
Abstract
Thioredoxin (TRX) is a small, multifunctional protein with a redox-active site and multiple biological functions that include reducing activity for reactive oxygen intermediates. We assayed TRX and TRX mRNA by immunohistochemical methods and hybridization experiments in the rat brain after middle cerebral artery (MCA) occlusion. During ischemia, the immunoreactivity for TRX decreased; it disappeared after MCA occlusion in the ischemic regions. It rapidly decreased and nearly disappeared at 4 and 16 hours after MCA occlusion in the lateral striatum and frontoparietal cortex, respectively. On the other hand, in the perifocal ischemic region, the penumbra, TRX immunoreactivity began to increase 4 hours after MCA occlusion and continued to increase until 24 hours after occlusion. In hybridization experiments, TRX mRNA decreased and nearly disappeared 4 hours after MCA occlusion in the lateral striatum. In the frontoparietal cortex, it decreased until 24 hours after MCA occlusion. In the perifocal ischemic region, TRX mRNA began to increase 4 hours after MCA occlusion and continued to increase until 24 hours. Northern blot analysis showed that total TRX mRNA in the operated hemispheres was induced from 8 hours and increased until 24 hours after the surgical procedures. We previously reported that recombinant TRX promotes the in vitro survival of primary cultured neurons. We now suggest that TRX in the penumbra has neuroprotective functions and that decreased levels of TRX in the ischemic core modify neuronal damage during focal brain ischemia.
Thioredoxin (TRX) is a small multifunctional protein with a redox-active disulfide/dithiol within the conserved active site sequence: -Cys-Gly-Pro-Cys- (Holmgren, 1985, 1989). It is an intracellular enzyme that performs a variety of activities as a hydrogen donor for ribonucleotide reductase, the key enzyme in the biosynthesis of deoxyribonucleotides (Holmgren, 1985, 1989) and other intracellular molecules. We previously described adult T-cell leukemia–derived factor (ADF) as an interleukin-2 receptor–inducing factor in human lymphotropic virus-1–transformed cells (Tagaya et al., 1988, 1989). We cloned ADF cDNA and found a close relation between ADF and TRX (Tagaya 1988, 1989; Yodoi and Uchiyama, 1992), suggesting that ADF is a human counterpart of TRX. Thioredoxin is a stress-inducible protein whose expression is enhanced by various types of stress such as viral infection, exposure to ultraviolet light, x-ray irradiation, and hydrogen peroxide (H2O2) (Nakamura et al., 1994; Ohira et al., 1994; Sachi et al., 1995). Also, TRX is a scavenger of reactive oxygen intermediates (ROI), and recombinant TRX (rTRX) has protective activity against cytotoxicity induced by H2O2, cis-diamminedichloroplatinum (II) (CDDP), or tumor necrosis factor-α), in which the generation of ROI seems to be involved in the cytotoxic mechanism (Matsuda et al., 1992; Nakamura et al., 1994; Sasada et al., 1996). Furthermore, TRX regulation of various intracellular molecules by thiol redox control includes the reduction of oxidized ribonucleotide reductase and the degradation of insulin, as well as an activity for the redox regulation of transcription factors such as NF-κB or AP-1 (Abate et al., 1990; Okamoto et al., 1992; Schenk et al., 1994). These data suggest that TRX plays several important biological roles both in intracellular and extracellular compartments.
Ischemia induces severe neuronal damage; however, the mechanisms of ischemic neuronal damage remain to be elucidated. Experimental evidence suggests that oxygen-free radicals are major factors involved in such damage (McCord, 1985; Kitagwa et al., 1990; Lundgren et al., 1991; Morooka et al., 1994; Dirnagl et al., 1995). A decline in superoxide dismutase (SOD) activity and immunoreactivity during ischemia has been reported (Chan et al., 1988; Liu et al., 1994; Kato et al., 1995). The amelioration of ischemic damage by treatment with SOD has been shown, and transgenic mice overexpressing SOD sustain a lesser degree of focal cerebral ischemic injury (Imaizumi et al., 1986; Kinouchi et al., 1991; Sutherland et al., 1991; Chan et al., 1994; Yang et al., 1994). Several free radical scavengers (e.g., glutathione, ascorbic acid, α-tocopherol, and ubiquinone) significantly decrease brain damage during focal ischemia (Abe et al., 1983; Hara et al., 1990; Sato et al., 1992; Villalobos et al., 1994). Furthermore, stress proteins and neurotrophic factors induced during ischemia have been shown to play an important role in neuroprotection (Kawagoe et al., 1992; Welsh et al., 1992; Takeda et al., 1993; Kinouchi et al., 1994; Iihara et al., 1994; Soriano et al., 1995; Li et al., 1995).
We have reported the neuroprotective effect of rTRX: the survival of primary cultured neurons from murine cortex and striatum is promoted by rTRX (Hori et al., 1994). Others also have found that TRX functions like a neurotropic factor (Endoh et al., 1993). We previously reported astroglial TRX induction in the CA-1 region during forebrain ischemia in gerbils (Tomimoto et al., 1993). Based on these considerations, we hypothesize that TRX, which can function as a cellular redox regulatory protein through its ROI-scavenging activity, plays an important role during brain ischemia. In the current study, we subjected rats to middle cerebral artery (MCA) occlusion, investigated the distribution and changes of TRX by immunohistochemical methods, and assayed TRX mRNA by hybridization experiments to shed light on the role of TRX in neuronal damage after focal ischemia.
