
Abstract
Interleukin-1β (IL-1β) has been implicated in ischemic brain damage. The site of action of IL-1β in such damage is not known, but we have demonstrated previously that injection of the interleukin-1 receptor antagonist (IL-1ra) in the striatum but not the cortex of rats inhibits damage caused by permanent middle cerebral artery occlusion. The present study investigated the site of action of IL-1β on ischemic damage by examining the effects of intracerebroventricular, striatal, or cortical injection of recombinant IL-1β at the onset of permanent middle cerebral artery occlusion in the rat. Intracerebroventricular injection of IL-1β (2.5 ng) significantly increased infarct volume in the striatum (35%, P < 0.0001) and in the cortex (44%, P < 0.0001) compared with vehicle treatment. Direct injection of IL-1β into the striatum also increased infarct volume in both the striatum (36%, P < 0.0001) and the cortex (38%, P < 0.0001), whereas injection of IL-1β into the cortex failed to affect infarct volume in either the striatum or the cortex. Cortical injection of a higher dose of IL-1β (20 ng) also failed to affect ischemic damage in either the striatum or the cortex. Injection of IL-1β into the striatum contralateral to the infarction had no effect on striatal damage in the ischemic hemisphere, but did increase cortical damage by 18% (P < 0.0001). In separate groups of animals, IL-1β (2.5 ng) was injected into either the striatum or the cortex, and body temperature was recorded continuously in conscious free-moving animals by remote telemetry. Injection of IL-1β at either site failed to influence body temperature, suggesting that exacerbation of brain damage by striatal injection of IL-1β is not caused by effects on body temperature. These results imply that IL-1β exacerbates ischemic damage by specific actions in the striatum where it can influence damage at distant sites in the cortex.
The interaction of neural and immune systems in neurodegeneration has become an important issue in our understanding of the processes of ischemic brain damage. The cytokine interleukin-1β (IL-1β) is induced rapidly after the onset of ischemia (Yabuuchi et al., 1994a; Hopkins and Rothwell, 1995; Davies et al., 1998), initiating a cascade of events that could participate in the development of ischemic and excitotoxic damage (Rothwell and Relton 1993; Yamasaki et al., 1995). It has been proposed that IL-1β can act on neurons, glia, and the vasculature through a variety of mechanisms (see Rothwell and Hopkins, 1995; Betz et al., 1996) to influence ischemic brain damage. Although IL-1β alone is not neurotoxic in normal brain, exogenous application of IL-1β by intracerebroventricular administration at the onset of ischemia markedly increases infarct volume (Relton and Rothwell, 1992; Yamasaki et al., 1992; Loddick and Rothwell, 1996).
Inhibition of the actions of IL-1β by the injection of the naturally occurring interleukin-1 receptor antagonist (IL-1ra), inhibits ischemic brain damage by up to 70% (Relton and Rothwell 1992; Loddick and Rothwell, 1996) and also reduces damage caused by traumatic brain injury, excitotoxins, hypoxia, and heat stroke (Lawrence and Rothwell, 1994; Martin et al., 1994; Garcia et al., 1995; Lin et al., 1995; Toulmond and Rothwell, 1995). Unlike many neuroprotective agents tested in focal cerebral ischemia (see Muir and Lees, 1995), IL-1ra reduces damage in both the cortex and striatum (Relton and Rothwell, 1992; Loddick and Rothwell, 1996). Our recent studies now provide evidence that IL-1β and IL-1ra may have site-specific actions in the brain in both ischemic and excitotoxic brain damage (Allan et al., 1995; Stroemer and Rothwell, 1997).
These data, as well as the knowledge that after cerebral ischemia the greatest induction of IL-1β mRNA is in the striatum (Yabuuchi et al., 1994a) and that IL-1β is present in both the cortex and dorsal striatum (Davies, Loddick, Stroemer, Rothwell, unpublished data), suggest that a major site of action of endogenous IL-1β in the pathology of ischemic brain damage is in the striatum. To further test this hypothesis, we studied the effects of localized injections of recombinant IL-1β into either the lateral cerebral ventricle, the striatum, or the cortex at the onset of permanent focal ischemia in the rat. Injection of biotinylated IL-1ra (similar sized and sequence to IL-1β) into either the striatum or cortex shows that it readily diffuses throughout the injected region (Toulmond, Luheshi, Rothwell, unpublished data). Therefore, direct injection of IL-1β should result in a fairly consistent concentration throughout the striatum or cortex. Because IL-1β is a potent pyrogen and increases of temperature can exacerbate ischemic brain damage, body temperature was measured by remote radiotelemetry in rats injected with IL-1β into either the striatum or the cortex.
METHODS
All experiments were performed on male, Sprague-Dawley rats (Charles River, Maidstone, U.K.) weighing 230 to 270 g. Animals were housed at 21°C with ad libitum access to food and water. Guide cannulae were implanted stereotaxically into the cortex (coordinates, 2.7 mm anterior, 3.0 mm lateral, 2.4 mm ventral to bregma) or striatum (0.3 mm posterior, 4.0 mm lateral, 5.5 mm ventral to bregma), using coordinates from Paxinos and Watson (1986), of rats under halothane anesthesia (4% induction, 2% to 3% maintenance in oxygen) 7 days before the induction of ischemia.
