Kochanek et al, 1987), and the filament model of middle cerebral artery occlusion (MCAo) (Matsuo et al, 1994). Yet procedures associated with these models may exacerbate the inflammatory response. For instance, the air embolism model has been shown to cause injury to endothelial cells (Garcia et al, 1981), whereas in electron microscopy studies, the filament model of stroke has been shown to cause physical damage to the endothelium, potentially increasing the expression of adhesion molecules and promoting neutrophil infiltration (Nishigaya et al, 1991). However, the endothelin-1 (ET-1) model of MCAo is less invasive than the other models of cerebral ischemia and is not likely to damage the endothelium or surrounding structures (Macrae, 1992; Sharkey et al, 1993). Furthermore, the ET-1 model allows focal ischemia to be performed in conscious rats, removing the confounding influence of anesthetics that have been shown to be neuroprotective (Sharkey et al, 1993; Zuo, 2001). In fact a number of anesthetics, including ketamine, have been shown to affect the neutrophil inflammatory response after cerebral ischemia (Zilberstein et al, 2002).
Thus, we investigated and quantified the time course of neutrophil infiltration after ET-1-induced MCAo firstly, using the MPO activity assay in accordance with previous studies, and secondly, using two different antibodies, so that double-label immunofluorescence could be used to specifically identify and separate both neutrophils and macrophages. All experiments in this study were conducted using the same animals to gain a clearer understanding of the contribution neutrophils may make to neuronal injury, and to allow neutrophil infiltration to be correlated with changes in infarct volume.
Materials and methods
Animals
A total of 51 adult male Long-Evans rats weighing 280 to 300 g were purchased from Monash University Animal Services for use in this study. Rats were group-housed (four rats to a cage) under diurnal lighting with temperature maintained between 18°C and 24°C and given free access to food and water, until surgery, whereupon rats were housed in separate cages. All animal experiments were performed in accordance with the Prevention of Cruelty to Animals Act 1986, under the guidelines of the National Health and Medical Research Council for the Care and Use of Animals for Experimental Purposes in Australia.
Surgical Preparation
Rats were anesthetized using pentobarbitone sodium (60 mg/kg) by intraperitoneal injection. Using the method of Sharkey et al (1993) a 23-gauge stainless-steel guide cannula was stereotaxically inserted into the piriform cortex 2 mm dorsal to the right middle cerebral artery (MCA) using the coordinates of Callaway et al (1999) (0.2 mm anterior, −5.2 mm lateral, and −5.9 mm ventral, relative to Bregma, according to a stereotaxic atlas (Paxinos and Watson, 1986). The cannula was secured in place with dental cement, and two small screws inserted into the skull, to assist the dental cement to adhere to the skull. The incision was closed with sutures. Rats were housed individually after surgery with access to food and water available ad libitum and allowed to recover for at least 3 days before the induction of cerebral ischemia.
Induction of Focal Cerebral Ischemia
Cerebral ischemia was induced in conscious rats by occlusion of the right MCA by an ET-1 injection (120 pmol in 6 μl of saline over 6 mins; American Peptide Company, Sunnyvale, CA, USA) via a 30-gauge injector needle that protruded 2 mm beyond the end of the previously implanted guide cannula. The injector was held in place by a polyethylene-tubing cuff, and the animal was placed into a clear Plexiglas box for video recording during the induction of ischemia (Callaway et al, 1999). Characteristic indications of MCAo included clenching or failure to extend the contralateral forelimb and counterclockwise circling. Rats that did not exhibit these behavioral signs were deemed not to have had any occlusion of the MCA and were excluded from any further study.
Brain Tissue Processing
Rats were anesthetized and transcardially perfused with 250 mL of physiological saline (0.9%) at a perfusion pressure of 100 mmHg, which removes blood components from the vasculature (Barone et al, 1991), at 0 (n = 3), 1 (n = 5), 2 (n = 5), 3 (n = 5), 7 (n = 5), or 15 days (n = 5) after ischemia. Rats in the 0-day group were killed immediately after ischemia. Brains were removed and using a brain matrix were cut into six 2 mm coronal slices from the olfactory bulbs. Slices were frozen in isopentane over dry ice and stored at −80°C. Coronal cryostat sections (16 mm thick) were then cut, using a Leica cryostat, from each of the six slices, slide mounted, and used for both image analysis and immunohistochemistry. Approximately 10 sections were taken from each of the slices, with the majority of the brain remaining in the slices used in the biochemical assay for MPO activity.
Determination of Infarct Volume
The infarct area was determined in unstained sections using the ballistic light method, developed by Callaway et al (2000), which has been shown to permit rapid and reliable measurements of infarct damage. The infarct area was measured, using a computerized image analysis system (MCID M4 image analyzer, Imaging Research Inc, St Catherines, Ontario, Canada), by tracing around the area of damage in each brain section, which appeared dark in contrast to nonlesioned areas. Infarct volume was determined by integrating the cross-sectional area of damage at each stereotaxic level and the distance between each of the levels according to the method of Osborne et al (1987). Correction for edema of ischemic area was calculated using the formula: (area of normal hemisphere/area of ischemic hemisphere) area of ischemic lesion (Leach et al, 1993).
Experimental Protocols
Experiment 1: time course of neutrophil infiltration, examined using myeloperoxidase: In the initial part of this study, to investigate the infiltration of neutrophils using MPO, rats were divided into 4 groups (0, 1, 2, and 3 days after induction of ischemia).
Myeloperoxidase Activity Assay
The remaining slices of the brain, not used for histology and immunohistochemistry, were used to determine MPO activity as described previously by Barone et al (1991). For each individual rat, the ischemic and nonischemic hemispheres (from the six slices) were pooled separately for the assay, and wet weight was then recorded. The tissues were homogenized (1:20, wt/vol) in 5 mmol/L phosphate buffer (pH 6; 4°C) and centrifuged at 30,000g for 30 mins (4°C). The supernatant was discarded and the pellet washed again as described above. After decanting the supernatant, the pellet was extracted by suspension in 0.5% hexadecyltrimethylammonium bromide (Sigma Chemical Co, St Louis, MO, USA), in 50 mmol/L potassium phosphate buffer (pH 6, 25°C) for approximately 2 mins at an original wet weight to volume ratio of 1:10. Three freeze-thaw cycles were then performed with sonications (10 secs, 25°C) between cycles. After the final cycle, the tissues were incubated at 4°C for 20 mins and centrifuged at 12,500g for 15 mins (4°C). A 0.1 mL sample of the supernatant was mixed with 2.9 mL of 50 mmol/L phosphate buffer (pH 6.0) containing 0.167 mg/mL o-dianisidine dihydrochloride (Sigma Chemical Co, St Louis, MO, USA) and 0.0005% hydrogen peroxide. The change in absorbance was measured at 460 nm using a spectrophotometer, with absorbance recorded at 15 secs intervals over a 3-min period. One unit of MPO activity is defined as the amount that degrades 1 mmol of peroxide/min at 25°C. Tissue MPO activity was calculated using human MPO (DakoCytomation, Botany, Australia) as a standard, with units of MPO activity for each tissue sample being normalized on the basis of grams per wet weight of tissue (Yang et al, 1998).
Experiment 2: time course of neutrophil infiltration using double-label immunofluorescence for neutrophils and macrophages: In the second part of the study, the time course for neutrophil infiltration into brain tissue was examined using double-label immunofluorescence for both neutrophils and macrophages. In this part of the study, an additional 2 groups were included, 7 and 15 days after cerebral ischemia. Double-label immunofluorescence was conducted on slide mounted coronal sections from rats at 0, 1, 2, 3, 7, and 15 days after ischemia.