MATERIALS AND METHODS
Induction of focal ischemia
Male Wistar rats weighing 300 to 400 g were placed under general anesthesia with a mixture of 2% halothane in 30% oxygen and 70% nitrous oxide. An 18.3 mm–long embolus, made from a 4-0 nylon thread, was inserted into the internal carotid artery through a small incision in the external carotid artery, so that the MCA was obstructed (Longa et al., 1989). A heat lamp was used during surgery to maintain the animals rectal temperature between 37.5° and 38.5°C. On awakening from the anesthesia, the operated rats exhibited ischemic symptoms such as paresis of the left limbs and circling movement. For the control, rats were operated in the same way but no embolus was inserted.
Fixation and histologic preparation
The rats were perfused transcardially with phosphate-buffered saline (PBS) followed by a cold fixative solution (4% paraformaldehyde and 0.1 mmol/L PBS) at 2 hours (n = 4), 4 hours (n = 4), 16 hours (n = 4), and 24 hours (n = 4) after surgery. Their brains were removed, fast-frozen in liquid nitrogen, and cut into 20 μm-thick coronal sections with cryostat. Then the sections stained with cresyl-violet to assess the neuronal damage.
Immunohistochemistry
The sections were immunohistochemically stained with polyclonal antibodies raised against a synthetic polypeptide of the C-terminal 10–amino acid polypeptide of mouse TRX (Tomimoto et al., 1993); a Vectastain elite ABC kit (Vector Laboratories, Burlingame, CA, U.S.A.) was used. The specificity of the staining was confirmed by the disappearance of specific staining when nonimmunized rabbit IgG was substituted for the primary antibody. The sections were washed for 5 minutes in 0.01 mmol/L PBS, pH 7.2, followed by 1 hour of preincubation with normal goat serum diluted 1:50. The sections then were incubated overnight at 4°C with anti-mouse TRX antibody (2 μg/mL). After 15-minute rinses in three changes of PBS, the sections were incubated with biotinylated second antibody for 2 hours. After 3 rinses in PBS (as earlier), the sections were incubated with avidin–biotin–peroxidase complex for 1 hour at room temperature. Color development was for 5 minutes at room temperature in a substrate medium containing 0.05% 3,3′-diaminobenzidine and 0.02% H2O2 in Tris-HCl buffer (pH 7.6). Each section then was dehydrated, washed in xylene, and mounted on glass slides.
In situ hybridization
The antisense RNA probes were made from mouse TRX cDNA. The antisense RNA was prepared from SP6 RNA polymerase and labeled with digoxigenin (DIG)-uridine 5′-triphosphate according to the manufacturer's instructions (Boehringer Mannheim Biochemica, Mannheim, Germany) and used as probes for in situ hybridization, which was performed according to the method of manufacturer's recommendations. The tissue sections were placed on slides, deparaffinized, treated with proteinase K (10 μg/mL for 15 minutes at 37°C), and acetylated (20% acetic acid for 1 minute). They then were washed with 2 × saline sodium citrate (SSC), dehydrated, and air-dried for 30 minutes. Hybridization was performed at 50°C for 16 to 24 hours in a humid chamber with 25 μL/section prehybridization solution (5 × SSC, 5 × Denhardt's solution, 50% deionized formamide, 250 μg denatured salmon sperm DNA, and 4 mmol/L ethylene diamine tetraacetic acid) containing 10 ng/μL RNA probe. After hybridization, the sections were washed twice at 42°C in 2 × SSC, and once in 0.2 × SSC and 0.1 × SSC for 15 minutes per wash. Sections then were blocked (30 minutes), incubated with alkaline phosphatase–conjugated anti-DIG antibody (1 hour), and detected with a color solution containing 337.5 μg/mL nitroblue tetrazolium salt and 175 μg/mL 5-bromo-4-chloro-3-indolyl-phosphate (10 to 30 minutes) according to the manufacturer's instructions (DIG labeling and detection kit, Boehringer Mannheim Biochemica). In some experiments, tissue sections were hybridized with DIG-labeled sense-RNA probes, which resulted in no detectable signals.
Northern blot analysis
The rats were decapitated at 1, 2, 4, 8, 16, or 24 hours after MCA occlusion (n = 3 at each time point). The control rats were killed 8 hours after the sham operation. Each of the cerebral hemispheres was fast-frozen in liquid nitrogen. Total RNA was prepared with an RNA isolation reagent (TRIzol Reagent, Life Technologies, Inc., Gaithersburg, MD, U.S.A.), and poly(A)+RNA was isolated using Oligotex-dT30 Super (Nihon Roche Co. Ltd., Tokyo, Japan). RNA samples (20 μg/lane) were separated on a 1% formaldehyde/agarose gel, transferred to a nylon membrane (Hybond N+, Amersham International, U.K.), and hybridized with an [α-32P]dCTP–labeled mouse TRX cDNA probe. The radioactivity of each sample was quantified using an NIH-image analyzer.
