
Abstract
Cytochrome P450 metabolism of arachidonic acid produces the potent vasoconstrictive metabolite, 20-hydroxyeicosatetraenoic acid (20-HETE). Recent studies have implicated 20-HETE as a vasoconstrictive mediator in hemorrhagic stroke. The purpose of this study was to determine the effect of the 20-HETE inhibitor, HET0016, on lesion volume and cerebral blood flow (CBF) after temporary middle cerebral artery occlusion (MCAO) in rats. Plasma pharmacokinetics and tissue concentrations of HET0016 were determined after a 10 mg/kg intraperitoneal dose. Separate rats were treated with HET0016 or vehicle before 90mins of MCAO. Lesion volume was assessed by 2,3,5-triphenyl-tetrazolium-chloride and cerebral flow was determined using laser Doppler flow. The effect of MCAO on in vitro microsomal formation of mono-oxygenated arachidonic acid metabolites was also determined. Results show that HET0016 has a short biologic half-life, distributes into the brain, and is associated with a 79.6% reduction in 20-HETE concentration in the cortex. Lesion volume was greatly reduced in HET0016-treated (9.1%±4.9%) versus vehicle-treated (57.4%±9.8%; n = 6; P < 0.001) rats. An attenuation of the observed decrease in CBF was observed in HET0016-treated (180 mins 89.2%±6.2%; 240 mins 88.1%±5.7% of baseline flow) versus vehicle control (180 mins 57.6%±19.0%; 240 mins 53.8%±20.0% of baseline flow; n=6; P < 0.05). Brain cortical microsomal formation rate of 20-HETE was also reduced at 24 h in the ipsilateral hemisphere after MCAO. These data support a significant role for 20-HETE in the pathogenesis of ischemic stroke.
Keywords
Introduction
The primary therapeutic target in ischemic stroke is to reduce damage in the ischemic penumbra. Studies in experimental animal models have identified the ischemic penumbra and have showed that the viability of this region is dependent on the degree of residual cerebral blood flow (CBF) remaining during and after cerebral ischemia (Ayata et al, 2004; Coyle 1987; Symon et al, 1976). Currently, the clinical use of thrombolytics is the only proven therapeutic approach to restore flow in patients with ischemic stroke, and has been shown to improve outcome when administered within 0 to 6 h of symptom onset (Furlan et al, 1999). However, even after restoration of flow, a period of delayed reduction in CBF has been observed in both global and temporary focal ischemia models (Hossmann 1997; Karibe et al, 1994; McHedlishvili et al, 1997). Although decreasing ischemic duration with thrombolytics is an established therapeutic modality, progress has been limited regarding the development of novel therapeutic interventions that target other important mediators of cerebrovascular regulation.
Studies in models of temporary middle cerebral artery occlusion (MCAO) and global ischemic injury have reported a reduction in the diameter of cerebral microvessels and have attributed this reduction to a pathologic mechanism of increased cerebrovascular resistance (Dawson et al, 1999; Fischer et al, 1977; Hossmann 1997; McHedlishvili et al, 1997); however, the key cellular vasoconstrictive regulators of postischemic cerebrovascular resistance are largely unknown.
One potential mediator of these effects is via the terminal hydroxylation of arachidonic acid by cytochrome P450 enzymes to form 20-hydroxyeicosatetraenoic acid (20-HETE), which produces cerebral vasoconstriction (Gebremedhin et al, 2000; Harder et al, 1994). 20-HETE is one of the most potent vasoconstrictive eicosanoids in existence and is known as a counter-regulator of nitric oxide in the cerebromicrovasculature (Hercule et al, 2003; Sun et al, 2000). In addition to the vasoconstrictive effects, inhibition of 20-HETE has also been shown to play an important role in mediating the angiogenic effects of vascular endothelial growth factor (VEGF) (Amaral et al, 2003; Chen et al, 2005). Animal and clinical studies have evaluated the role of 20-HETE in the pathogenesis of hemorrhagic and ischemic stroke. Studies evaluating the effect of specific inhibitors of 20-HETE synthesis or 20-HETE antagonists have shown a significant improvement in CBF in a rat model of subarachnoid hemorrhage (Kehl et al, 2002; Yu et al, 2004). These studies also showed that 20-HETE levels are increased in the cerebrospinal fluid of rats after hemorrhagic stroke. Recently, our laboratory showed that 20-HETE is present in the cerebrospinal fluid of patients with subarachnoid hemorrhage who developed vasospasm (Poloyac et al, 2005). Collectively, these studies suggest that 20-HETE may play a role in the pathogenesis of cerebrovascular disease.
Based on the data implicating 20-HETE as a pathogenic mediator of ischemic damage, it was the purpose of this study to determine if inhibition of 20-HETE formation in the rat cerebral cortex would lessen neuronal damage after temporary focal ischemia with reperfusion. We also evaluated the effect of 20-HETE inhibition on CBF in this injury model. We report that the specific 20-HETE formation inhibitor, HET0016, decreases brain 20-HETE tissue concentrations, produces a highly significant reduction in lesion volume after MCAO, and attenuates the injury-mediated reduction in CBF in a rat temporary focal ischemia model of thromboembolic stroke.
Materials and methods
All experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with their guidelines. We used male Sprague—Dawley rats (250 to 300 g) for the completion of these studies. Three experiments were conducted. In the first experiment, we evaluated the tissue pharmacokinetics of HET0016 after administration of a single intraperitoneal dose and determined the tissue concentrations of HET0016 and 20-HETE after four daily intraperitoneal doses. In the second experiment, we evaluated the effects of HET0016 on lesion volume and CBF after MCAO. Finally, in the third experiment, we evaluated the effects of MCAO on the in vitro formation of 20-HETE in rat brain microsomes.
Pharmacokinetic and Tissue Concentration Analysis
Permanent jugular vein cannuli were surgically placed 3 days before treatment. Rats were then administered a single intraperitoneal 10 mg/kg dose of HET0016 and 0.3 mL blood samples were obtained from the femoral access site at 0, 5, 10, 30, 60, 90, 120, 150, and 180 mins. Pharmacokinetic analysis was conducted using WinNonlin (Pharsight, Mountain View, CA, USA). The compartmental pharmacokinetic analyses were conducted using uniform weighting and estimation based on clearance and volume parameterization of the compartmental structure using a Gauss-Newton minimization method. Models were compared for fitness using the Akaike Information Criterion. In the tissue concentration experiment, rats received HET0016 (10 mg/kg per day) intraperitoneally for 4 days. Sixty minutes after the fourth dose, rat brains were briefly perfused and rats were killed. Both the cerebral cortex and kidney tissue were harvested. All tissue was immediately homogenized, extracted, and analyzed by high-performance liquid chromatography-mass spectrometry as described below.
Middle Cerebral Artery Occlusion
Male, Sprague—Dawley rats (250 to 300 g) were randomized to receive either HET0016 10 mg/kg dissolved in 10% lecithin saline or vehicle (10% lecithin saline) intraperitoneally once daily on days 1 to 6 of the study (n = 6 per treatment group). On day 3 of the study, rats received either MCAO for 90 mins followed by reperfusion or sham surgery using isoflurane anesthesia as described previously (Shimizu et al, 2001). Briefly, rats were intubated using a 14G catheter and maintained with 3% isoflurane, 70/30 N2O/O2 throughout surgery. An incision was made in the neck exposing the left common carotid artery. The external carotid artery was isolated and ligated using 5-0 silk (Ethicon, Somerville, NJ, USA). Middle cerebral artery occlusion was achieved by inserting 4-0 nylon suture (with tip coated with silicon 1.5 to 2.0 mm in diameter) via the stump of the external carotid artery into the internal carotid artery a distance of 1.5 cm from the bifurcation of the common and internal carotid arteries. The wound was closed and the rat recovered from anesthesia. After 90 mins, the rat was reanesthetized and the suture removed, initiating reperfusion.