Double-labelled Immunofluorescence for Neutrophils and Macrophages
Slide-mounted sections were fixed in 4% paraformaldehyde for 30 mins. All sections were subject to a preblock in 10% normal goat serum (NGS), 0.5% bovine serum albumin and 0.3% Triton X-100 in phosphate-buffered saline (PBS; 0.1 mol/L, pH 7.4) for 1 h at room temperature, and then washed in PBS. All washes mentioned herein were 3 × 10 mins. Sections were then incubated overnight at 4°C in rabbit polyclonal polymorphonuclear neutrophil (PMN) antisera at 1:100,000 (Accurate Chemicals, Westbury, NY, USA) for neutrophils and mouse ED1 monoclonal antibody at 1:300 (Accurate Chemicals, Westbury, NY, USA) for macrophages in PBS containing 2% NGS and 0.3% Triton X-100. Tissue sections were washed and subsequently incubated with Alexa Fluorophore 488 nm donkey antimouse at 1:500 (Invitrogen, Mount Waverley, Australia) and Texas Red Fluorophore 595 nm goat antirabbit at 1:500 (Vector Laboratories, Burlingame, CA, USA) in PBS containing 2% NGS and 0.3% Triton X-100 for 90 mins at room temperature. Sections were again washed in PBS, before being coverslipped with antifade mountant (Vector Laboratories, Burlingame, CA, USA). In control experiments, primary antibodies were omitted to verify the absence of uncontrolled secondary antibody binding.
Quantification of Immunohistochemistry
The resulting sections were then examined under an Olympus BH-2 bright field photomicroscope, with immunoreactive (IR) cells being counted in a 20 × field for each brain section, using UTHSCSA Image Tool (version 3) (The University of Texas, San Antonio, TX, USA). The total number of neutrophils was estimated by counting the PMN IR neutrophils in the ischemic and nonischemic hemispheres of the cortex and striatum across six stereotaxic levels (4.2 to −5.2 mm relative to Bregma; Paxinos and Watson, 1986). The total number of neutrophils in each brain was calculated by integrating the PMN IR cells counted in the six predetermined stereotaxic levels and the distance between each of the levels according to the method of Osborne et al (1987). The total number of PMN IR cells that were also colocalized with ED1 IR cells were counted in a similar manner.
Confocal Imaging
Slide-mounted sections stained with both PMN antisera and ED1 monoclonal antibody (see above) were imaged with a Leica TCS-NT (Argon laser, Leica Microsystems, Wetzlar, Germany) inverted confocal microscope using 100 × /1.0 oil immersion lens. In dual-channel imaging, photomultiplier sensitivities were set to a level at which bleed-through effects from one channel to the other were negligible. For simultaneous imaging of PMN and ED1, immunostaining controls were used to adjust the photomultiplier levels separately for PMN and ED1, and the same settings were used during imaging of double-immunostained sections.
Statistical Analyses
Data are expressed as mean ± s.e.m. unless stated otherwise. Infarct volume was analyzed by analysis of variance (ANOVA) followed by Student-Newman-Keuls test. Twoway repeated measures ANOVA followed by Tukey's method was used for analysis of neutrophil infiltration and neutrophil-macrophage dual-labelling, whereas the post hoc test for analysis of the MPO activity was the Student-Newman-Keuls test. Linear regression with the Deming correction (Cornbleet and Gochman, 1979) was used to analyze the correlation between neutrophil infiltration and ischemic damage in the cortex and striatum. A value of P < 0.05 was considered to be significant.
Results
As shown in Figure 1, the infarct volume determined from measurements using the ballistic light method (Callaway et al, 2000) indicates the temporal progression and resolution of the ischemic lesion over the first 15 days after ischemia. The infarct volume increased significantly up to 3 days after ischemia, where the maximum infarct volume was found. After this point, infarct volume significantly decreased up to 15 days (Figure 1). The progression of the infarct volume from 0 to 15 days was similar for both the cortex (Figure 1A) and striatum (Figure 1B), although a greater increase in the infarct volume relative to the striatum was seen in the cortex at 3 days. The volume of damage in the cortex was almost four times greater than that of the striatum.
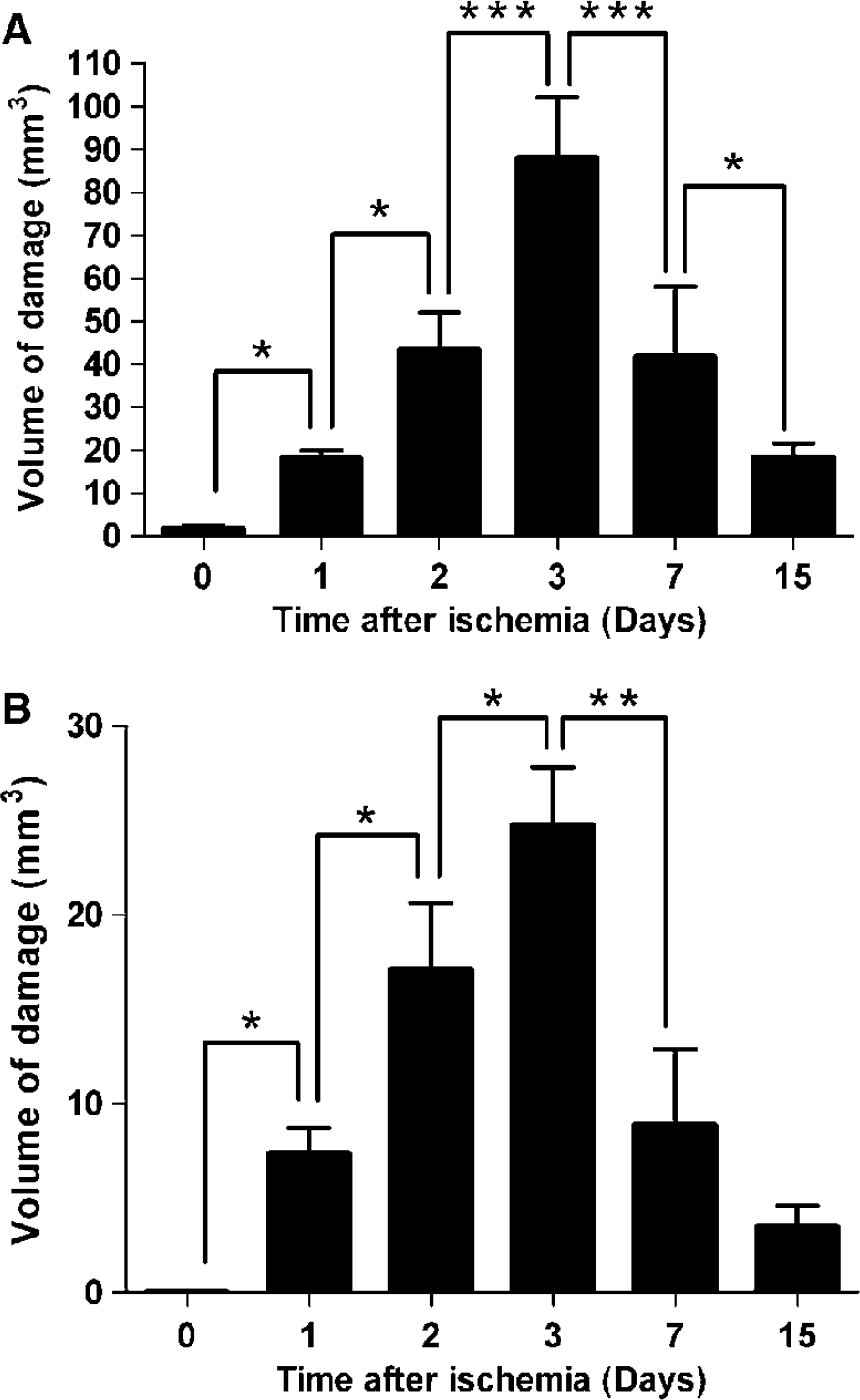
Total infarct volume in both rat cortex (
Experiment 1: Time Course of Neutrophil Infiltration, Examined using myeloperoxidase
Myeloperoxidase activity assay: To quantify neutrophil accumulation after cerebral ischemia, we initially used the MPO activity assay. This method has been used extensively in previous studies, and is reported to be reliable for detecting neutrophil infiltration (Barone et al, 1991). The time points 0,1, 2, and 3 days after ischemia were chosen because previous studies suggest neutrophil infiltration into the ischemic region of the brain occurs early after ischemia-reperfusion. Myeloperoxidase activity was negligible when measured at 0 h after ischemia in the ischemic hemisphere (ipsilateral), similarly the MPO activity in the nonischemic hemispheres (contralateral) at each time point was also negligible (Figure 2). In contrast, MPO activity in the ischemic hemispheres in rat brain homogenates taken from rats killed at 1, 2, and 3 days after ischemia showed MPO activity significantly increased with time (Figure 2). The increases in MPO activity (units MPO/g wet weight of tissue) seen at 1 day (0.2 ± 0.04 U/g wet tissue) and 2 days (0.4 ± 0.04 U/g wet tissue) after cerebral ischemia was approximately linear but was less at 3 days (0.5 ± 0.03 U/g wet tissue).