RESULTS
Histopathologic findings
Ischemic changes were limited to the territories supplied by the MCA, that is, the lateral part of the striatum and the corresponding frontoparietal cortex. The anatomical areas we investigated are depicted in Fig. 1A.
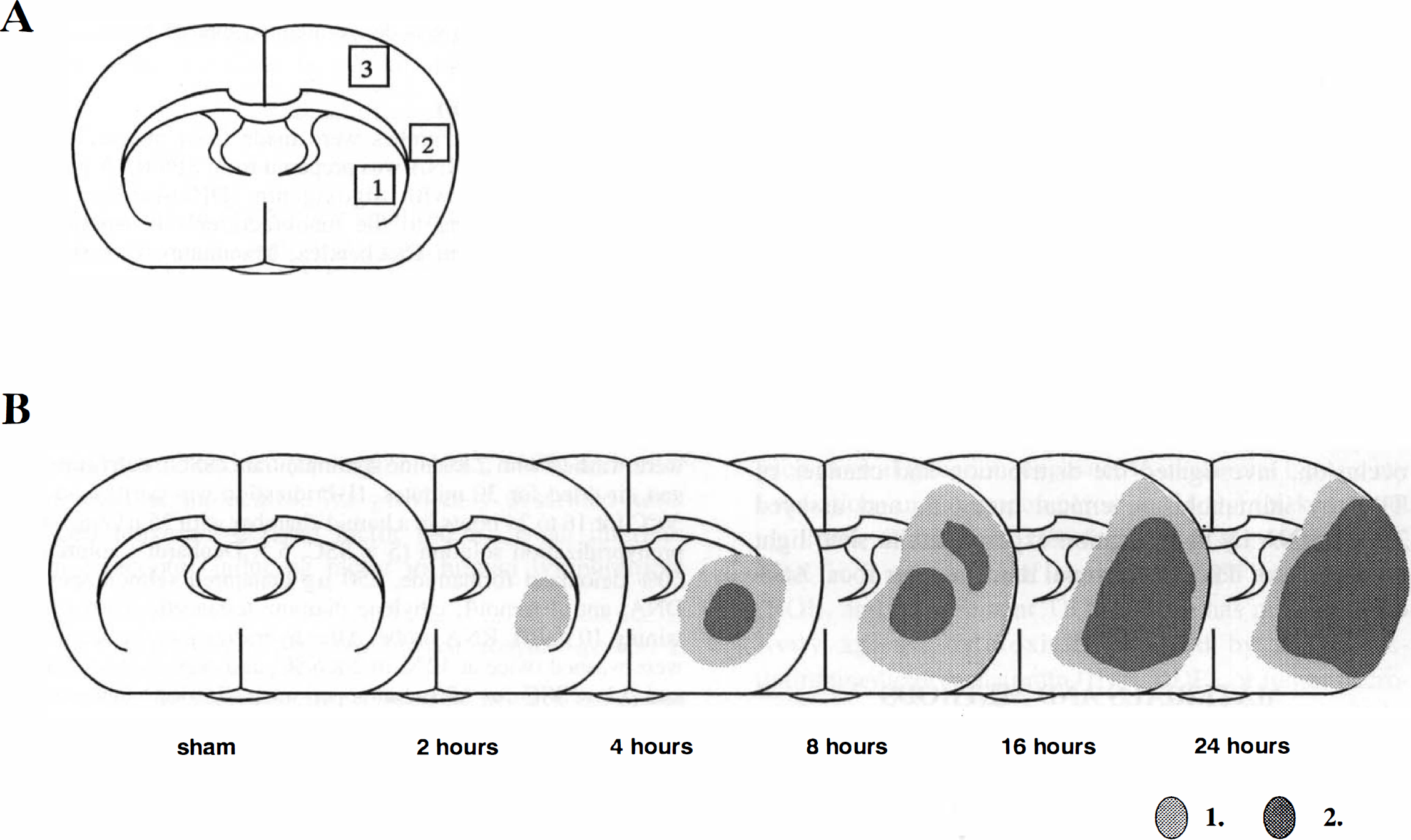
Diagrams showing the anatomical areas we studied and the neuronal damage during ischemia in coronal sections at the level of the anterior commissure.
Striatum. The lateral part of the striatum was weakly stained with cresyl violet 4 hours after MCA occlusion. Most of the medium-sized neurons in the affected area lost their normal morphologic features and appeared to be shrunken compared with the contralateral intact neurons. There were widespread, intense perineuronal vacuolations with crescent neuronal outlines. The ischemic area was well demarcated. These changes became more obvious at 16 hours after MCA occlusion (Fig. 1B).
Cortex. Most neurons in the frontoparietal ischemic cortex supplied by the MCA did not show striking pathologic changes at 4 hours after the ischemic insult; a few neurons showed ischemic changes and early perineuronal vacuolations. At 16 hours after occlusion, neuronal cell bodies were shrunken and triangular, and perineuronal vacuolation was obvious (Fig. 1B).