Lesion Volume Determination
Rats were killed 72 h after the end of suture removal and lesion volume was assessed by 2, 3, 5-triphenyl-tetrazolium chloride (TTC) staining. The isolated rat brains were put into the matrix and were sliced into 1 mm sections. The sections were immersed in 3% TTC (Sigma, St Louis, MO, USA) for 30 mins at room temperature. The sections were transferred to formalin and photographed. Infarct volume was measured using image analysis (MCID, St Catharines, Ontario, Canada). To minimize the effect of edema on the quantification of infarct size, the method of Swanson et al (1990) was used. The percentage infarct volume was calculated by dividing infarct volume by contralateral hemisphere volume.
Laser Doppler Flow and Physiologic Assessments
Continuous assessment of laser Doppler flow was conducted via drilling a burr hole over the parietal cortex distal to the site of core ischemic damage. Laser Doppler flow probe (Perimed, Jarfalla, Sweden) was used to estimate blood flow at various time points during isoflurane anesthesia. A laser Doppler blood flow probe was used during MCAO to confirm occlusion and measurements were obtained for 4 h after reperfusion. For physiologic assessments, one femoral artery and one femoral vein were cannulated for arterial blood gas determinations, and blood pressure monitoring. Mean arterial pressure (MAP) was measured by transducers from Power Lab, ADI instruments (Colorado Springs, CO, USA) and blood gas determinations by blood gas analyzer from Radiometer Copenhagen (Westlake, OH, USA). The contralateral temporalis muscle temperature and rectal temperature of the animals was measured during the experiments and animals were placed on a heating pad during experimentation. Temperature was monitored with a Physitemp Probe.
Tissue 20-Hydroxyeicosatetraenoic Acid, 12-Hydroxyeicosatetraenoic Acid, and HET0016 Concentrations
Tissue samples were homogenized in buffer (0.12 mol/L phosphate buffer with 5 mmol/L MgCl2 and 0.113 mmol/L butylated hydroxytolune) immediately after dissection and were centrifuged at 10,000g for 30 mins for isolation of the S9 fraction. Samples, duplicate standards, and duplicate quality controls were spiked with deuterated (d6)-20-HETE and N1-(4-butyl-2-methylphenyl) acetamide as the internal standards for 20-HETE/12-HETE and HET0016, respectively. Samples, standards, and quality controls were then extracted using hydrophilic-lipophilic balance (HLB) solid-phase extraction cartridges (Oasis, Waters, Milford, MA, USA) as described previously (Poloyac et al, 2004). HETE metabolites were separated via reverse-phase HPLC (Waters, Milford, MA, USA) with a 5 μm Beta Basic-18 (150 × 2.1; Thermo Hypersil, Bellefonte, PA, USA) column. Metabolite quantification was completed with a MSQ single quadrupole mass spectrometer (Thermo-Finnigan, San Jose, CA, USA) using negative electrospray and singleion mode detection at mass m/z 319.5 for 20-HETE and 12-HETE; m/z 325.5 for deuterated (d6)-20-HETE; positive electrospray and single-ion-mode detection at m/z 207 for HET0016; and m/z 206 for N1-(4-butyl-2-methylphenyl) acetamide. Chromatographic conditions achieved separation of HET0016 and N1-(4-butyl-2-methylphenyl) acetamide to prevent mass overlap in the quantitative analysis. 20-HETE, 12-HETE, and HET0016 concentrations were quantified from the standard curve of the ratio of the analytes to internal standard peak areas. Data acquisition and analysis were performed with Xcalibur software version 1.4 (Thermo-Finnigan, San Jose, CA, USA).
Subcellular Fractionation, Cytochrome C Oxidase, and Cytochrome C Reductase Assessment
Before being killed, animals were anesthetized with ketamine/xylazine. Brain tissue was harvested and homogenized in 50 mmol/L Tris buffer containing 150 mmol/L KCl, 0.1 mmol/L dithiothreitol, 1 mmol/L ethylenediami-netetraacetic acid (EDTA) and 20% glycerol, pH 7.4, as described previously (Tindberg et al, 1996). Brain tissue was harvested and subcellular fractions were obtained via differential centrifugation performed at 4°C. Briefly, aliquots of homogenates were retained for enzyme activity determination and the remainder was centrifuged at 1,000g for 10 mins to obtain the nuclear pellet. The 1,000g supernatants subsequently centrifuged at 10,000g for 15mins or 120,000 g for 60 mins in an L8-70 Ultracentrifuge (Beckman Coulter, Fullerton, CA, USA). Pellets were suspended in 20 mmol/L Tris buffer containing 250 mmol/L sucrose at pH = 7.4. The cytosolic fraction was obtained as the supernatant from the 120,000g spin. Inner membranes (mitoplasts) were isolated from the crude mitochondrial pellets via treatment with digitonin as described previously (Anandatheerthavarada et al, 1997). Microsomal purity was estimated by the activity of nicotinamide adenine dinucleotide phosphate –cytochrome C reductase as described previously (Guengerich, 1984). The purity of mitochondrial and isolated inner-mitochondrial membranes was estimated by cytochrome c oxidase activity as described previously (Hevner et al, 1993). Subcellular fractions were obtained from five animals per data point. Subcellular fraction formation rate of 20-HETE was evaluated via in vitro incubations with 1,000 mg of total protein per incubation as described previously by our laboratory (Bolcato et al, 2003).
Brain Micosomal Incubations after Middle Cerebal Artery Occlusion
Rats were killed at various time points after MCAO for brain regional microsomal preparation. Brain tissue was dissected from the contralateral and ipsilateral hemisphere. Hemispheric, forebrain, and cortical tissue was homogenized in 50 mmol/L Tris buffer containing 150 mmol/L KCl, 0.1 mmol/L dithiothreitol, 1 mmol/L EDTA, and 20% glycerol, pH 7.4, as described previously (Tindberg et al, 1996). Brain tissue was harvested and microsomes were obtained via differential centrifugation performed at 4°C (n = 4 per dissection/treatment group). Microsomal incubations containing 250 μg of total protein, 1 μmol/L NADPH, 100 μmol/L arachidonic acid in 0.12 mol/L phosphate buffer with 5 mmol/L MgCl2, pH = 7.4, were performed for 60 mins at 37°C. In addition, rat brain cortical microsomes were extracted from untreated animals. These microsomes were then incubated in the presence of vehicle control, high concentration HET0016 (0.61 nmol/mL), or low concentration HET0016 (0.05 nmol/mL). The formation rate of 20-HETE and epoxyeicosatrienoic acid (EET) metabolites was determined in these microsomal incubates to evaluate HET0016 specificity.
Statistical Analysis
Significant differences between HET0016 and vehicle treatment groups for single measurement parameters were assessed by an unpaired Student's t-test. Significant differences for laser Doppler measurements were determined via repeated measures one-way analysis of variance with Bonferroni's post hoc analysis. For all statistical tests, a P < 0.05 was considered significant.