Cerebral ischemia was induced by a proximal, permanent occlusion of the middle cerebral artery by electrocautery as described by Tamura et al. (1981). Anesthesia was induced by 4% halothane in oxygen, and maintained at 2% to 3% halothane during the procedure. The total period from induction of anesthesia to recovery and consciousness was approximately 30 minutes. Body temperature was maintained during the surgical procedure by heated pads and thereafter by placing animals under heat lamps.
Either vehicle (saline) or human recombinant IL-1β (2.5 ng/μL saline, 18 IU/ng protein, endotoxin content 2.5 EU/mg protein, Dupont de Nemours, Glenolden, PA, U.S.A.) was infused at a rate of 1 μL for 2 minutes through an injection cannula placed within the guide cannula. The same volume (1 μL) was injected in all regions. The injection cannula was withdrawn 2 minutes after the end of the infusion.
Sham-operated animals were preimplanted with guide cannulae. The same surgical protocol was followed until the coagulation of the middle cerebral artery, then the coagulating forceps were placed around the artery, but no current was passed. The animals were then returned to the surgical protocol and subsequent sacrifice and staining.
Infarct volume was determined 24 hours after ischemia by staining 500-μm fresh brain sections with 2% triphenyltetrazolium chloride staining (Sigma Chemical Co., St. Louis, MO, U.S.A.). This procedure stains for mitochondrial viability giving a clear representation of the lesion (Bederson et al., 1986). Injection sites were verified during the staining protocol. To correct lesion volume for the effects of edema, lesion areas were charted onto stereotaxic maps (Paxinos and Watson, 1986), which were quantified by automated image analysis (SeeScan, Cambridge, U.K.). Total lesion volumes were calculated by integration of the areas of infarction.
To determine the effects of IL-1β on core body temperature, further groups of rats were implanted with guide cannula in the ipsilateral striatum or cortex and temperature-sensitive radiotransmitters (Data Sciences International, St. Paul, MN, U.S.A.) were inserted into the peritoneal cavity under halothane anesthesia, 5 to 7 days before IL-1β injection. During the injection, animals were anesthetized briefly with halothane (4% induction, 3% maintenance), placed on a heating pad, and injected with 1 μL of either saline or IL-1β (2.5 ng) for 2 minutes; then the injection cannula was removed. Animals were allowed to recover from anesthesia, and when conscious were returned to home cage (22°C) for at least 18 hours before sacrifice. Data from individual animals were compared at the time of onset of stroke. Body temperature data were compared as the area under the curve for each group.
All data are expressed as group means ± standard deviation. Statistical differences between lesion volumes in the striatum and cortex of saline and IL-1β-treated rats were assessed using Student's t test with probability of less than 5% for statistical significance. Temperature data and areas of damage were analyzed by multiple analysis of variance. Probabilities are two-tailed with a probability of less than 5% for statistical significance.
RESULTS
Sham-operated animals showed little or no damage (<3 mm3) as a result of the implantation of the injection cannula in the territory of the middle cerebral artery. Rats subjected to middle cerebral artery occlusion exhibited damage in the striatum and the cortex that was consistent with the areas supplied by the middle cerebral artery and similar to that reported in our earlier studies (Loddick and Rothwell, 1996; Stroemer and Rothwell, 1997). Rats subjected to middle cerebral artery occlusion also exhibited some behavioral dysfunction, noticeably in postural reflexes with twisting of the forepaw contralateral to the infarction; however, there were no gross differences between treated and control groups.
Effects of intracerebroventricular injection of IL-1β on ischemic damage
Intracerebroventricular injection of IL-1β significantly increased (35%) ischemic damage in the striatum (34.2 ± 2.8 mm3) when compared with vehicle-injected animals (25.3 ± 1.7 mm3; n = 10, P < 0.0001). Damage in the cortex was also increased (by 44%) in IL-1β-treated animals (138.0 ± 6.9 mm3) compared with vehicle treated animals (95.4 ± 6.9 mm3; n = 10, P < 0.0001); this exacerbation was apparent in both the frontal and parietal cortical brain regions (Fig. 1). The difference between the areas of damage was statistically significant in the striatum and cortex (P < 0.0001 in both regions).