Most neurons in the perifocal ischemic cortex did not show striking pathologic changes at 24 hours after the insult; only a few neurons had ischemic changes and early perineuronal vacuolations (Fig. 1B). Anatomical areas in which 30% to 100% of the neurons showed marked ischemic changes (obvious perineuronal vacuolation or axonal swelling) also are shown in Fig. 1B.
Immunohistochemical staining
Immunoreactivity for TRX was observed in many neurons in the normal striatum and cortex, although it was relatively weak in the striatum. Vascular endothelial cells also were weakly immunoreactive for TRX. Astroglia and microglia were not immunoreactive for TRX in any regions of the brain. No specific staining was observed in control specimens.
Striatum. In the controls, most medium-sized and a few large neurons in the lateral segment of the striatum showed immunoreactivity for TRX. Immunoreactivity was severely reduced in neurons in the lateral segment of the striatum at 4 hours, and completely lost at 16 hours after the ischemic insult (Figs. 2A through D, and 3A and B).
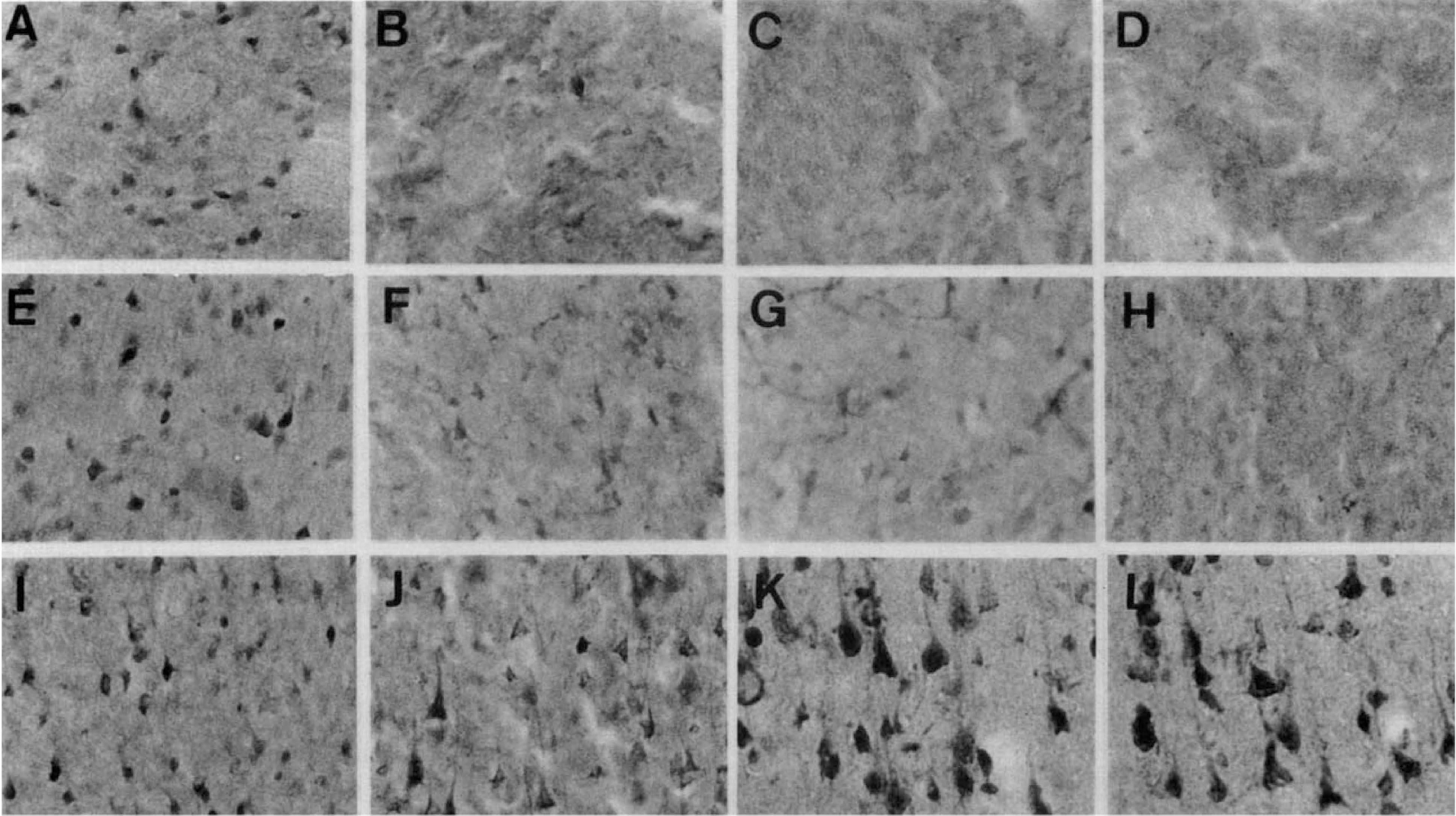
Immunohistochemical staining for TRX in rat lateral striatum (