Results
HET0016 Pharmacokinetics after Intraperitoneal Administration
The plasma concentration profile of HET0016 (10 mg/kg) was determined after intraperitoneal administration (Figure 1). The plasma concentration profile was best described by a one-compartment model with first-order absorption and elimination. The pharmacokinetic estimates (best-fit model) show that HET0016 has a large volume of distribution of 18.7 ± 7.8 L/kg. The clearance of HET0016 was 224 ± 131 mL/min per kg and the half-life was 0.84±0.55 h (Table 1). These estimates show that HET0016 has a short in vivo half-life with a large distribution.
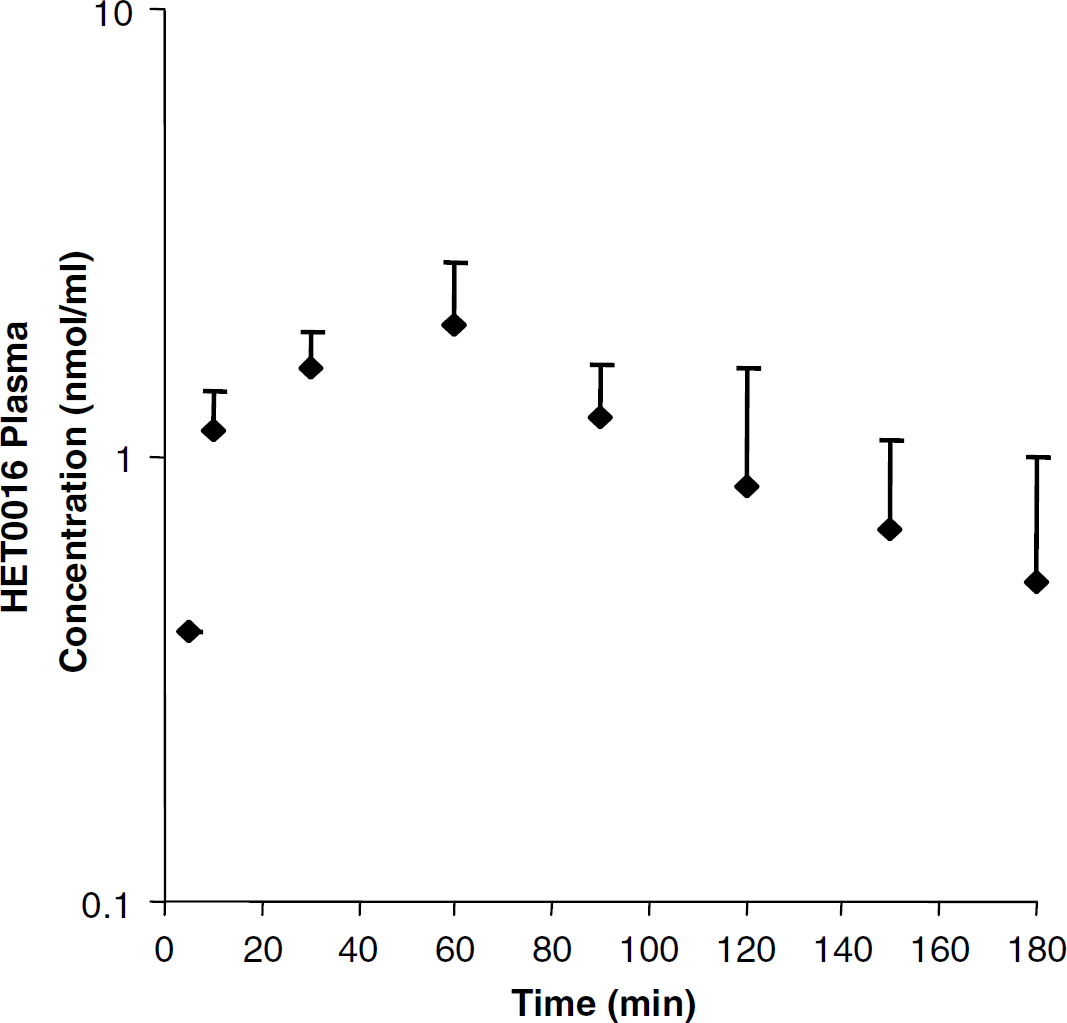
The in vivo plasma profile for HET0016 after a single intraperitoneal (10 mg/kg) dose. Blood samples were obtained for 3h after administration and plasma HET0016 concentrations were determined. Each data point is presented as the mean concentration 7s.d. (n = 3).
HET0016 plasma pharmacokinetic parameter estimates after a single intraperitoneal 10 mg/kg dose (n=3 rats)
Tissue Concentrations of HET0016 and the Effect of HET0016 on Cortical 20-Hydroxyeicosatetraenoic Acid and 12-Hydroxyeicosatetraenoic Acid Concentrations
HET0016 plasma concentrations as well as brain and kidney tissue concentrations were determined. Animals were treated with HET0016 10 mg/kg intraperitoneally daily for 4 days and were killed 60 mins after the last dose. Concentrations in the plasma, brain cortical, and kidney tissue were 2.53±0.99, 0.20±0.08, and 0.05±0.04 mmol/L, respectively (Figure 2A; n = 6). As depicted in Figure 2B, brain cortical tissue 20-HETE concentrations were 6.97±2.6 ng/g wet tissue weight in vehicle control animals and 1.42±0.19 ng/g wet tissue weight in HET0016-treated animals; thereby, showing a 79.6% decrease in tissue 20-HETE concentration with HET0016 treatment (n = 6; P < 0.001). No differences in cortical tissue 12-HETE concentrations were observed between HET0016 and vehicle-treated animals (Figure 2C).