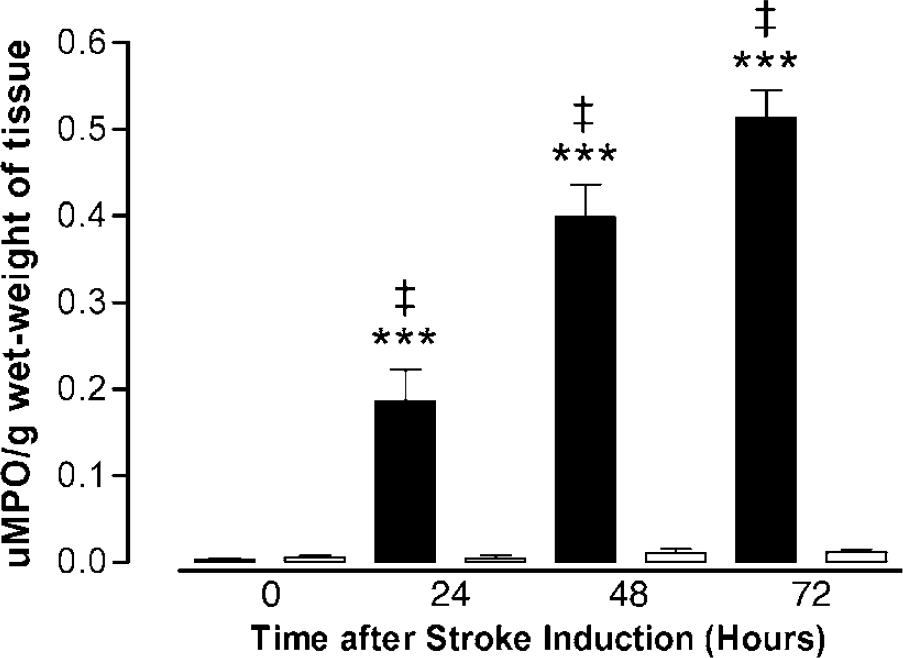
Time course of myeloperoxidase (MPO) activity as a measure of neutrophil infiltration, measured at 0, 1, 2, and 3 days after focal ischemia in the ischemic and nonischemic hemispheres. Values are mean ± s.e.m., expressed as units of MPO activity per gram wet-weight of brain tissue, for n = 3 to 4 in each group. Myeloperoxidase activity after ischemia increased significantly with time. ***P < 0.0001 ischemic hemisphere versus nonischemic hemisphere for each time (1 to 3 days); ‡P < 0.05 MPO activity versus adjacent time point in the ischemic hemisphere, ANOVA. (▪: ischemic hemisphere and □: nonischemic hemisphere)
Experiment 2: Time Course of Neutrophil Infiltration using Double-Label Immunofluorescence for Neutrophils and Macrophages: We used double-label immunofluorescence to visualize neutrophils (stained red) and macrophages (stained green) in the brain at 0 to 15 days after cerebral ischemia, using a PMN polyclonal antisera and ED-1 monoclonal antibody, respectively. Confocal imaging of double-immunostained sections confirmed that macrophages were engulfing neutrophils in the ischemic brain, as demonstrated by the localization of apoptotic neutrophils within macrophages seen in the merged image of Figure 3.
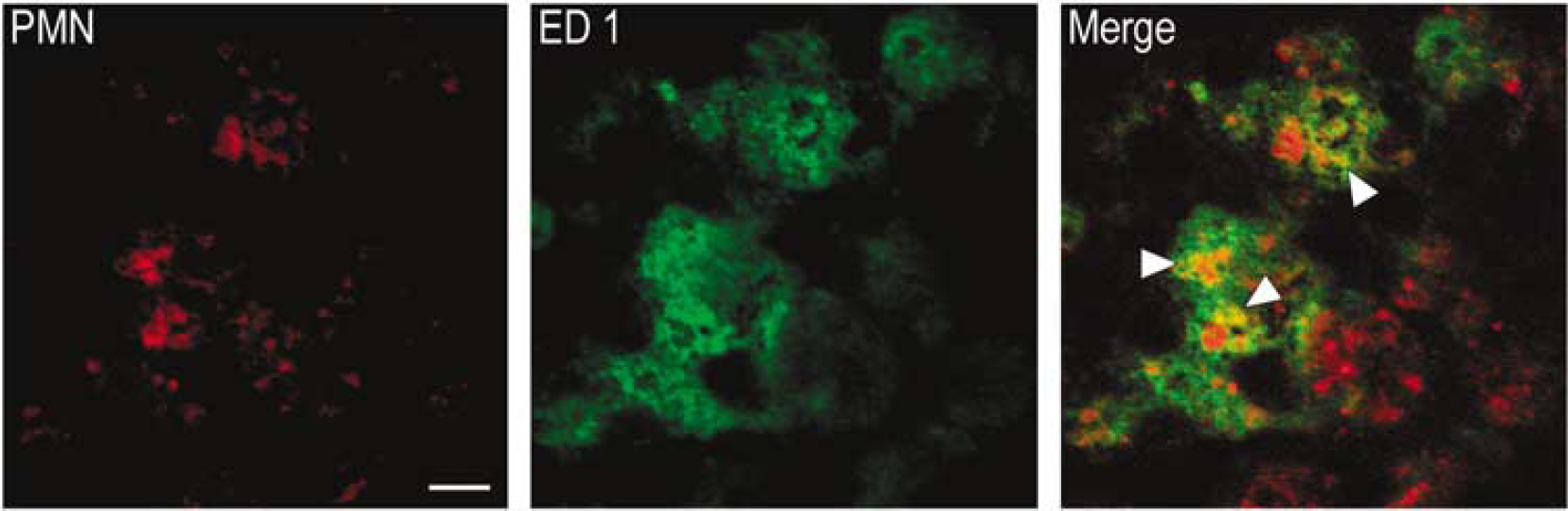
Confocal imaging of double immunostained sections shows colocalization of PMN (red) IR bodies within ED1 (green) IR cells in the cortex at 15 days after cerebral ischemia. This image represents macrophage (green) engulfment of apoptotic neutrophils (red), indicated by the arrows. Scale bar represents 25μm. PMN, polymorphonuclear neutrophil; IR, immunoreactive.