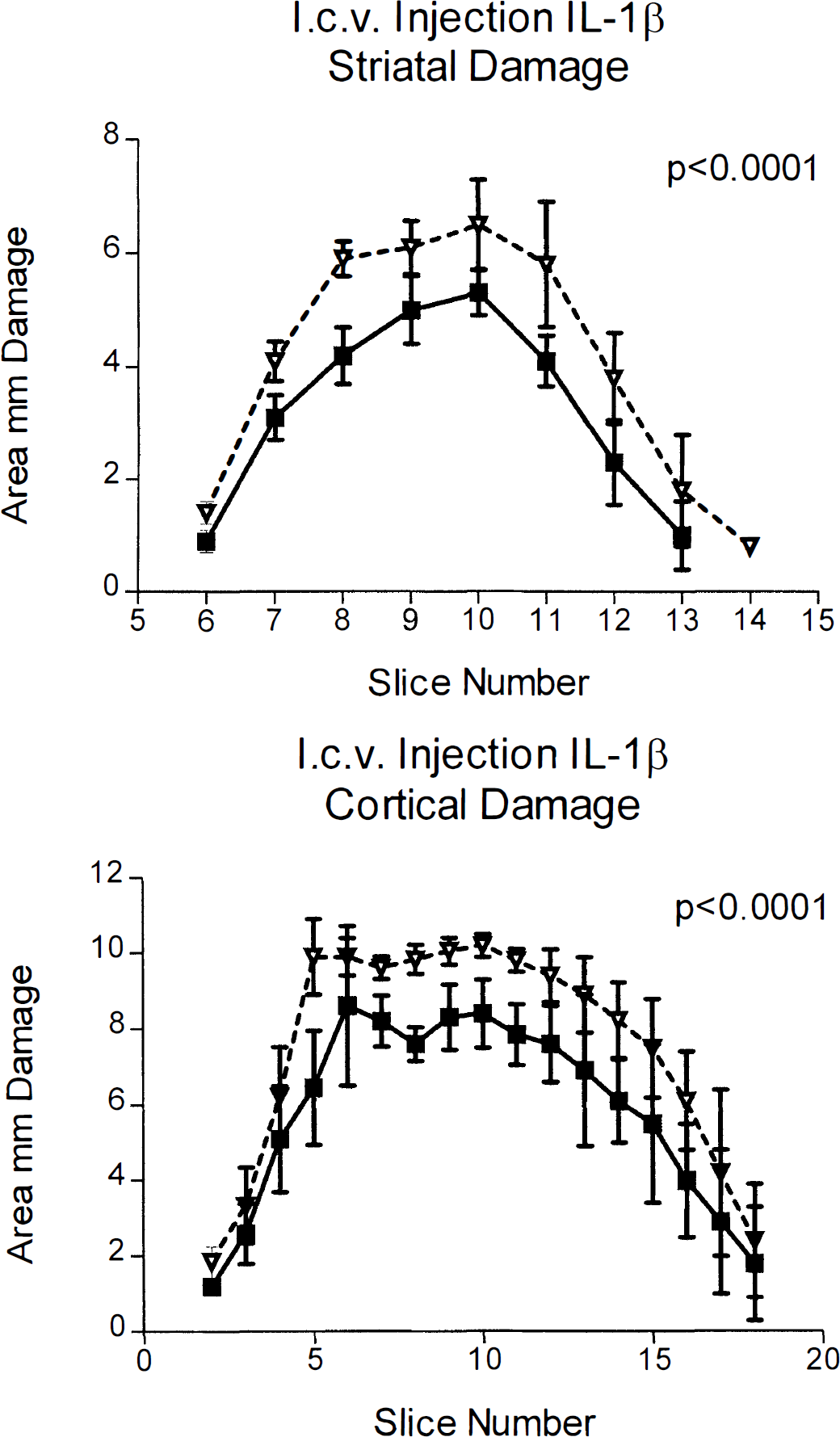
Infarct areas (mm2) measured 24 hours after middle cerebral artery occlusion in rats that received intracerebroventricular (l.c.v.) injection of IL-1β (2.5 ng) or vehicle. Filled squares represent saline-injected animals, open triangles show IL-1β-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Animals injected with IL-1β showed significantly more damage in the striatum (P < 0.0001) and the cortex (P < 0.0001) when compared with vehicletreated animals. Data shown as means ± SD, n = 10.
Effects of striatal injection of IL-1β on ischemic damage
Animals subjected to striatal injection of IL-1β also showed an increase (36%) in the volume of damage in the striatum (35.8 ± 2.4 mm3) compared with vehicle injected animals (26.2 ± 3.2 mm3; n = 11, P < 0.0001). Similarly, infarct volume in the cortex was increased (38%) in IL-1β (striatal) -injected animals (127.8 ± 6.8 mm3) compared with vehicle-treated animals (92.3 ± 4.3 mm3; n = 11, P < 0.0001), particularly in the more caudal regions of the parietal cortex (Fig. 2). Again, patterns of damage (areas) between groups were statistically significant in both the striatum and the cortex (P < 0.0001 both regions).
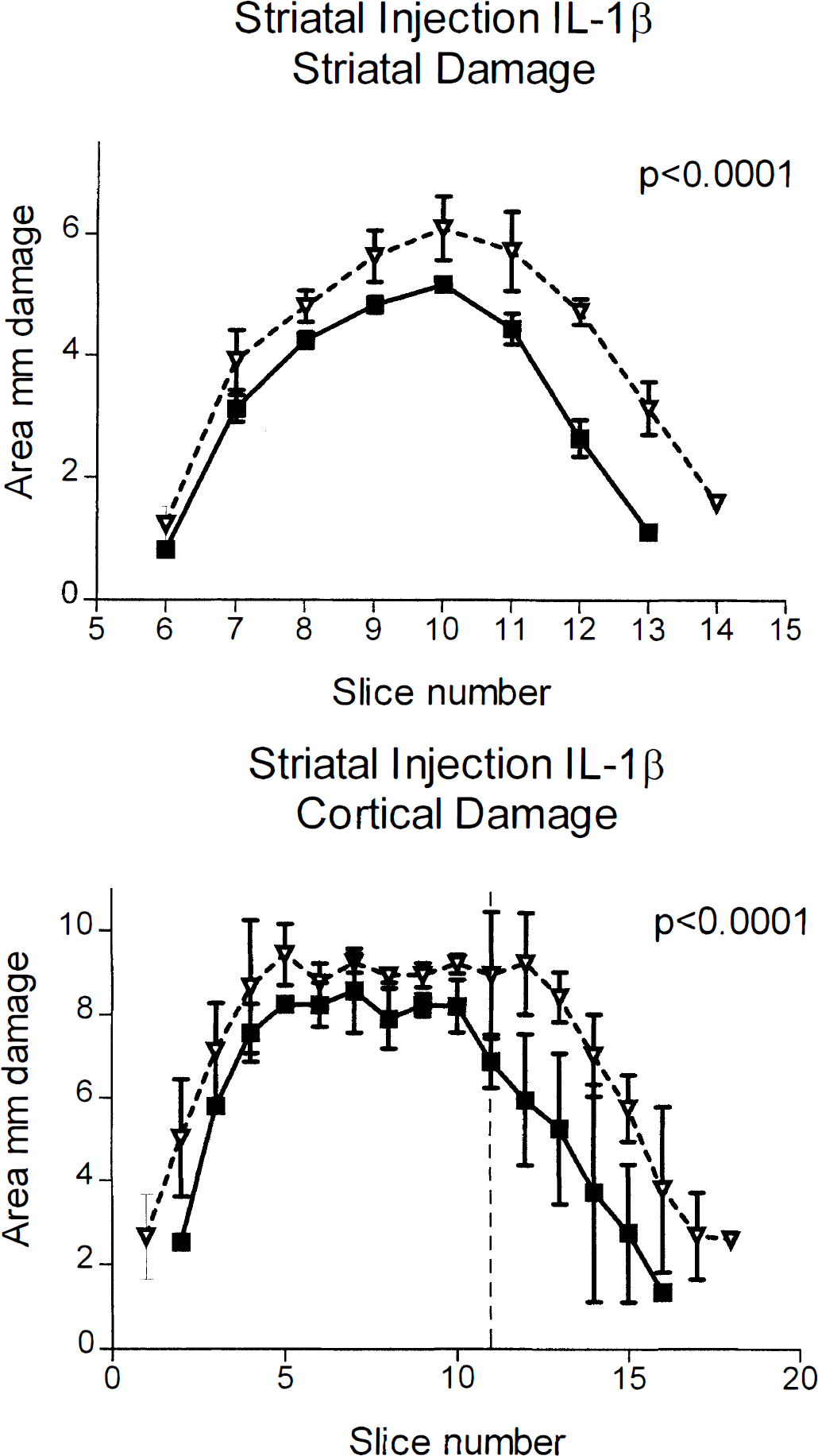