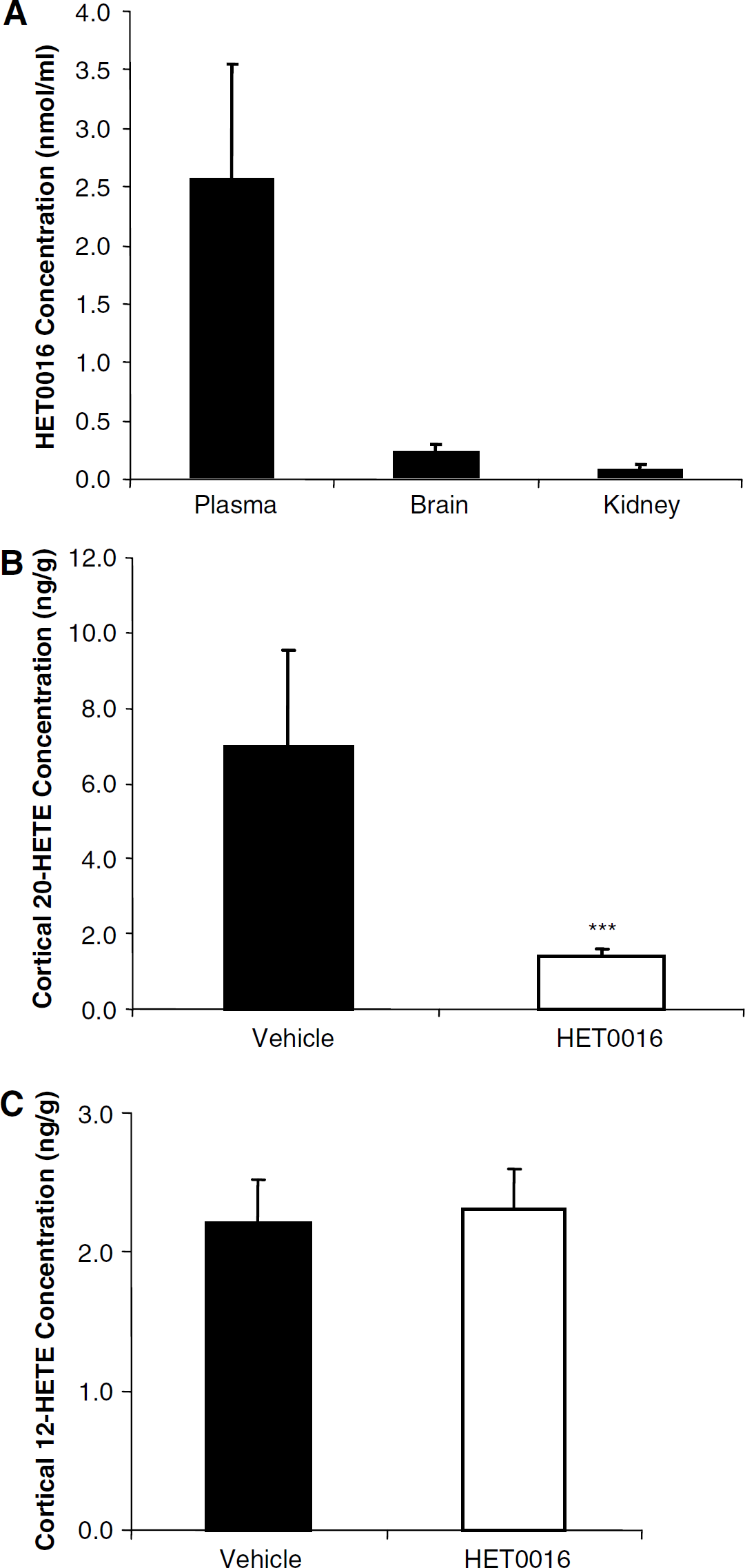
HET0016 and 20-hydroxyeicosatetraenoic acid (20-HETE) tissue concentrations in the rat brain cortex. (
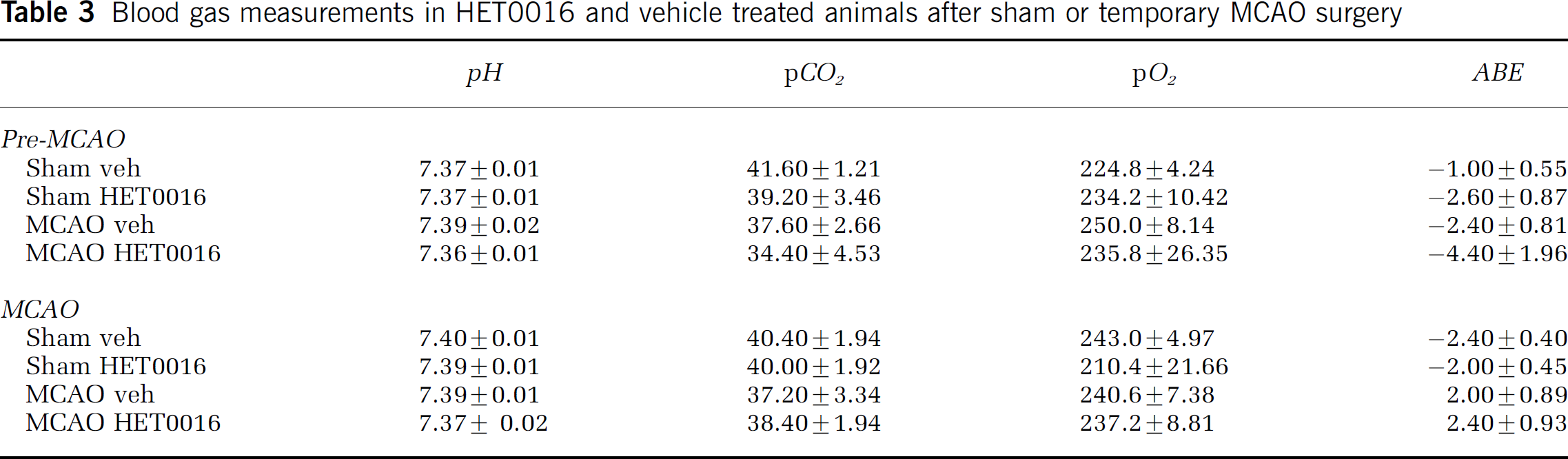
Effect of HET0016 on Physiologic Measures and Lesion Volume after Middle Cerebal Artery Occlusion
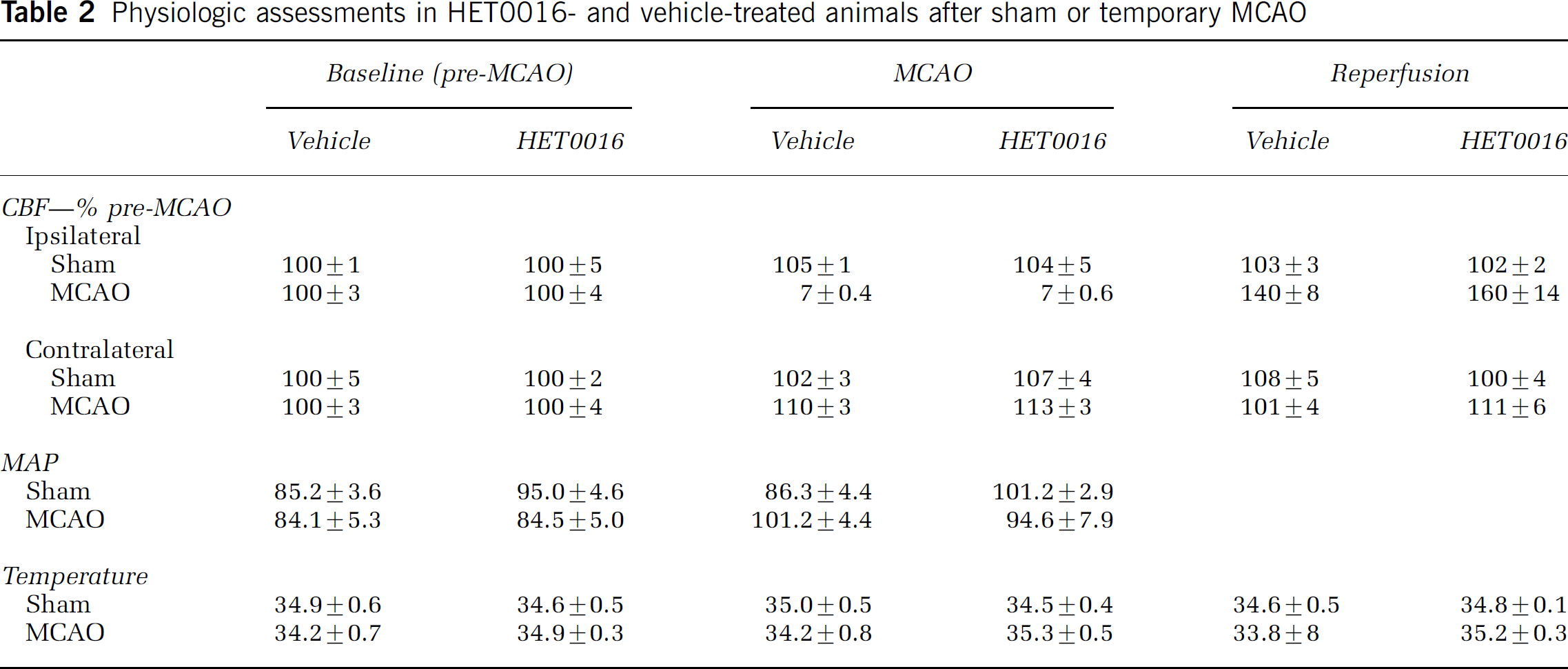
The effect of HET0016 versus vehicle control pretreatment on lesion volume after MCAO was evaluated. Physiologic measurements of CBF, MAP, temperature, as well as, arterial pH, pCO2, pO2, and base excess showed no significant differences between vehicle- and HET0016-treated animals. Data are presented in Tables 2 and Table 3. After MCAO, rats received daily doses of HET0016 or vehicle and lesion volume was determined by TTC staining at 72 h after MCAO. As depicted in Figure 3, an 84% reduction in hemispheric infarct volume was observed in HET0016-treated (9.1%±4.9%) as compared with vehicle control (57.4%± 9.8%) animals (n = 6, P ± 0.001).