Cell counting of red IR cells was performed in the cortex and striatum to quantify the number of infiltrating neutrophils and the number of neutrophils that were colocalized with macrophages (seen in representative image in Figure 3 as the yellow color resulting from merged images) and the results are shown in Figures 4 and 5. The total number of neutrophils infiltrating the cortex of the ischemic hemisphere at the selected stereotaxic levels over time can be seen in Figure 4A. It was observed that the greatest accumulation of neutrophils occurred at level 0.3 mm relative to bregma, which corresponds to the location of the infarct core, where ET-1 was injected to cause MCAo. Figure 4A also shows that the number of neutrophils that are also colocalized with macrophages increases with time. This is better seen in Figure 4B that shows the percentage of the total number of infiltrating neutrophils that were colocalized with macrophages increased with time after ischemia. The percentage of neutrophils detected in the brain that were also colocalized with macrophages increased from almost 0 at day 0% to ∼50% by 3 days, and by 15 days the number of neutrophils colocalized with macrophages was ∼85%. Interestingly, at 7 days the percentage of neutrophils colocalized with macrophages was significantly decreased from 3 days to ∼20%. Additionally, the merged image (Figure 3) clearly shows multiple neutrophils colocalized with macrophages.
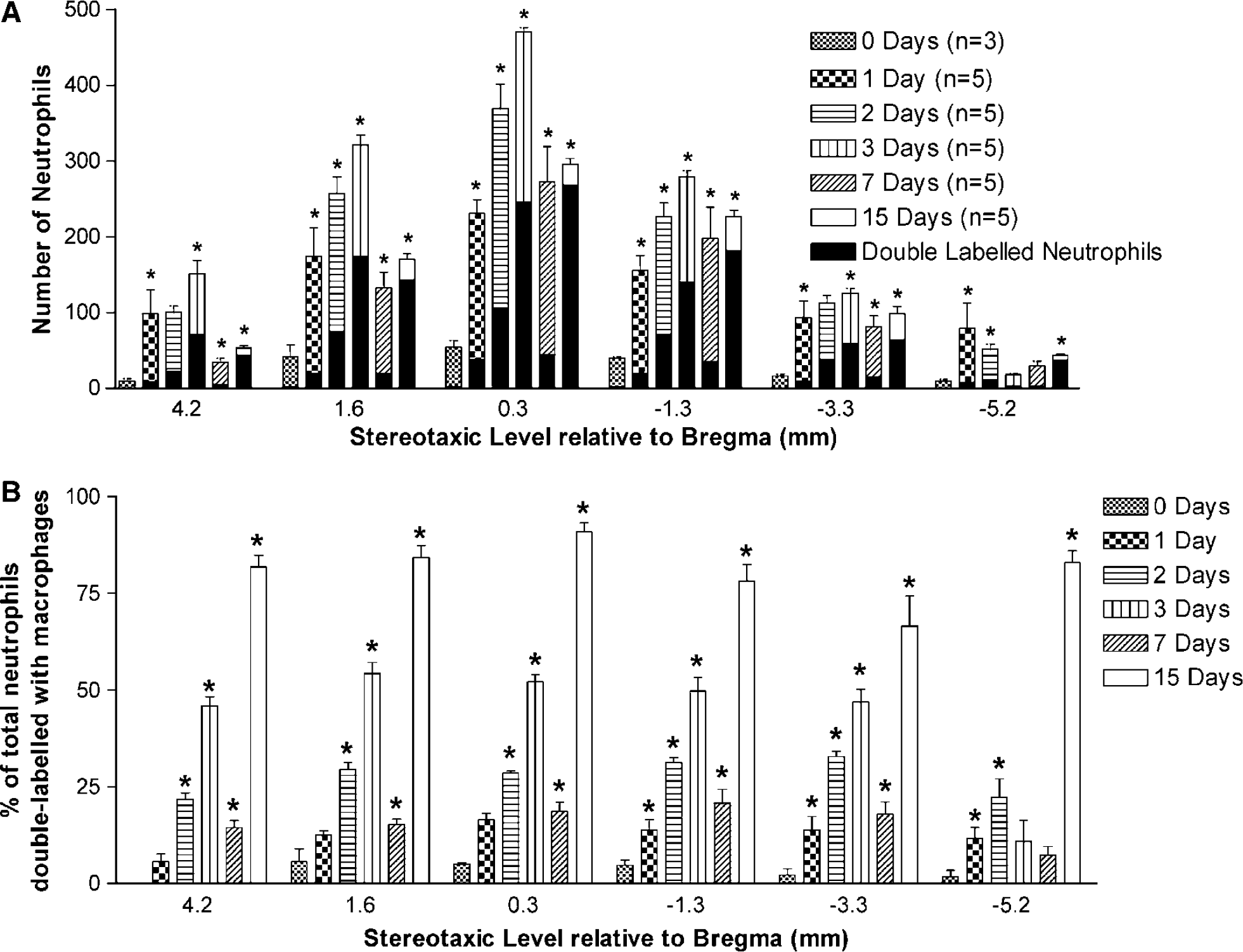
Neutrophil and neutrophil–macrophage cell counting conducted in the cortex. (
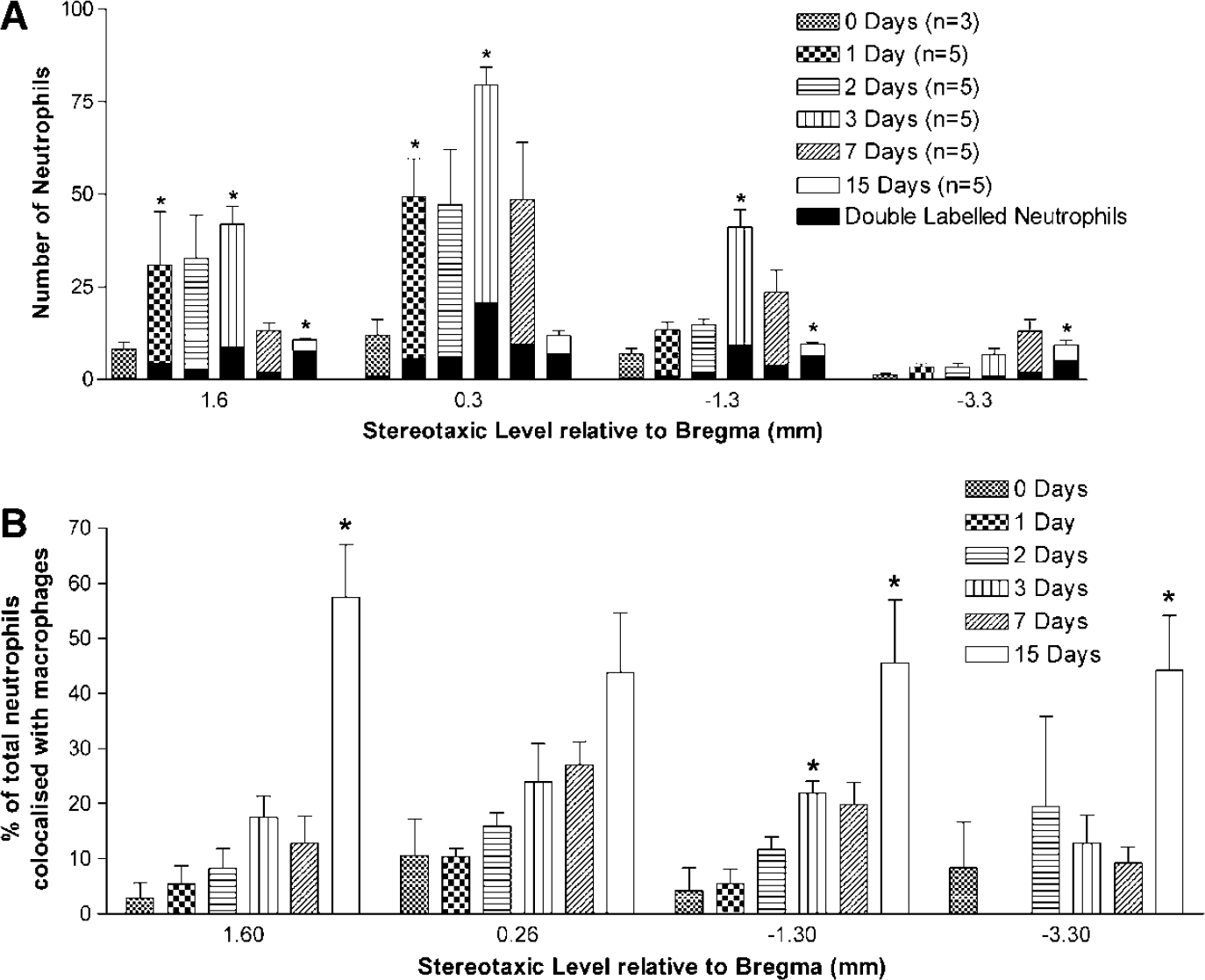
Neutrophil and neutrophil-macrophage cell counting conducted in the striatum. (
The number of neutrophils infiltrating into the striatum of the ischemic hemisphere (Figure 5A) was five times less than the number infiltrating the cortex. In accordance with results for cortical regions, peak neutrophil infiltration was also seen at 3 days for the striatum. The percentage of the total number of infiltrating neutrophils that were engulfed by macrophages in the striatum (Figure 5B) was not as great as that seen in the cortex, with only 25% of neutrophils engulfed by macrophages at 3 days and only 60% at 15 days. The result at 7 days in the striatum was also surprising, because in contrast to the result observed in the cortex (significantly decreased colocalization) there was no significant decrease in neutrophil-macrophage colocalization. Although a small trend towards a decrease in colocalization could be observed, it was not significantly different from 3-day values. The maximal accumulation of neutrophils into the ischemic region at 3 days was, however, the same in both the cortex and striatum (Figures 4A and 5A).