Infarct areas (mm2) measured 24 hours after middle cerebral artery occlusion in rats that received cortical injection of IL-1β (2.5 ng) or vehicle. Filled squares represent saline-injected animals, open triangles show IL-1β-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Vertical dashed line is the location of the injection cannula. Animals injected with IL-1β showed significantly more damage in the striatum (P < 0.0001) and the cortex (P < 0.0001) when compared with vehicle-treated animals. Data are shown as means ± SD, n = 11.
Effects of cortical injection of IL-1β on ischemic damage
Animals injected with IL-1β into the cortex showed almost identical volumes of ischemic brain damage as their vehicle-treated cohorts in both the striatum (vehicle 27.2 ± 1.8 mm3, IL-1β 26.4 ± 2.1 mm3; n = 10, not significant) and the cortex (vehicle 93.4 ± 5.1 mm3, IL-1β 91.2 ± 7.3 mm3; n = 10, not significant) (Fig. 3). The areas of damage were not statistically different between groups in either region.
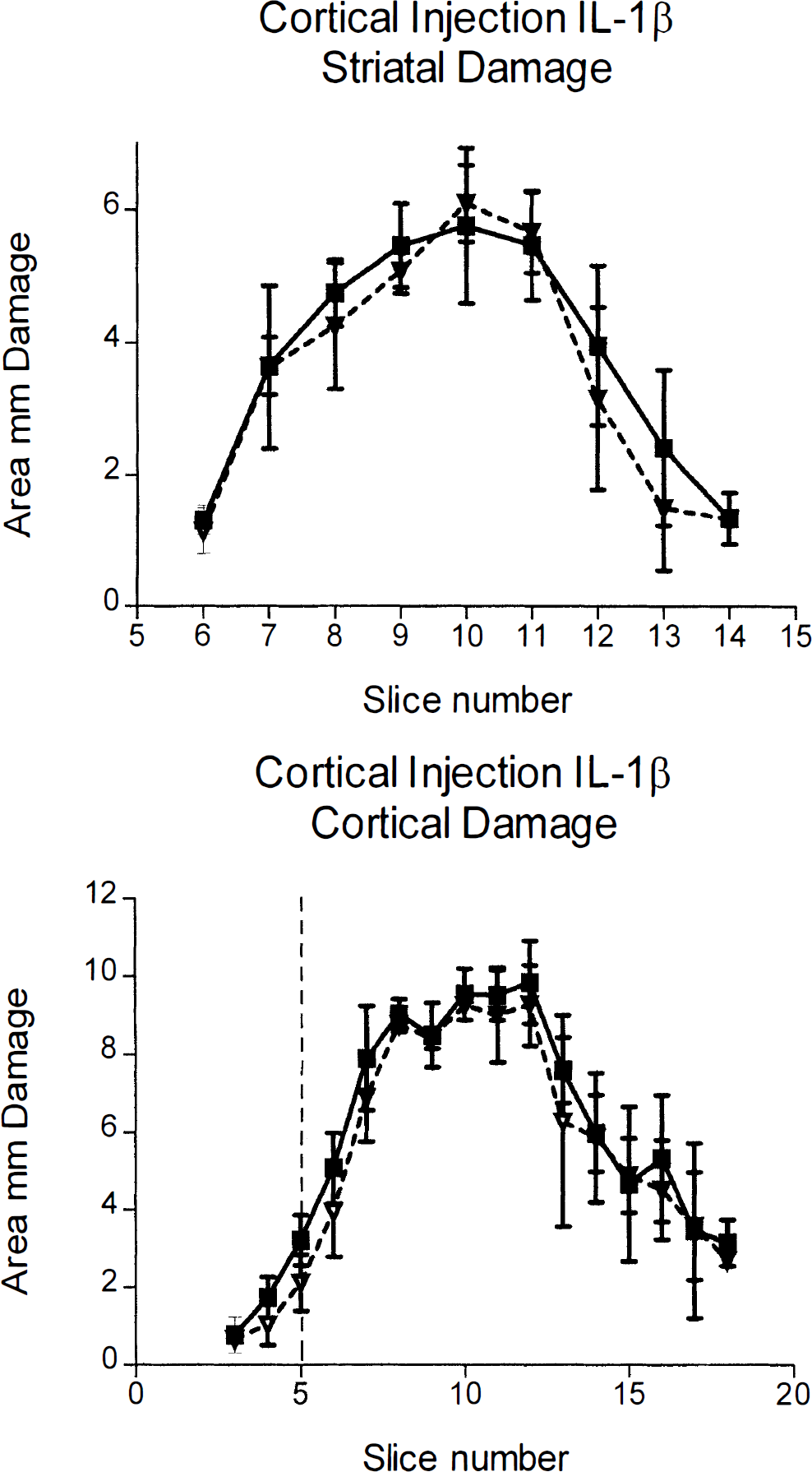
Infarct areas (mm2) measured 24 hours after middle cerebral artery occlusion in rats that received cortical injection of IL-1β (2.5 ng) or vehicle. Filled squares represent saline-injected animals, open triangles