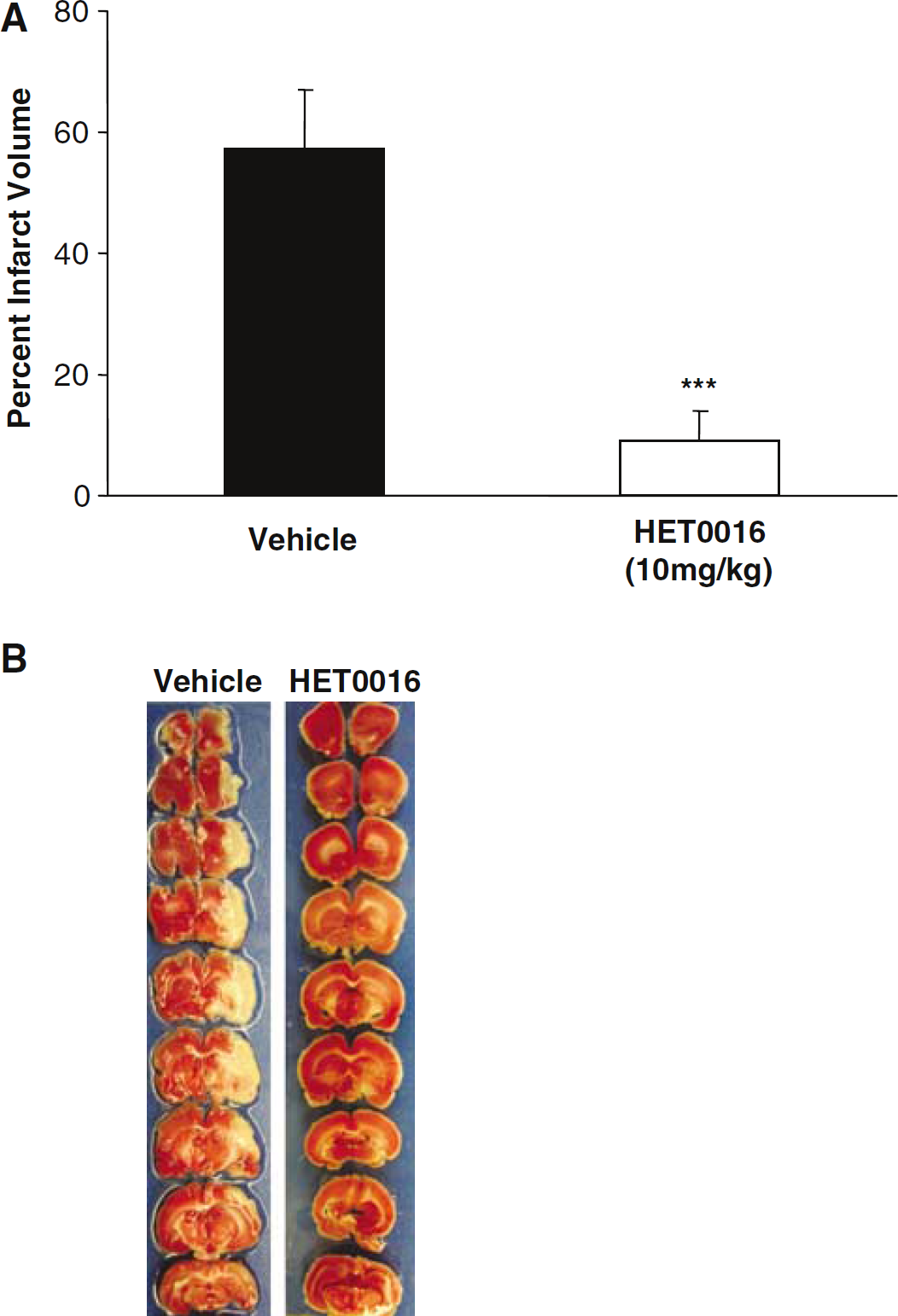
Effect of HET0016 pretreatment on lesion volume after temporary focal ischemia. The figure depicts 2, 3, 5-triphenyltetrazolium chloride (TTC)-stained rat brain sections. Tissue was obtained from 10% lecithin vehicle pretreated and 10 mg/kg HET0016 in 10% lecithin-pretreated animals. Animals received daily intraperitoneal injections of HET0016 or vehicle beginning 3 days before temporary focal ischemia (90 mins occlusion) and continuing until being killed at 72 h after reperfusion. (
Effect of HET0016 and Middle Cerebal Artery Occlusion on Laser Doppler Cerebral Blood Flow
A more detailed evaluation of the effects of HET0016 on CBF was evaluated. Vehicle-treated animals showed a transient (60 mins) period of postischemic hyperperfusion followed by sustained (at least 3 h) reduction in CBF after MCAO. The reduction in CBF was attenuated in HET0016-treated animals as compared with vehicle control at the 180 mins (57.6%±19.0% vehicle versus 89.2%±6.2% HET0016) and 240 mins (53.8%±20.0% vehicle versus 88.1%±5.7% HET0016) time points after MCAO (Figure 4).