To investigate whether any correlation existed between neutrophil infiltration and ischemic damage, we compared the total number of infiltrating neutrophils (those not engulfed by macrophages) with the infarct volume (Figure 6). For both the cortex (Figure 6A) and striatum (Figure 6B) a significantly positive correlation existed between neutrophil infiltration and infarct volume (r2 = 0.86 and 0.80, respectively).
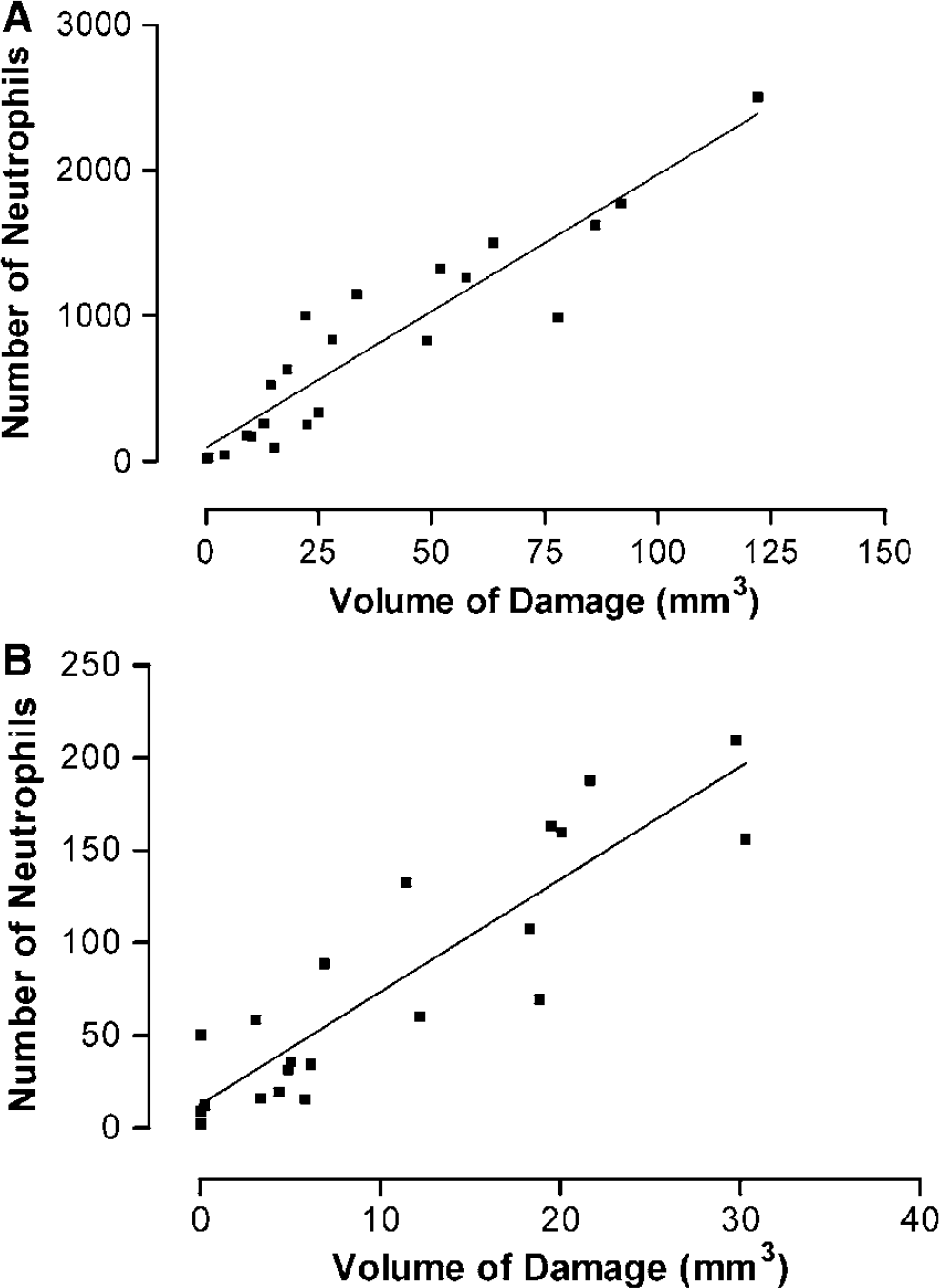
Correlation between the volume of damage (abscissa) in the right hemisphere for each rat with the total number of infiltrating neutrophils (ordinate) (i.e., neutrophil-positive cells that are not colocalized with macrophages). (
Discussion
The present study is the first to investigate neutrophil infiltration into the ischemic brain in conscious rats. The benefits of using the ET-1 model of focal cerebral ischemia over other models is that the ET-1 model does not damage mechanically the cerebral vasculature (McAuley, 1995), which could increase adhesion molecule expression and disrupt the blood–brain barrier, and being performed in conscious rats removes the confounding influence of anesthetics, which have been shown to affect neutrophil function and accumulation (Miller et al, 1996; Zilberstein et al, 2002). This study also examined the interaction between neutrophils and macrophages for up to 15 days after cerebral ischemia. Being the first study to investigate neutrophil infiltration in the ET-1 model of focal cerebral ischemia, we examined the temporal profile of neutrophil infiltration in this model using the assay for MPO activity, thus allowing comparisons with previous studies. Our results show that engulfment of neutrophils by macrophages (seen as fluorescent colocalization) occurs as early as 1 day after ischemia and increases with time, with approximately 50% of neutrophils colocalized with macrophages by 3 days and 85% to 90% by 15 days, supporting the notion that the common marker for neutrophil accumulation, myeloperoxidase, is not sufficient for studies of neutrophil infiltration, even at early time points. Interestingly, at 7 days there was a decrease in the number of double-labelled cells, indicating the time course of neutrophil infiltration may not be as simple as previously thought. Furthermore, in our study we were able to perform multiple experiments in the same rat enabling direct correlations to be drawn between neutrophil accumulation and ischemic damage.