show IL-1β-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Vertical dashed line is the location of the injection cannula. There was no difference between vehicle- and IL-1β-treated animals. Data are shown as means ± SD, n = 10.
Effects of contralateral striatal injection of IL-1β on ischemic damage
Animals that received injections of IL-1β into the striatum contralateral to the infarction exhibited a slight, but not statistically significant, increase in the volume of damage in the striatum compared with the vehicle-injected animals (vehicle 26.2 ± 1.9 mm3, IL-1β 27.4 ± 1.3 mm3; n = 10, not significant). However, there was a statistically significant increase in the areas of damage in IL-1β-treated rats (P < 0.02). There was a statistically significant increase (18%) in volume of cortical damage (113.6 ± 5.9 mm3) compared with the vehicle-treated animals (96.3 ± 4.7 mm3n = 10, P < 0.0001) (Fig. 4), with a statistically significant difference between the curves of the areas of damage (P < 0.0001).
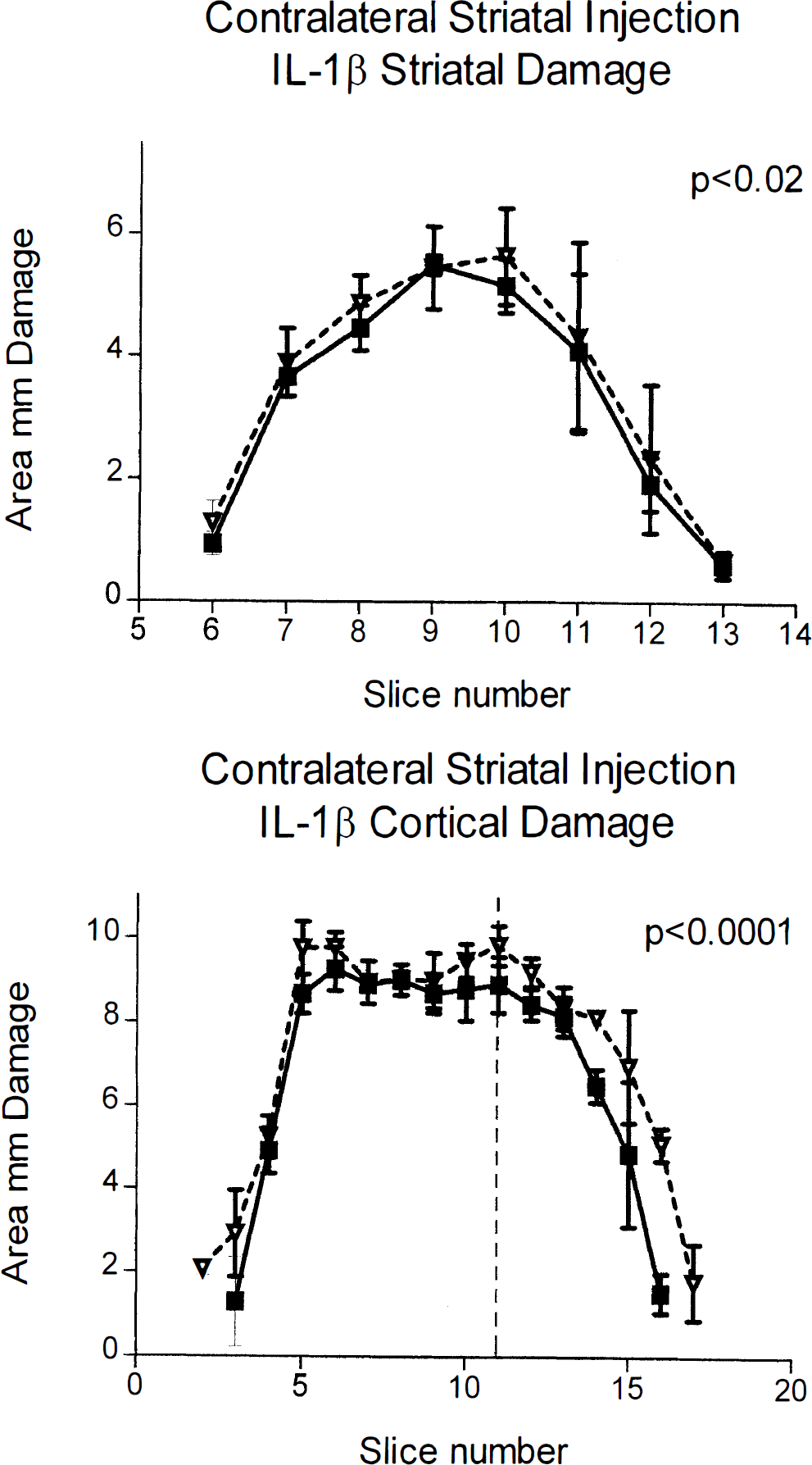
Infarct areas (mm3) measured 24 hours after middle cerebral artery occlusion in rats that received contralateral injection of IL-1β (2.5 ng) or vehicle. Filled squares represent saline-injected animals, open