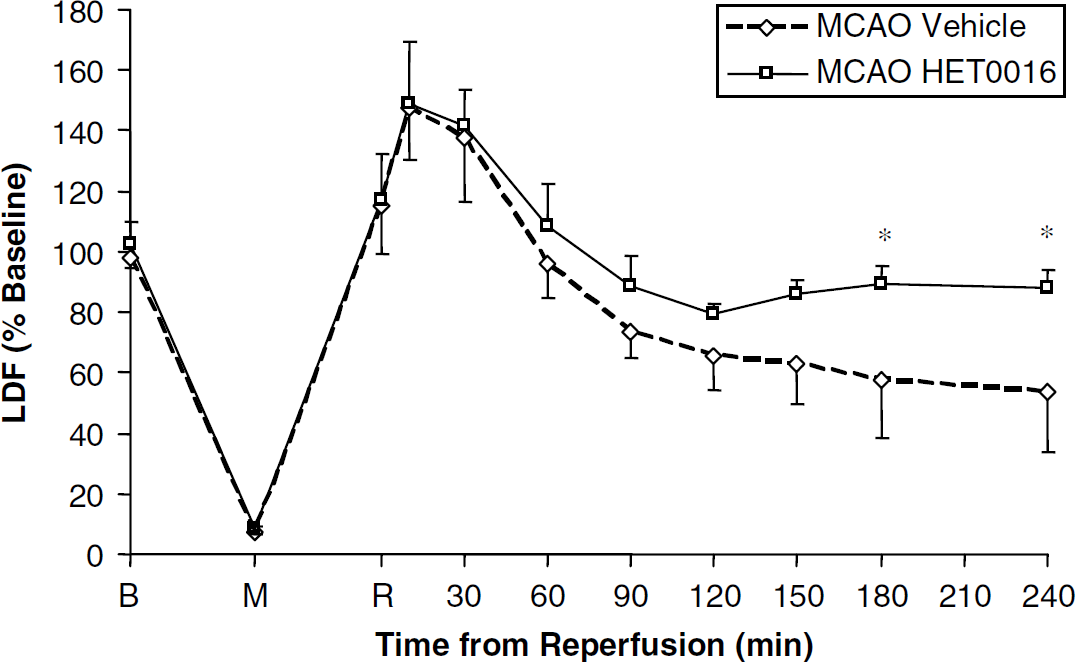
Effect of HET0016 pretreatment on regional cerebral blood flow (rCBF) after temporary focal ischemia. The figure depicts laser Doppler flow (LDF) values presented as percent of combined averaged baseline values. Rats received daily intraperitoneal injections of HET0016 or vehicle beginning 3 days before temporary focal ischemia (90 mins occlusion) and 1 h before baseline LDF measurement. Baseline, middle cerebral artery occlusion (MCAO), and reperfusion points are represented by B, M, and R, respectively. Data are mean±s.d. from n = 6 per treatment group. Significant differences are denoted by *P ± 0.05.
20-Hydroxyeicosatetraenoic acid Microsomal Formation Rate after Middle Cerebal Artery Occlusion
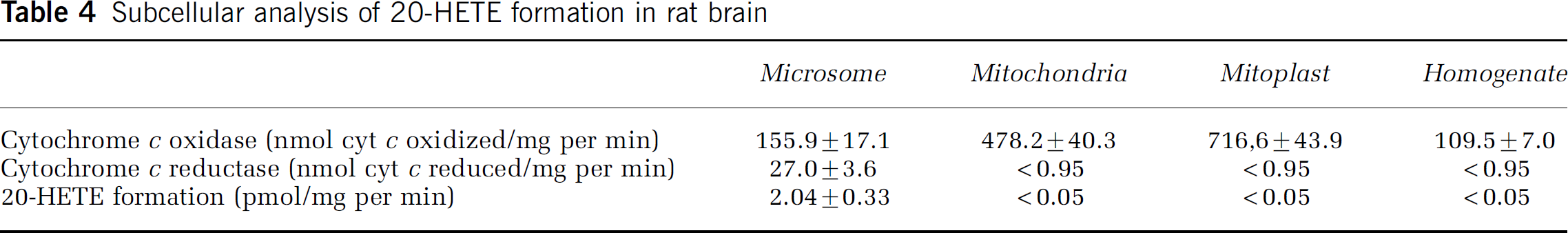
Identification of the subcellular fraction(s) responsible for 20-HETE formation was completed in untreated rat brain hemispheric isolates. As anticipated, microsomes showed the greatest cytochrome c reductase activity; whereas, mitochondria and mitoplasts had the greatest cytochrome c oxidase activity. In vitro incubations with these same subcellular fractions show that detectable levels of 20-HETE were only observed in microsomes with a formation rate of 2.047 0.33 pmol/mg per min (Table 4). Based on the microsomal localization of 20-HETE formation, subcellular microsomes were isolated from rat brain hemispheric, forebrain, and cortical tissue at 24h after sham or MCAO surgery (as described in the Materials and methods section). A reduction in the rat cortical microsomal 20-HETE formation rate was observed in the MCAO (1.5±0.3 pmol/mg per min) as compared with shams (3.5±0.6pmol/mg per min) (Figure 5A; n =4; P < 0.01). The cortical microsomal formation rate was also different between the contralateral (3.4±1.5 pmol/ mg per min) and ipsilateral (1.5±0.3 pmol/mg per min) hemisphere at 24 h after ischemia (Figure 5B; n = 4; P±0.05). Significant differences were not observed at 1, 3, 6, or 12 h after MCAO (data not shown).
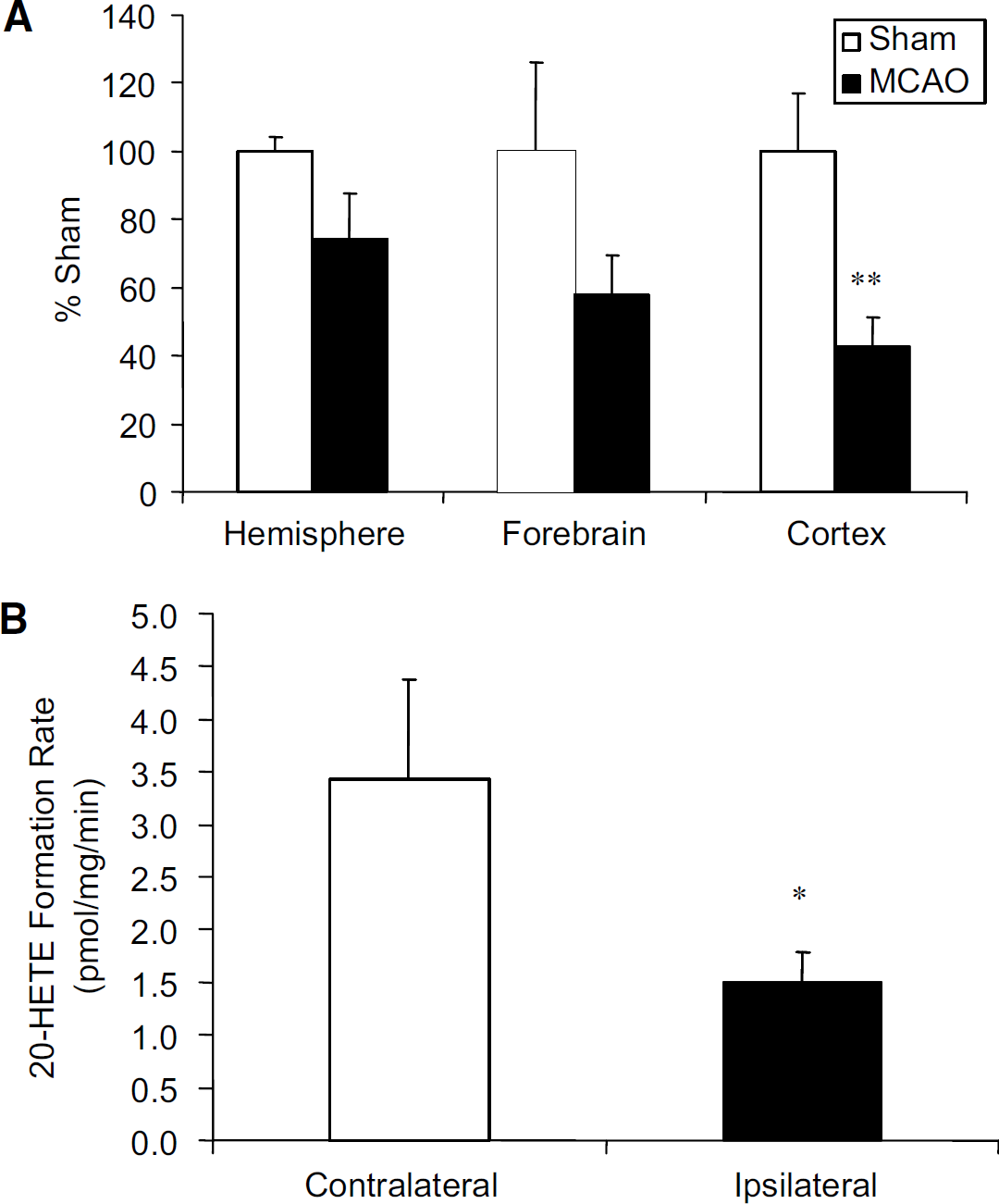
Effect of temporary focal ischemia on the in vitro rat brain microsomal 20-hydroxyeicosatetraenoic acid (20-HETE) formation rate. Rat brain ipsilateral and contralateral hemispheric, forebrain, and cortical microsomes were isolated from rats killed at 24 h after reperfusion. (
Brain Cortical Microsomal Formation of 20-Hydroxyeicosatetraenoic Acid and Epoxyeicosatrienoic Acids in the Presence of HET0016
The in vitro formation of 20-HETE and several EET metabolites was evaluated in the presence of HET0016. HET0016 concentrations evaluated were 0.61 and 0.05 nmol/mL. These concentrations were selected such that the range of concentrations observed in brain cortical tissue (i.e., 0.2 nmol/mL) was included (see Figure 2A). As depicted in Figures 6A-C, no significant difference in brain cortical microsomal formation of 5,6-EET, 8,9-EET, or 11,12-EET was observed. These same incubations showed complete inhibition of 20-HETE formation at the 0.61 nmol/mL concentration and a 95% reduction in 20-HETE formation at the 0.05 nmol/mL concentration (Figure 6D).