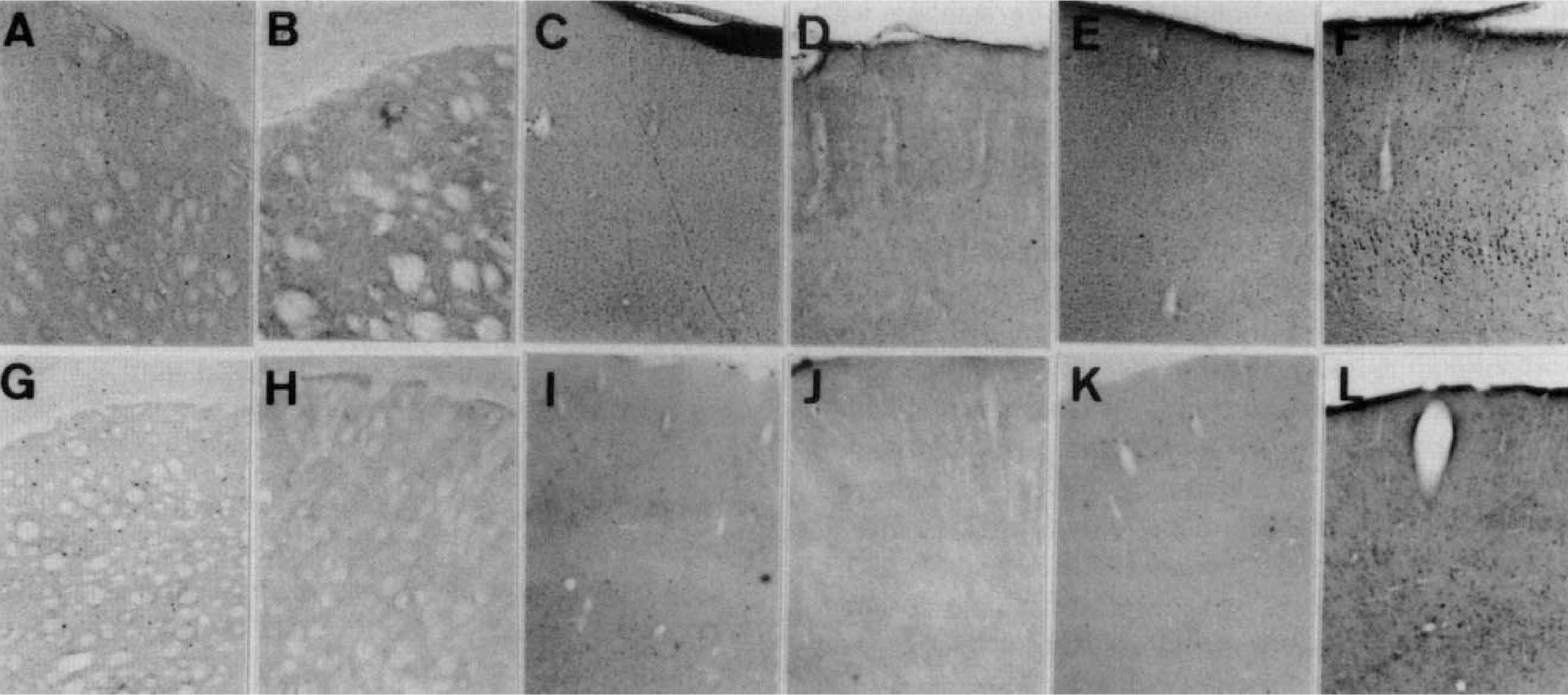
Low-power views of immunohistochemical and hybridization experiments to detect TRX (
Cortex. In the normal cortex, weak immunoreactivity against TRX was observed in most nonpyramidal and a few pyramidal neurons. In the frontoparietal ischemic cortex, a reduction in the immunoreactivity was observed in most of the cortical neurons at 4 hours after the insult; it was almost completely lost at 16 hours (Figs. 2E through H, and 3C and D). A few neurons in the ischemic cortex were obviously immunoreactive.
In the perifocal ischemic cortex, immunoreactivity for TRX was induced at 4 hours after MCA occlusion; it increased until 24 hours after occlusion. Pyramidal neurons were intensely immunoreactive (Figs. 2I through L, and 3E and F).
In situ hybridization
The TRX mRNA signals were observed in the normal striatum and cortex. No specific signals were seen in the control specimens.
Striatum. The TRX mRNA signal was strongly reduced in the lateral segment of the striatum at 4 hours and almost completely lost at 8 hours after the ischemic insult (Figs. 3G and H, and 4A through C).