triangles show IL-1β-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Vertical dashed line is the location of the injection cannula. Animals injected with IL-1β showed significantly more damage in the striatum (P < 0.02) and the cortex (P < 0.0001) when compared with vehicle-treated animals. Data are shown as means ± SD, n = 10.
Effects of injection of IL-1β on body temperature
Injection of IL-1β into either the striatum or the cortex did not affect core body temperature (Fig. 5). Striatal injection of IL-1β caused a slight acceleration in the return of body temperature to normal during recovery from anesthesia. There was no significant difference between the two groups, with the maximal difference being 0.5°C at anytime.
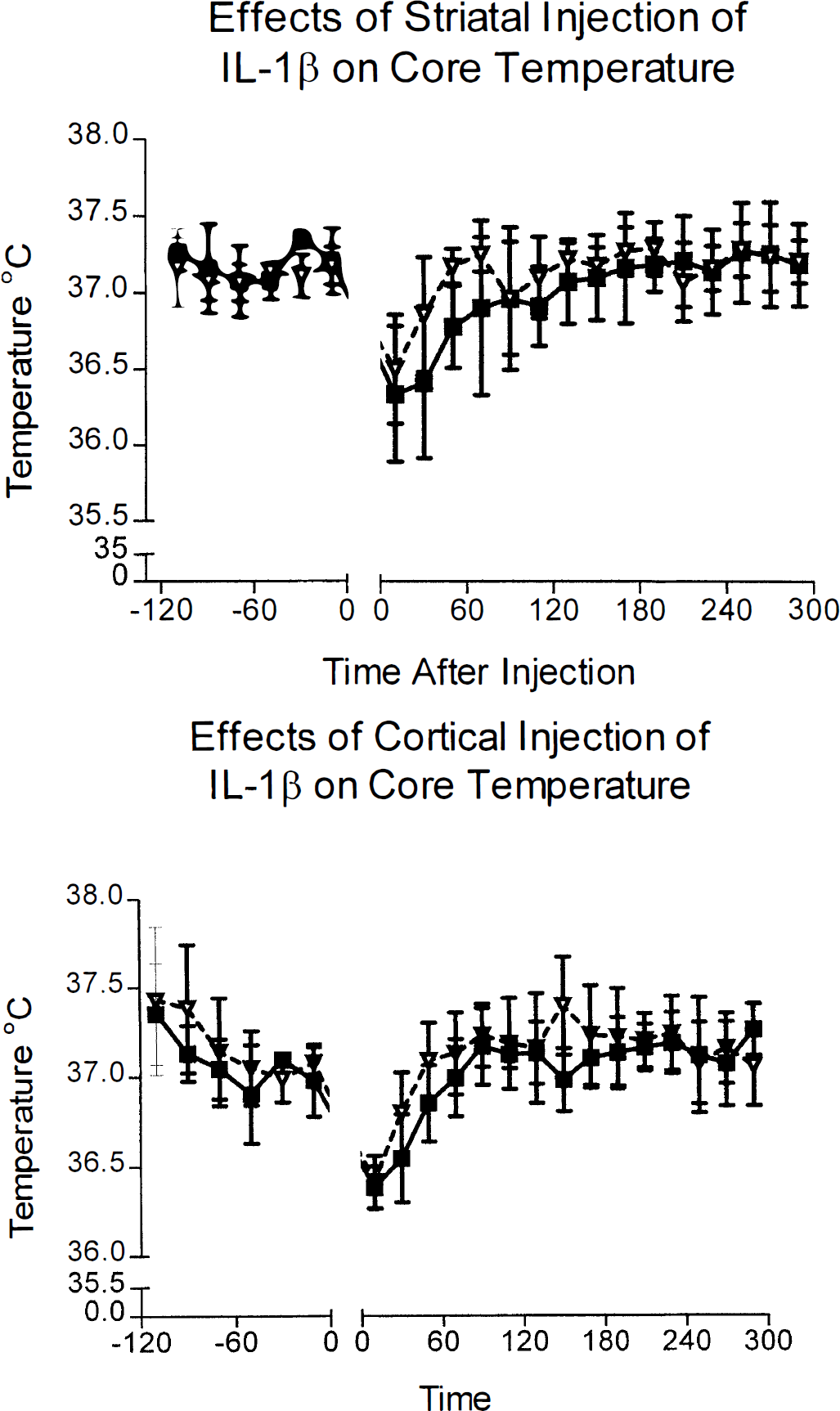
Core temperatures of saline- and IL-1β-treated animals. Top: Body temperatures of animals with striatal injection of vehicle (filled squares) or IL-1β (open triangles). Data are shown as means ± SD, n = 8. Bottom: Body temperatures of animals with cortical injection of either vehicle (filled squares) or IL-1β (empty triangles). There was no significant difference in temperature between vehicle- and IL-1β-treated groups with either the striatal or cortical injection site.