The role of neutrophils in the progression of ischemic damage has been extensively studied in transient focal cerebral ischemia by the use of the MPO activity assay since Barone et al (1991) first used it to indirectly quantify neutrophil accumulation in the brain. In fact, the use of MPO, whether in the biochemical activity assay or as an immunomarker, has become a predominant method used to measure neutrophil infiltration (Barone et al, 1992; Batteur-Parmentier et al, 2000; Beray-Berthat et al, 2003a; Chopp et al, 1996; Feuerstein et al, 1998; Jiang et al, 1995; Kitagawa et al, 1998; Matsuo et al, 1994; Zhang et al, 1995). Myeloperoxidase has traditionally been described as a useful marker for neutrophil infiltration within damaged tissue, because it is primarily localized in neutrophils (Barone et al, 1991), but it has also been shown to be expressed in monocytes and macrophages (Kochanek and Hallenbeck, 1992) although in much smaller amounts (Bos et al, 1978; Zhang and Chopp, 1997). Activated macrophages can even acquire MPO from the ischemic environment and by phagocytosis of apoptotic neutrophils (Locksley et al, 1987; Neill et al, 1985; Zeller et al, 1988).
From the results obtained, using both immunohistochemistry and the biochemical assay, we observed that neutrophil infiltration increased over time with the progression of the ischemic injury, which is in accordance with previous findings but with different methods of producing focal ischemia (Barone et al, 1995; Feuerstein et al, 1998; Matsuo et al, 1994). Results from the biochemical assay in the present study indicated that MPO activity was 20% lower than values obtained by Barone et al (1991), suggesting that the type of model used to induce cerebral ischemia may partially determine the inflammatory response observed in the rats (Beray-Berthat et al, 2003b; Emerich et al, 2002; Hayward et al, 1996; Macrae, 1992). It has been proposed that other models, including the filament model, may affect the progression of injury in rats subjected to cerebral ischemia by mechanically damaging the endothelium and surrounding structures (Macrae, 1992; McAuley, 1995). This could account for the decreased neutrophil infiltration observed in the ET-1 model in comparison to infiltration seen in other models of cerebral ischemia. However, variability in the severity of the ischemia injury induced with the different models cannot be discounted, as previous studies investigating neutrophil infiltration using the filament model have shown an increased infarct volume when compared with the ET-1 model (Matsuo et al, 1994; Prestigiacomo et al, 1999; Zhang et al, 1995). In fact, Sharkey et al (1994) commented on the results of Dawson et al (1993), which showed that the volume of ischemic damage was reduced by approximately 25% when compared with models that surgically occlude cerebral vessels. This reduction in ischemic damage using the ET-1 model may also account for the 20% reduction in MPO activity seen in the present study.
The ET-1 model may provide a more attractive alternative to the existing models of cerebral ischemia, as it is performed in conscious rats without the use of anesthetics, which have been reported to be neuroprotective (Sharkey et al, 1993; Zuo, 2001), making it highly relevant to the clinical situation of stroke. Furthermore, anesthetics including ketamine, pentobarbital, propofol, and halothane have been shown to interact with neutrophils by modulating functions such as chemotaxis, phagocytosis, the accumulation and activation of neutrophils, and the release of superoxide anion (Mikawa et al, 1998; Miller et al, 1996; Nishina et al, 1998; Zilberstein et al, 2002). Thus, the accumulation of neutrophils and progression of the neutrophil inflammatory response after cerebral ischemia can be investigated using the ET-1 model without the confounding effects of anesthetics. However, ET-1 has also been reported to be involved in exacerbating ischemic damage, as seen by studies using ET-1 antagonists that reduce infarct formation after ischemia (Barone et al, 2000; Dawson et al, 1999; Gupta et al, 2005; Matsuo et al, 2001). Moreover, in a recent study by Lo et al (2005) in which transgenic mice overexpressing ET-1 were generated, an increase in brain edema was observed after cerebral ischemia. Additionally, studies have shown that ET-1 can enhance neutrophil activation and adherence to the vascular endothelium (Caramelo et al, 1997; Jeremy et al, 1999), which for the purposes of this study could have affected the results obtained and must be considered, but given that we saw a 20% reduction in MPO activity compared with previous studies the affect of ET-1 on the neutrophil inflammatory response in this study appears to be negligible. In contrast to these studies, ET-1 has been shown not to activate neutrophils (Kopprasch et al, 1995) and in a human study by Haapaniemi et al (2000) ET-1 levels did not correlate with patient outcomes after ischemic stroke. Furthermore, previous studies have established the usefulness of ET-1 for the production of focal cerebral ischemia (Adkins et al, 2004; Sharkey et al, 1994).
Much research has been conducted into the role of macrophages in attenuating the neutrophil inflammatory response in the periphery (Coxon et al, 1996; Meszaros et al, 1999, 2000). In the case of the brain, it has been demonstrated that neutrophil infiltration occurs over the first 3 days after MCAo, which our results with the MPO assay substantiate. Macrophages then replace neutrophils as the dominant inflammatory cell in the ischemic region at later time points (Barone et al, 1995; Feuerstein et al, 1998). However, the interaction between neutrophils and macrophages after cerebral ischemia has not been investigated. Studies have shown that the MPO activity assay can detect both neutrophil and monocyte-macrophage infiltration into the brain after ischemia (Barone et al, 1995; Feuerstein et al, 1998), which suggests that MPO alone may not be selective enough to quantify neutrophil infiltration. However, Zhang et al (2001) using an antibody against MPO to immunohistochemically measure inflammatory cells, reported that macrophages typically did not exhibit MPO immunoreactivity. In contrast, the studies by Bayir et al (2005) and Schopfer et al (2003) showed that MPO could be immunohistochemically detected within macrophages. Given the controversy surrounding the role of neutrophils after cerebral ischemia (Emerich et al, 2002; Hayward et al, 1996), it is important to investigate all of the physiologic factors that may be involved in the neutrophil response. Therefore, along with using MPO as a marker for neutrophils, we additionally performed double-label immunofluorescence to examine the influence of macrophages on the inflammatory response of neutrophils using two different antibodies to discriminate between the two cell types.