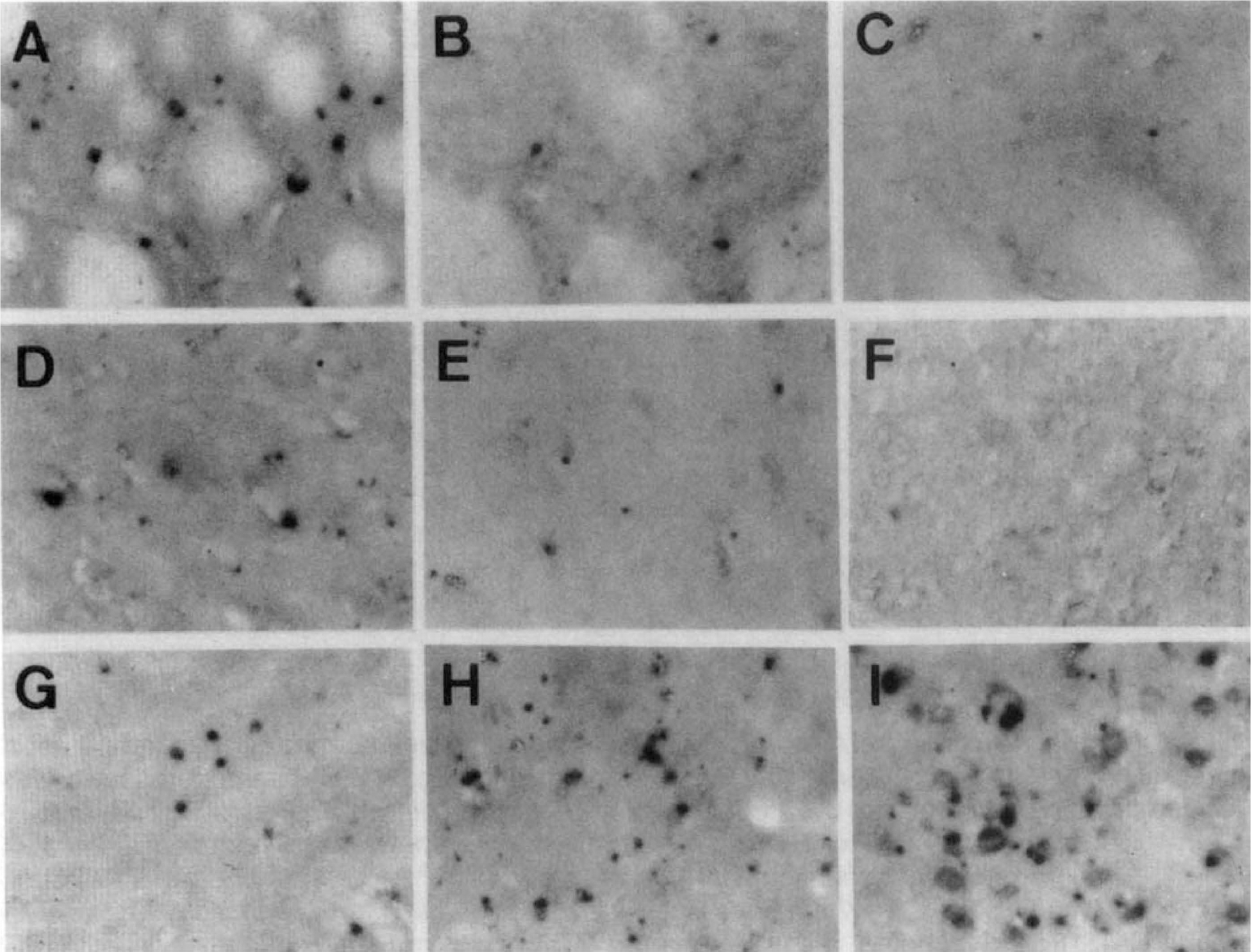
Hybridization experiments to detect TRX mRNA in the rat lateral striatum (
Cortex. In the normal cortex, weak signals for TRX mRNA were detected. In the frontoparietal ischemic cortex, TRX mRNA was reduced until 24 hours after MCA occlusion (Figs. 3I and J, and 4D through F).
In the perifocal ischemic cortex, TRX mRNA signals were induced at 4 hours and increased until 24 hours (Figs. 3K and L, and 4G through I).
Northern blot analysis
Northern blot analysis revealed TRX mRNA as a single 1.3-kilobase band (Fig. 5A). The TRX mRNA in the operated hemisphere was induced at 8 hours and increased until 24 hours after the surgical procedure (Fig. 5).