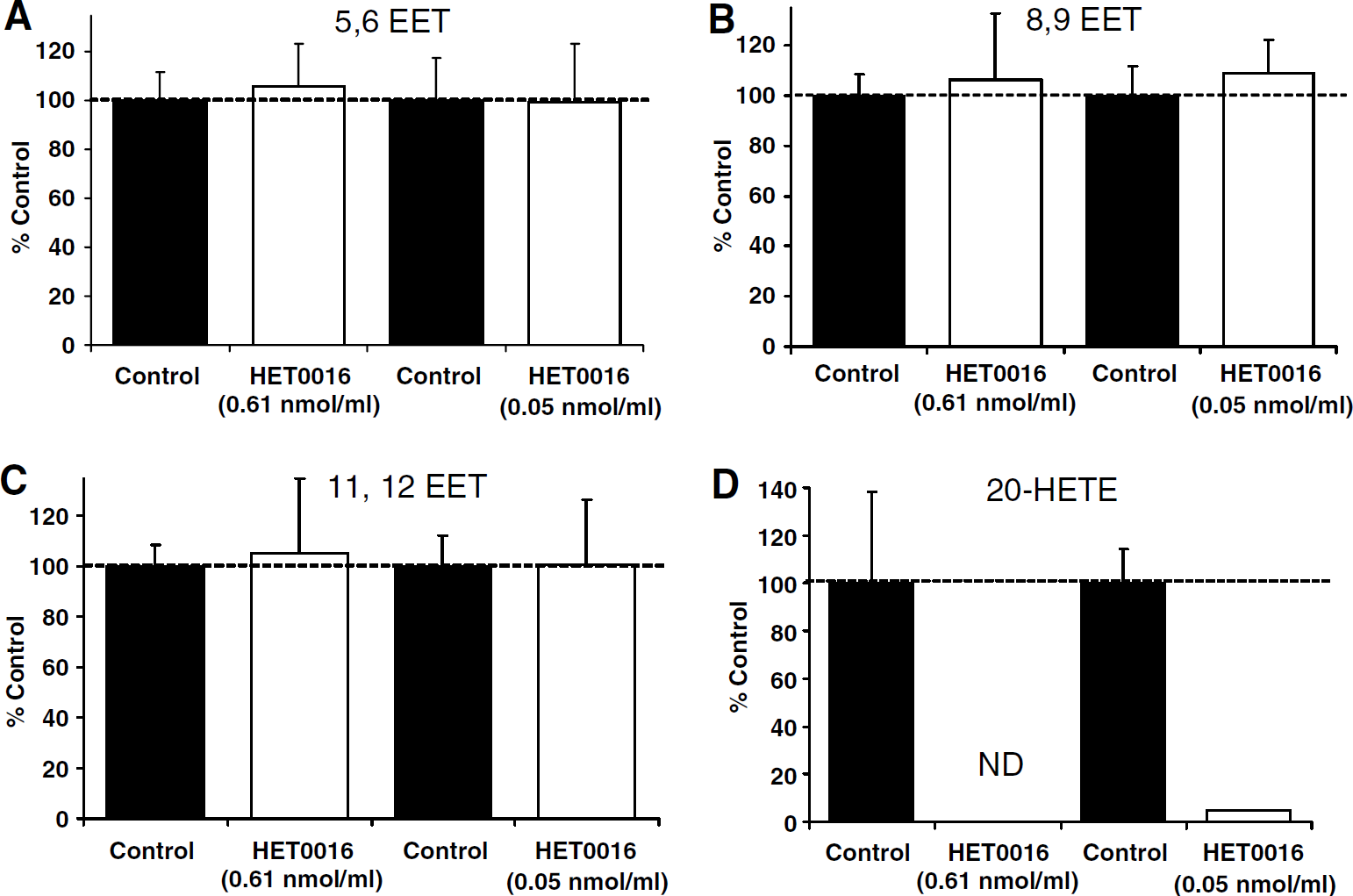
Effect of HET0016 on rat brain microsomal in vitro formation of 20-hydroxyeicosatetraenoic acid (20-HETE) and epoxyeicosatrienoic acid (EET) metabolites. Cortical brain microsomes were prepared from untreated animals. Microsomes were incubated in the presence of vehicle control, 0.61 nmol/mL HET0016, or 0.05 nmol/mL HET0016. Metabolites were separated chromatographically and identified by mass and elution of authentic standard. (
Physiologic assessments in HET0016- and vehicle-treated animals after sham or temporary MCAO
Blood gas measurements in HET0016 and vehicle treated animals after sham or temporary MCAO surgery
Subcellular analysis of 20-HETE formation in rat brain
Discussion
These experiments resulted in three novel findings. First, HET0016 administered at a 10mg/kg intraperitoneal pretreatment dose significantly reduces brain cortical 20-HETE tissue concentrations and dramatically reduces lesion volume after MCAO. Second, HET0016 attenuates the reduction in CBF observed after MCAO. Third, MCAO significantly decreases the in vitro formation of 20-HETE in rat brain cortical microsomes at 24 h after insult. From these data we conclude that 20-HETE is an important pathogenic mediator after temporary focal ischemia and that further evaluation of the role of 20-HETE in stroke pathogenesis represent an important area of future study.