Zhang and Chopp (1997), showed that MPO immunohistochemistry is more sensitive than the MPO biochemical assay for detecting neutrophil accumulation. Subsequent studies by Phillips et al (2000) and Williams et al (2003) examined neutrophils and macrophages at 24 and 72 h after cerebral ischemia using standard hematoxylin and eosin staining to examine cell morphology. In the present study, the more powerful tool to specifically identify infiltrating inflammatory cells, double-label immunofluorescence was used. Double-label immunofluorescence provides a method that can decrease the substantial amount of time taken to identify the different inflammatory cell types seen when using standard morphologic staining, by labelling two different antigens, thus separating neutrophils and macrophages.
Our results using double-label immunofluorescence show that large-scale emigration of neutrophils into the ischemic region occurs during the first day after cerebral ischemia and peaks at 3 days, in accordance with observations made when we used MPO alone as a marker to quantify neutrophil infiltration. In fact, these results are in agreement with previous findings by Matsuo et al (1994) and Feuerstein et al (1998) who have shown neutrophil infiltration to occur over the initial 3 days after ischemia and then decrease with time. Our study also showed that neutrophil infiltration, although decreased after the peak at 3 days, persists for at least 15 days, which would appear to contradict previous findings that form the basis for the currently accepted theory regarding the temporal profile of neutrophil accumulation into the ischemic brain region.
However, our results also show that macrophages in the ischemic brain region can engulf neutrophils, as previous results in the periphery have demonstrated (Coxon et al, 1996; Meszaros et al, 1999). Induction of apoptosis and then phagocytosis of neutrophils by macrophages is the major pathway for clearance of activated neutrophils in inflammatory sites in the periphery (Meszaros et al, 1999, 2000). Studies by Meszaros et al (1999) in vitro, demonstrated that macrophage engulfment of neutrophils was extremely impaired whereas the neutrophils remained active and viable, such that the frequency of macrophage phagocytosis of neutrophils only increased when the levels of neutrophil apoptosis increased. Meszaros et al, (1999) demonstrated that macrophages use distinct receptor patterns (expressed on apoptotic neutrophils) to recognize and ingest only neutrophils undergoing apoptotic cell death. Furthermore, Meszaros et al (2000) demonstrated that macrophages could actually induce apoptosis in viable neutrophils through a constitutive effector mechanism requiring both intercellular binding and membrane-bound TNF-α. Indeed, the apoptosis of neutrophils and their clearance by macrophages is considered to be beneficial because recognition and engulfment of neutrophils prevents the lytic release of cytotoxic and immunogenic intracellular contents into the surrounding microenvironment (Meszaros et al, 2000). Moreover, persistent neutrophil-rich inflammatory sites have been associated with increased tissue destruction, and in disorders of apoptosis where phagocytosis of neutrophils is impaired, persistence of chronic inflammatory conditions has been reported (Cox et al, 1995; Haslett et al, 1994; Meszaros et al, 2000). Thus, the inducement of apoptosis and subsequent removal of neutrophils by macrophages is considered to be critical in the resolution of the inflammatory response and in preventing further exacerbation of the ischemic injury (Meszaros et al, 2000; Savill, 1997).
The present results suggest that the clearance of neutrophils from the ischemic lesion is achieved via macrophage phagocytosis of apoptotic neutrophils. The colocalization we observed in the present study would also explain the reduction in neutrophils that is seen over time in previous studies (Barone et al, 1995; Matsuo et al, 1994), as the majority of neutrophils present at the later times after cerebral ischemia have been engulfed by macrophages. Our results show that the entry of macrophages into the ischemic region marks the onset of neutrophil engulfment, so that by 3 days after ischemia the number of neutrophils colocalized with macrophages in the cortex is approximately 50% and by 15 days this colocalization has increased to approximately 85%. The result seen for the 7-day group in the cortex is interesting, with the amount of neutrophils colocalized with macrophages being approximately 25%. This was surprising given the 3 and 15 days result, suggesting that perhaps a second wave of neutrophil infiltration is occurring around 7 days. This time point corresponds to the proposed peak in monocyte-macrophage accumulation demonstrated by Barone et al (1995) and it is possible that any additional neutrophil infiltration has been masked by the accumulation of macrophages, when MPO was used as the marker for neutrophils in previous studies.
Previous studies have shown that at 3 days afterischemia MPO activity increases dramatically, peaking at 5 days, before decreasing to preischemic values by 15 days (Barone et al, 1995). The increase and peak in MPO activity at 5 days seen by Barone et al (1995) has previously been demonstrated histologically to be the result of a dramatic increase in monocyte and macrophage accumulation within the ischemic brain, and corresponds to a time when neutrophils were reported to be decreasing (Clark et al, 1993, 1994). Barone et al (1995) unequivocally demonstrated that macrophages are a source of MPO, and suggested that macrophages may have acquired additional MPO activity through phagocytized neutrophils in the tissue or alternatively, that MPO is available after the lysis of neutrophils. It is likely that both of these events are occurring, however, the first event is most likely the predominant mechanism, with macrophages having been shown previously to engulf apoptotic neutrophils in the periphery and can also induce neutrophil apoptosis before phagocytizing them. To investigate whether neutrophil infiltration was in fact biphasic, and that previous studies using MPO activity as a marker may have masked the secondary infiltration of neutrophils, additional time points would need to be examined, such as days 4 and 6 and also time points between 7 and 15 days.
The results seen in the striatum appear different to those of the cortex. The peak in neutrophil infiltration at 3 days is still present but is less pronounced than the peak seen in the cortex. The number of infiltrating neutrophils then decreases in a similar manner to that proposed by previous studies, so that by 15 days the number of cells that are solely neutrophil-positive is extremely small. Additionally, the results from the striatum did not show the second wave of neutrophil infiltration that was seen in the cortex at 7 days, and although, a similar trend in the percentage of neutrophils colocalized with macrophages at 7 days was observed, it was not statistically significant. The percentage of neutrophils that were colocalized with macrophages in the striatum across all the levels of the brain over the 0 to 15 days, was approximately 20% to 30% lower than that seen in the cortex. This includes the 15-day group whose percentage of 50% to 60%, may indicate that neutrophils are still entering the striatum at 15 days, although in much smaller numbers when compared with the cortex. The accumulation of neutrophils in the ischemic brain at later times, seen in this study, is consistent with clinical studies that have shown that neutrophil accumulation in human brains after stroke can persist for over 30 days (Akopov et al, 1996).
The significance of the difference between neutrophil infiltration and accumulation in the cortex, and striatum is not known. Previous studies have shown that differences exist between cerebral regions in the extent that neutrophils are capable of contributing to ischemic damage (Batteur-Parmentier et al, 2000; Beray-Berthat et al, 2003a, b; Kitagawa et al, 1998; Yenari et al, 1998). Beray-Berthat et al (2003a) showed that neutrophils only contributed to ischemic damage in the cortex and not the striatum at 48 h after ischemia, when neutrophils in blood were depleted using vinblastine. The intrinsic variability in the accumulation of neutrophils that we observed across brain regions suggests that the rate of ischemic damage is different for the striatum than for the cortex. Indeed, the striatum is more vulnerable than the cortex to ischemic stress (Abe et al, 1988), as seen by a more pronounced reduction in cerebral blood flow in this region, which is thought to occur because striatal arteries are end arteries and do not allow collateral blood flow (Tamura et al, 1981). Thus, infarct maturation may occur before the infiltration of neutrophils, meaning neutrophils may not contribute to infarct formation in the striatum (Beray-Berthat et al, 2003a).