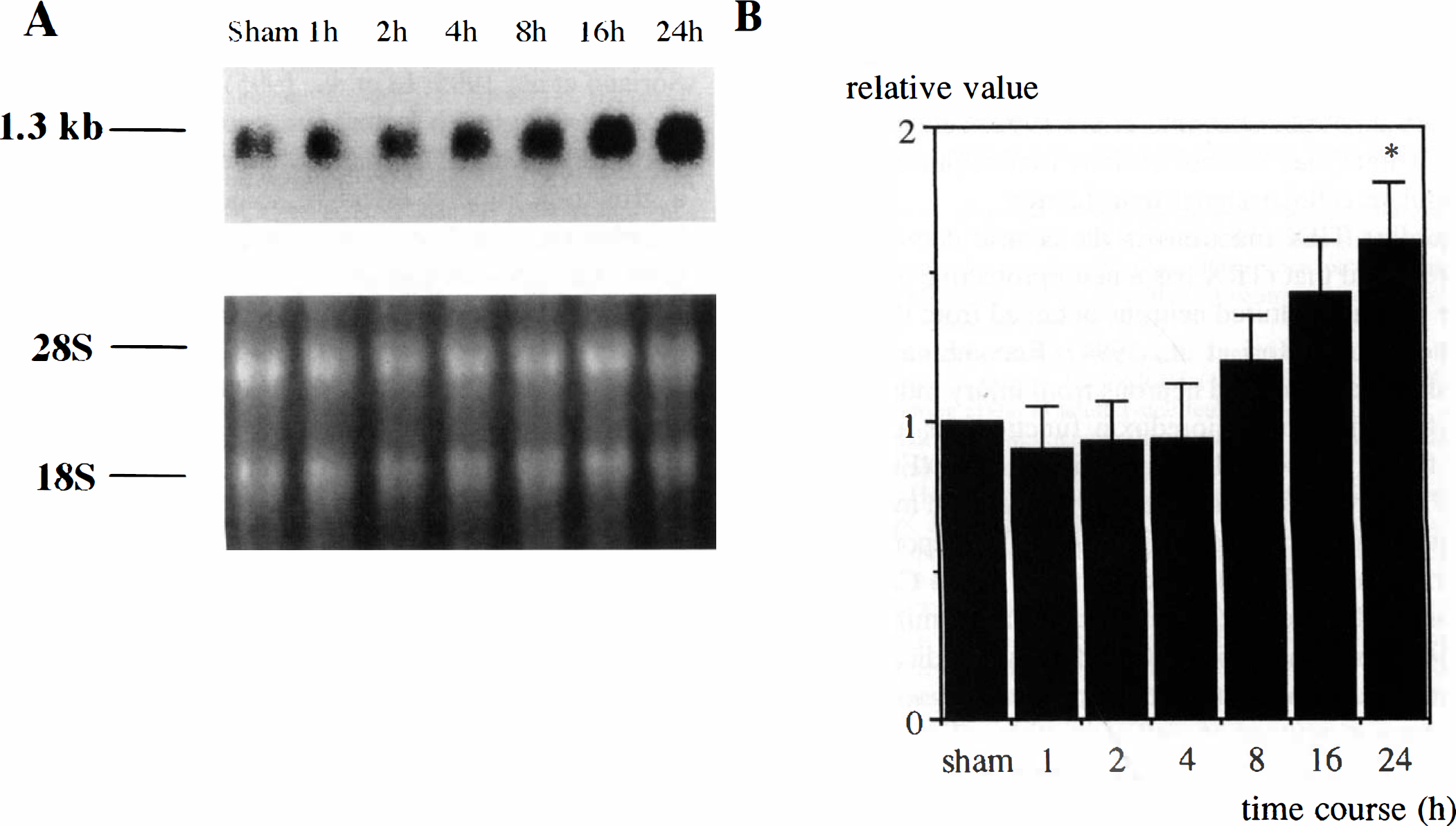
Northern blot and densitometric analyses of TRX mRNA in the ischemic hemisphere after MCA occlusion.
DISCUSSION
Using our rat focal ischemia model, we demonstrated the corresponding nature of immunoreactivity and mRNA signals for TRX in the infarct areas and in perifocal ischemic regions, the penumbra. Among the ischemic regions, the lateral striatum was the most vulnerable area in our model. This is the case because the blood flow in the lateral lenticulostriate arteries is impaired to a greater degree than in the medial lenticulostriate arteries (Zhang et al., 1994). Since these are end arteries with few collaterals, the blood flow is lowest in the lateral striatum, and it is likely that energy depletion is greatest there. In the lateral striatum, the immunoreactivity for TRX and TRX mRNA were quickly reduced and had almost disappeared by 4 hours after occlusion, when the striatal neurons were undergoing a degenerative process. In our model, ischemia is moderate in the frontoparietal cortex compared with the lateral striatum (Takagi et al., 1993), since this area is perfused by collateral channels. Memezawa and others (1992) report that in rats, this region is salvaged after 30 minutes of occlusion of the MCA. In the frontoparietal cortex, immunoreactivity for TRX and TRX mRNA were reduced and disappeared later than in the lateral striatum. Interestingly, these findings show that the reduction of immunoreactivity for TRX and TRX mRNA occurred in parallel with neuronal damage. On the other hand, in the perifocal ischemic region, immunoreactivity for TRX and TRX mRNA were induced at 4 hours after MCA occlusion and increased until 24 hours after occlusion. This region is perfused by collateral channels from the anterior cerebral artery and called the penumbra. Most neurons in the penumbra escaped death, suggesting that the induced TRX plays an important role in neuroprotection. The terms perifocal and penumbral might be stretched in various directions. We used these terms to mean “perifocal MCA territory” and “penumbral when ischemia has completed.”