Cortical injection of IL-1β also slightly accelerated the return to normal body temperature during recovery from anesthesia. Interleukin-1β-injected animals exhibited modest increases in body temperature (up to 0.3°C) at 2 to 3 hours and 6 to 7 hours after injection, but neither increase was statistically significant. There were no differences in body temperatures between vehicle- or IL-1-treated groups during the rest of the study (Fig. 5).
DISCUSSION
Administration of the naturally occurring IL-1ra by injection into the cerebral ventricles or peripherally, or by acute gene transfer (adenovirus) into the brain, markedly inhibits ischemic brain damage in the rat (Relton and Rothwell, 1992; Betz et al., 1995; Loddick and Rothwell, 1996; Yang et al., 1997). Our recent studies have shown that injection of IL-1ra into the striatum ipsilateral to ischemia reduces damage in the striatum and the cortex, but direct injection of IL-1ra into the ischemic cortex does not affect infarct volume in either region (Stroemer and Rothwell, 1997). These data suggest site-specific actions of IL-1ra (and here IL-1β) in neurodegeneration.
In the present study, animals injected intracerebroventricularly with IL-1β showed markedly increased ischemic damage (44%) throughout the frontal and parietal regions of the cortex and also in the striatum (35%). This observation is consistent with our previous studies (Relton and Rothwell, 1992) showing an exacerbation of ischemic brain damage with intracerebroventricular injection of IL-1β and also that the lesion did not grow with time (Loddick and Rothwell, 1996). Striatal injection of IL-1β also increased ischemic damage throughout the cortex (38%), with the greatest increase in the more caudal parietal cortex and throughout the striatum (36%).
Injection of IL-1β into the cortex did not affect ischemic damage in the striatum or the cortex (Fig. 3). This lack of change complements our earlier study demonstrating that IL-1ra is ineffective when administered to the cortex at the onset of cerebral ischemia (Stroemer and Rothwell, 1997). It is possible that IL-1β may influence ischemic damage when injected into specific cortical sites or at larger doses. However, we have tested the effects of IL-1β injected into other cortical sites in ischemic and excitotoxic brain damage (0.7 mm anterior, 5.0 mm lateral, 3.5 mm ventral to the bregma) and have seen no effects on either form of damage (data not shown). Injection of a higher dose of IL-1β (20 ng) into the cortex at the onset of ischemia also did not affect damage in either the striatum (29.0 ± 2.0 mm3, vehicle; 27.2 ± 3.2 mm3, IL-1β; n = 5, not significant) or the cortex (120.6 ± 6.6 mm3, vehicle; 110.8 ± 11.3 mm3, IL-1β; n = 5, not significant). These results suggest that injection of IL-1β into the cortex does not influence ischemic damage in either the cortex or the striatum.
The mechanisms of these site specific effects of IL-1 (and IL-1ra) are unclear and do not relate to the known distribution of IL-1 type 1 receptors (IL-1R1) in the brain. The type 1 receptor is believed to be the “active” interleukin-1 receptor, with the type 2 receptor acting as a “decoy”, which does not induce signaling (Sims et al., 1993). IL-1R1 mRNA expression has been reported in the cortex and other brain regions, but not in the striatum (Ericsson et al., 1995; Gayle et al., 1997, Yabuuchi et al., 1994b). This paradox suggests that either the neurons from other regions possess receptors on their striatal terminals that have an effect on damage, or that a novel or atypical interleukin-1 receptor mediates IL-1 actions in the striatum.
Injection of IL-1β into the striatum contralateral to the infarction produced a 20% increase in cortical damage compared with vehicle-injected animals. Again, this was seen mainly in the parietal cortex (Figure 4). There was no significant change in the total volume of striatal damage in the infarcted hemisphere of IL-1β-treated animals. The areas of striatal damage were slightly different, suggesting that contralateral injection of IL-1β may have had some minor effect on the distribution and volume of ischemic damage in the ipsilateral striatum. Injection of IL-1β into the striatum or cortex of normal rats (i.e., without ischemia) caused no damage.