Previous studies have commented that neutrophil infiltration occurs very early for a brief temporal window after which point neutrophil accumulation decreases (Barone and Feuerstein, 1999; Barone et al, 1995; Becker et al, 2001; Feuerstein et al, 1998), yet the results obtained in this study show that neutrophil infiltration into the brain increases early after ischemia, and, although reduced, is still occurring at the later times (7 and 15 days). Perhaps the large-scale accumulation of macrophages in the inflammatory site at later times is masking the infiltration of neutrophils, and the methods used have not been sensitive or selective enough to detect them. This could mean temporal differences in the pathophysiologic sequelae after ischemia between cerebral regions might not have been detected.
The area and severity of damage seen in this study after ET-1-induced MCAo progressively increased and peaked at 3 days after ischemia. By 3 days, it encompassed the majority of the ischemic hemisphere and was accompanied by an increased swelling of the ischemic hemisphere owing to edema, but after this increase cerebral infarct volume is reduced at 7 days and more so by 15 days. The increase in cerebral infarct volume, followed by the subsequent decrease after 3 days, is consistent with the findings of Clark et al (1993) who also showed an increase in cerebral infarct volume which decreased with time, and was almost completely resolved (that it could not be detected) by 30 days after ischemia. Clark et al (1993) demonstrated that the reduction in infarcted tissue corresponded to the time point at which macrophages became a dominant feature of the ischemic lesion, and was associated with a loss in hemispheric volume. Subsequently, it was postulated that the phagocytic activity of macrophages in the necrotic tissue is responsible for reducing the hemispheric volume by causing cavitation within the lesion, after cerebral ischemia (Clark et al, 1993). However, differences in the temporal progression of the cerebral infarct between the two studies could be attributed to the use of different models of ischemia or alternatively, differences in the strain of rat used.
Development of cerebral infarction after cerebral ischemia has been previously shown in planimetric studies to begin within hours of MCAo (Clark et al, 1993), a time that coincides with the appearance of neutrophils in the ischemic lesion (Barone et al, 1991; del Zoppo et al, 2001). Yet studies have failed to show a clear correlation between neutrophil infiltration and infarct formation (Beray-Berthat et al, 2003b; Emerich et al, 2002; Fassbender et al, 2002; Hayward et al, 1996; Soriano et al, 1999; Takeshima et al, 1992). This, in part, could be because infarct volume measurements and histology being performed in one group of animals whereas neutrophil accumulation is measured, using the MPO activity assay, in a separate group. Thus, direct correlations between infarct volume and neutrophil accumulation cannot be made, contributing to the controversy that exists over the role of neutrophils after cerebral ischemia, with some researchers showing neutrophils to be a principal mediator of secondary cell death responses (Beray-Berthat et al, 2003a; Chen et al, 1994; Chopp et al, 1996; Connolly et al, 1996, 1997; Jiang et al, 1995; Kitagawa et al, 1998; Prestigiacomo et al, 1999; Yenari et al, 1998; Zhang et al, 1995), whereas other investigators suggest that infiltrating neutrophils may only be passive bystanders to the injury (Emerich et al, 2002).
All the experiments in this study were performed in the same rats so that measurements of infarct volume could be directly related to neutrophil infiltration. Our results show that very robust correlations exist between neutrophil infiltration (i.e., the intact neutrophils, and not neutrophils engulfed by macrophages) and the volume of damage in both the cortex and striatum, suggesting a cause-effect relationship between the two might exist. These results do not show whether the ischemic damage is causing the infiltration of neutrophils, or whether the infiltration of neutrophils is exacerbating the ischemic damage. Similar correlations can be made using MPO, between neutrophils and volume of damage, this however is only true over the first 3 days after cerebral ischemia (unpublished observations). Previous studies have demonstrated that 3 days after ischemia, MPO activity increases dramatically, peaking at 5 days, before decreasing to preischemic values by 15 days (Barone et al, 1995). The peak in MPO activity at 5 days, because of monocyte-macrophage accumulation (Feuerstein et al, 1998), and the relationship between MPO and infarct volume is no longer maintained, and cannot be related to neutrophil accumulation. To investigate how neutrophil infiltration and ischemic damage are related, further experiments could investigate neutrophil infiltration in animals that are deficient in MPO or that have a cell specific deletion of MPO.
The activation of neutrophils can lead to the release of various proinflammatory mediators and ROS that can exacerbate ischemic damage (Witko-Sarsat et al, 2000). Recently, Yu et al (2004) have elegantly shown that neutrophils are clearly activated by adenosine when entering the ischemic region of the brain after cerebral ischemia. In an editorial of this paper, Iadecola (2004) commented that Yu et al (2004) were able to demonstrate a direct contribution of neutrophils to the ischemic damage. In addition, studies by Beray-Berthat et al (2003a) have shown that neutrophils contribute to the progression of the ischemic lesion (at least in the cortex). Given our results that show a direct correlation between neutrophil accumulation and ischemic damage, it may be argued, at least in part, that neutrophils do contribute to the progression of the ischemic lesion. However, cerebral ischemia may be inducing the expression of adhesion molecules in cerebral endothelial cells and hence facilitating the infiltration of neutrophils into the brain (del Zoppo et al, 2001). We have shown that macrophages engulf neutrophils that enter the brain in a manner similar to those in the periphery, so the direct contribution that neutrophils play in exacerbating oxidative stress and ischemic damage might only be small. Macrophages could limit the inflammatory response of neutrophils in the brain by inducing apoptosis in activated neutrophils (Meszaros et al, 2000) and then, via phagocytosis, engulf the apoptotic neutrophils (Meszaros et al, 1999) thus preventing, or at least minimizing the cytotoxic release of their inflammatory mediators.
Clearcut data in animal models shows a therapeutic effect of inhibiting both neutrophil adherence to the vascular endothelium and subsequent migration into inflammatory regions (for review see Ulbrich et al, 2003). However, the negative results seen in clinical trials with antineutrophil drugs such as enimolab and LeukArrest need to be interpreted carefully. Future clinical studies need to be designed that extensively investigate the entire inflammatory response of neutrophils from their infiltration and removal from inflammatory sites, to delineate whether antineutrophil strategies can actually be used safely and selectively.
This is the first study in a rat model of focal cerebral ischemia to examine infarct volume, MPO activity and double-label immunofluorescence in the same rats, allowing a greater understanding of neutrophil infiltration in relation to ischemic damage. Methodology utilized in this study allows correlations of neutrophil infiltration and infarct formation to be determined for the first time. Although this study is not definitive in the conclusion of neutrophil contribution to infarct formation after cerebral ischemia, it does show that a significant correlation between neutrophil infiltration and infarct formation exists in an unanesthetized rodent model of cerebral ischemia, indicating a possible relationship between the two. The early infiltration and accumulation of neutrophils into the ischemic region after cerebral ischemia, coupled with the phagocytosis of neutrophils by macrophages seen in this study indicates that using MPO alone as a marker for neutrophils is not selective enough, even at earlier times points. Therefore, it is essential that future studies consider the contribution of macrophages in the resolution of the neutrophil-induced inflammatory response.