The reduction in lesion volume is consistent with a recent publication that reported a reduction in lesion volume in the temporary focal ischemia model with a different newly characterized 20-HETE inhibitor, TS-011 (Miyata et al, 2005). Unlike other vasoactive arachidonic acid metabolites, 20-HETE is capable of being stored in the plasma membrane for later release by phospholipase A2-mediated cleavage (Carroll et al, 1997; Kaduce et al, 2004). Because of this potential for storage and release, we chose to pretreat rats with HET0016 for 3 days before MCAO to eliminate all potential stores of 20-HETE. However, the short biologic half-life of HET0016 coupled with the effects of acute inhibition reported by Miyata et al suggests that the observed effect is primarily attributable to the acute exposure before artery occlusion. Future studies are necessary to delineate the importance of the degree and timing of 20-HETE inhibition on the resultant damage after temporary focal ischemia.
We selected a dose of 10mg/kg administered intraperitoneally, which produced 1 h concentrations of 0.2 μmol/L (nmol/mL) and 2.53 μmol/L (nmol/mL) in the brain and plasma, respectively. Similar plasma concentrations of 2.8 μmol/L at 1 h after a 10mg/kg intravenous dose were reported previously (Kehl et al, 2002). However, the brain concentrations reported in the study by Kehl et al were 7.2 μmol/L as compared with 0.2 μmol/L reported in the present study. The extraction efficiency of HET0016 was confirmed in our determinations of brain concentration (data not shown) and brain concentrations were verified in six replicate animals. One possible reason for this difference in concentration is that the Kehl et al study administered HET0016 intravenously and we used the intraperitoneal administration route. It is likely that brain concentrations were not in pseudoequilibrium with the plasma concentrations at the 60 mins time point and that higher brain concentrations would be observed at later time points in our study. Previous in vitro studies report that HET0016 concentrations between 0.05 and 1 μmol/L produce selective inhibition of 20-HETE (Miyata et al, 2001). Concentrations greater than 1 μmol/L are known to also inhibit the formation of EETs. Our data showing significant inhibition of basal 20-HETE formation after the 10 mg/kg intraperitoneal dose also suggest that brain concentrations were sufficient for in vivo inhibition of 20-HETE synthesis.
Because of the fact that the 7.2 μmol/L brain concentrations observed in the Kehl et al study were above the determined selective concentration range, subsequent studies were conducted using a 1 mg/kg intraperitoneal or intraveneous doses of HET0016 (Amaral et al, 2003; Cambj-Sapunar et al, 2003). However, these subsequent studies using the 1 mg/kg dose did not reduce basal 20-HETE concentrations and resulted in more minor reductions in 20-HETE formation after insult. To identify a HET0016 dose capable of inhibiting 20-HETE formation in the rat brain cortex, we evaluated the single dose pharmacokinetics of HET0016 after a 10 mg/kg intraperitoneal dose. This evaluation determined that HET0016 has a short plasma half-life equal to 0.84 h. Based on this short half-life, we selected a dose of 10 mg/kg intraperitoneally to maintain concentrations in the brain sufficient for inhibition of basal 20-HETE formation. It is possible that the 10 mg/kg dose produced concentrations of HET0016 that were above the selective range (1 mmol/L) at later time points. However, the non-selective inhibition would be very short due to the short half-life of this compound in vivo. Furthermore, our extraction and analytical methods are capable of evaluating EET metabolites in rat tissues; however, we did not observe detectible EET metabolites in the rat cortex in the vehicle-treated animals. Neither tissue 12-HETE concentrations nor in vitroEET formation were altered by HET0016, thereby suggesting that the dosing regimen used in this study produced specific inhibition of 20-HETE formation. Future studies employing continuous infusion strategies for the administration of HET0016 will be necessary to target the selective concentrations for confirmation of the observations in this paper.
The second finding in this study was the attenuation of the injury-induced reduction in CBF after temporary focal ischemia in the HET0016-treated rats. This observation is consistent with the theory that 20-HETE acts as a vasoconstrictor in the rat cerebral microvasculature and that inhibition of 20-HETE formation improves microvascular flow. This vasoconstrictive effect is believed to occur via the effects of 20-HETE on microvascular tone via altering calcium and potassium flux (Gebremedhin et al, 1998; Zou et al, 1996). Although these results suggest an alteration in focal assessment of CBF, the use of the laser Doppler methodology is limited in that a regional map of CBF cannot be generated. Alternative methods allowing serial mapping of CBF such as perfusion magnetic resonance imaging (Hendrich et al, 2001) or laser speckle flowmetry (Ayata et al, 2004) should be used to directly address this important issue in future studies.
Although CBF was increased by HET0016 treatment, it is highly likely that other mechanisms of neuroprotection are responsible for the large reduction in lesion volume. 20-HETE has been recently shown to promote cellular magnesium efflux (Ikari et al, 2004). Thus, inhibition of 20-HETE may increase intracellular magnesium concentration, thereby mediating antiexcitotoxic and ATP preservation effects (Johnson and Ascher 1990; McIntosh et al, 1988; Nowak et al, 1984). Additional studies in experimental MCAO are needed to aid in determining the relative importance of these underlying mechanisms of effect. Furthermore, future studies will be important to delineate the role of 20-HETE relative to other known mediators of secondary injury after temporary focal ischemia.
20-HETE has also been implicated as a second messenger for angiogenesis mediated by VEGF (Amaral et al, 2003; Chen et al, 2005; Josko 2003). Studies in the rat MCAO model have shown that VEGF expression is induced by ischemia early after MCAO (Hayashi et al, 1997). However, studies evaluating the effects of acute VEGF administration on lesion volume have shown a paradoxical increase in lesion volume with intravenous VEGF administration (Kaya et al, 2005; Zhang and Chopp 2002) and decreased lesion volume with brain cortical (Hayashi et al, 1997) or intracerebroventricular administration (Kaya et al, 2005). Based on this paradoxical role of VEGF in damage after focal ischemia, the effect of systemic 20-HETE inhibition may be beneficial acutely, but may prevent the benefits of angiogenesis on vascular and neuronal regeneration and/or recovery if administered chronically after injury. Future studies will be essential to determine these acute and chronic effects.
Finally, we also showed that 20-HETE is formed by the microsomal fractions in the brain and that there is a significant reduction in the formation of 20-HETE by rat brain cortical microsomes in the ischemic hemisphere 24 h after MCAO. Furthermore, we showed that HET0016 at concentrations up to 0.61 nmol/mL specifically inhibits brain microsomal formation of 20-HETE without altering EET formation in vitro. The microsomal fraction in the brain consists of the smooth endoplasmic reticulum that is the known localization of the CYP4A and CYP4F isoforms involved in 20-HETE formation. As with other P450 isoforms, the expression of both CYP4A and CYP4F are known to be downregulated after activation of the acute phase response (Cui et al, 2001). This reduction in CYP isoform expression occurs after traumatic insults and is also known to be tissue and isoform specific (Kalsotra et al, 2003; Poloyac et al, 2000, 2004). In addition, the incubations performed in our study were conducted at saturating concentrations of substrate and cofactor as determined previously (Bolcato et al, 2003); therefore, the observed reduction in formation rate is due to a decrease in the content of functional enzymes in these microsomal fractions. The mechanisms of these effects after cerebral ischemia are largely unknown; however, functional downregulation may represent an adaptive cytokine-mediated response to limit neuronal damage after temporary focal ischemia.
In summary, our study shows that HET0016 administration produces a significant reduction in lesion volume in the rat MCAO model. The most likely mechanism of this effect is via the specific inhibition of 20-HETE in the rat brain cortex. These data are consistent with recent studies showing that 20-HETE is involved in the pathogenesis of ischemic stroke. Collectively, these studies support further study of the role of 20-HETE in the pathogenesis of neuropathologic disease states.