Central administration of IL-1β also results in a modest increase in heart rate and blood pressure (Morimoto et al., 1992), although it has been proposed that this is too small to affect ischemic damage (Betz et al., 1996). Intracerebroventricular administration of IL-1ra also has no significant effect on heart rate and blood pressure (Loddick and Rothwell, 1996). One possible mechanism of the exacerbation of damage by injection of IL-1β may be the induction of fever. The cytokine IL-1β is a potent pyrogen and is believed to be an important endogenous mediator in the febrile response (Dinarello, 1996). The dose used in this study was chosen to minimize hyperthermic effects; intracerebroventricular injection of 2.5 ng of IL-1β results in increases in body temperature of up to 1.5° to 2°C (Anforth, 1996), with a maximal pyrogenic dose at 5 ng. Although hyperthermia can increase ischemic brain damage in transient ischemia, it has only minimal effects on permanent ischemia (Morikawa et al., 1992). Injection of IL-1β (2.5 ng) into the striatum or cortex of normal rats did not affect body temperature (Fig. 5). Although we cannot exclude the possibility that changes in brain temperature independent of core temperature influenced ischemic damage, this lack of effect of IL-1β on core temperature suggests that the exacerbation of ischemic brain damage in response to IL-1 is not caused by hyperthermia. It should be noted that intracerebroventricular injection of IL-6, which causes marked fever, does not increase infarct volume in this stroke model (Loddick et al., 1997), but actually resulted in a reduction of ischemic brain damage.
Injection of IL-1ra into the striatum reduces ischemic damage in both the striatum and cortex, and the greatest reductions of damage coincide with the origins of corticostriatal projections to the injection sites (Stroemer and Rothwell, 1997). Inhibition of damage in the cortical projection origins was also reported in regional injection of IL-1ra into the contralateral striatum. Exacerbation of cortical damage by striatal injection of IL-1β, however, is not reflected in the origins of the corticostriatal projections (McGeorge and Faull, 1989); instead it is increased in the caudal parietal cortex. This lack of specific damage in the projection origins has several explanations. Interleukin-1β may diffuse more readily throughout the striatum than IL-1ra, thus affecting larger portions of the cortex; a higher local concentration of IL-1ra is necessary to inhibit ischemic damage; or injection of IL-1β could result in increased release of inflammatory agents and free radicals that then freely diffuse to cause damage in the cortex.
Injection of IL-1β into the contralateral striatum resulted in a smaller, but nevertheless consistent and statistically significant, increase in cortical damage, but no significant increase in the total volume of striatal damage. Projections to the contralateral striatum from about 13% of cortical neurons innervate the ipsilateral striatum, providing an anatomic pathway for specific damage to the cortex (McGeorge and Faull, 1989). We demonstrated previously that IL-1ra injected into the contralateral striatum reduces damage in the ischemic hemisphere in a pattern corresponding with the origins of the corticostriatal projections (Stroemer and Rothwell, 1997). This protection by IL-1ra did not follow a concentration gradient away from the injection site as would be expected if damage was being produced solely by the diffusion of toxins. In contrast, the exacerbation of damage in the IL-1β-treated animals was not specific to the origins of the projections, but as in the animals injected ipsilaterally, this may be caused by diffusion of IL-1β throughout the injected striatum.
Intracerebroventricular administration of IL-1β is likely to affect sites throughout the brain and cerebral vasculature owing to diffusion, and its mechanisms of actions may depend on a combination of neuronal, glial, and inflammatory effects (Rothwell and Hopkins, 1995). It is generally believed that substances injected into the striatum, having to pass through the corpus callosum, do not diffuse readily into the cortex. Interleukin-1β injected into the striatum or the cortex may have diffused into the ventricles and the CSF, although the coordinates used in this study were chosen to reduce the possibility of ventricular puncture, and any substantial diffusion of IL-1β in the ventricles would have caused hyperthermia, which was not seen in this study. The greater exacerbation of damage caused by intracerebroventricular administration may result from IL-1β diffusing more readily throughout the brain from the ventricles and perhaps affecting vascular elements.
The dose of IL-1β used in this study was chosen to exacerbate damage with minimal changes in core temperature. In our laboratory, increasing concentrations of IL-1β tend to have an “all or nothing” effect on neurodegeneration. The dose (2.5 ng) is sufficient to affect damage yet not cause severe hyperthermia. Preliminary studies in this laboratory have shown levels of total immunoreactive IL-1β as high as 17 ng/mg wet weight in the infarcted cortex at 24 hours after the onset of ischemia (Stroemer, Cartmell, and Rothwell, unpublished data), which suggests that the dose of 2.5 ng IL-1β (<1 picomole) injected in the present study is not excessively high and is also within the range of concentrations of IL-1β after cerebral ischemia as reported by others (Ianotti et al., 1993).
The exacerbation of cortical ischemic damage by caused IL-1β injection into the striatum after cerebral ischemia focuses some attention on the pathways and mechanisms that affect ischemic damage. The pathways between the cortex and striatum are primarily the corticothalamic and striatothalamocortical pathways, suggesting that the effects of IL-1β on the cortex are either retrograde or polysynaptic. Interleukin-1β is pleiotropic and may generate a variety of mediators, such as tumor necrosis factor and nitric oxide (Dinarello, 1996) The relationship between the production of IL-1β during ischemia and the mechanisms of action of IL-1 are not well understood, and although IL-1β does not damage normal neurons, it can apparently kill those that are under stress. These actions appear to be highly site specific and can influence neurodegeneration at distant sites in the brain.
CONCLUSION
Injection of IL-1β into either the striatum or the cerebral ventricles markedly exacerbates ischemic damage in both the striatum and cortex, whereas IL-1β injected into the cortex does not increase ischemic damage. Thus, we conclude that IL-1 has site- or regional-specific effects on ischemic brain damage and can influence neurodegeneration at distant sites.
Footnotes
Acknowledgements
The authors thank Anthea Hughes and Helen Anforth for technical support.