Thioredoxin is an endogenous redox regulating protein with a redox-active site. It has been reported to be a stress-inducible protein whose expression is enhanced by various oxidative stimuli (Holmgren, 1985, 1989; Yodoi and Uchiyama, 1992). Previous studies show that the expression of TRX is up-regulated after exposure to H2O2, x-ray irradiation, and ultraviolet irradiation (Nakamura et al., 1994; Ohira et al., 1994; Sachi et al., 1995). Not only can TRX function as a hydrogen donor for various proteins, but it also acts as a scavenger of ROI in cooperation with NADPH and TRX reductase (Holmgren, 1985). In vitro, rTRX efficiently reduces ROI, such as H2O2 and hydroxyl radicals. Moreover, a family of TRX-dependent peroxide reductase has been identified in yeast and mammalian cells; it directly reduces H2O2 and various alkyl hydroperoxides, with the help of hydrogen molecules provided by the TRX system. These findings suggest that TRX may play an important role in cellular defense mechanisms against oxidative stress. In fact, rTRX exhibited protective activity against H2O2-induced cytotoxicity in murine endothelial cells (Nakamura et al., 1994). In addition, in a human histiocytic lymphoma cell line, pretreatment with rTRX inhibits tumor necrosis factor-α–induced cytotoxicity; the generation of ROI seems to be involved (Matsuda et al., 1992). Moreover, endogenous TRX plays a protective role against CDDP-induced cytotoxicity by scavenging toxic ROI generated by CDDP (Sasada et al., 1996).
Besides its scavenging of intracellular ROI, we previously reported that TRX plays a critical role in the regulation of intracellular thiols, especially glutathione. Exogenous rTRX increased the intracellular glutathione level in some cell types (Iwata et al., 1994; Hori et al., 1994). Up-regulated intracellular glutathione may participate in the effect of TRX.
Thioredoxin appears to have a crucial role in the regulation of various enzymes and transcription factors, including NF-κB and AP-1, through its redox activity (Abate et al., 1990; Okamoto et al., 1992; Schenk et al., 1994). It thus may control various transcription factors and regulate cellular signal transduction.
Regarding TRX functions in the central nervous system, we found that rTRX has a neuroprotective effect on murine primary cultured neurons obtained from the striatum and cortex (Hori et al., 1994). Recombinant TRX rescued primary cultured neurons from injury induced by serum-free medium. Thioredoxin functions as a neurotropic factor for central cholinergic neurons (Endoh et al., 1993; Barde, 1994) and was up-regulated in mechanical brain injury (Lippoldt et al., 1995). We reported the proliferated astroglial induction of TRX in the CA-1 region during transient forebrain ischemia (Tomimoto et al., 1993). In combination, these findings indicate that TRX has a function in the cellular defense against oxidative stress in neurons as well as in other cell types, and may play a role during acute and delayed brain injury.
Many factors are involved in the development of neuronal damage during ischemia. The neurotoxicity of excitatory amino acids, an overload of intracellular Ca2+, the suppression of protein synthesis, and free radical formation are thought to play detrimental roles in the pathogenesis of ischemic injury.
The formation of oxygen-derived free radicals may occur in the electron transport chain, in metabolic processes such as free fatty acids, and in purine and nitric oxide (McCord, 1985; Kinuta et al., 1989; Kitagawa et al., 1990; Lundgren et al., 1991; Morooka et al., 1994; Dirnagl et al., 1995). These oxidants may destroy cellular and subcellular membranes and induce brain edema during ischemia and reperfusion. In this sense, oxygen-derived free radicals play an important role in neuronal death. This hypothesis is supported by the finding that SOD activity and immunoreactivity in decline in the ischemic area during ischemia (Chan et al., 1988; Liu et al., 1994; Kato et al., 1995).
Several antioxidants (e.g., glutathione, ascorbic acid, α-tocopherol, and ubiquinone) significantly decrease brain damage during focal ischemia (Abe et al., 1983; Hara et al., 1990; Sato and Hall 1992; Villalobos et al., 1994). In vitro, SOD and several antioxidants protect neurons from apoptosis (Yan et al., 1995). These findings reveal that ROI-scavenging systems are important for the survival of neurons during ischemia.
The decline of SOD immunoreactivity in the ischemic area is similar to the decline in immunoreactivity for TRX. Like SOD, TRX has ROI-scavenging activity, indicating that TRX functions as an ROI scavenger during brain ischemia.
Stress proteins such as heat shock proteins and several growth factors are induced in ischemic and periischemic lesions (Kawagoe et al., 1992; Welsh et al., 1992; Takeda et al., 1993; Kinouchi et al., 1994; Iihara et al., 1994; Soriano et al., 1995; Li et al., 1995). Induced heat shock proteins and growth factors enhance neuronal survival in vitro. Reportedly, TRX has protein refolding activity and a growth-promoting effect in some cell types such as lymphocytes, in liver carcinoma, vascular endothelial cells, and neurons (Mitsui et al., 1992; Makino et al., 1992; Nakamura et al., 1992, Endoh et al., 1993, Biquet et al., 1994, Gasdaska et al., 1994, Powis et al., 1994). These findings suggest that induced TRX may function not only as an ROI scavenger but also as a neurotropic agent.
In summary, the cellular redox state is modified by the expression of TRX in parallel with the development of neuronal injury. Neurons in the ischemic penumbra in which TRX is highly expressed tend to survive, whereas decreased neuronal TRX in ischemic leads to neuronal death. Thioredoxin and the redox state modified by TRX play a crucial role in neuronal death and survival during brain ischemia.