
Abstract
Glutamate excitotoxicity has been involved in the pathophysiology of epilepsy. Normal functioning of glutamate transporters clears the synaptically released glutamate to prevent excitotoxic neuronal death. Using densitometric immunohistochemical analysis, we examined the temporal expression of the neuronal glutamate transporter (EAAC1) in the lithium-pilocarpine rat model of temporal lobe epilepsy. During the acute period of lithium-pilocarpine-induced status epilepticus, EAAC1 transporter expression increased in the pyramidal neurons of cornus ammonis (CA)1, CA2 and CA3 (fields of the hippocampus), in dentate gyrus (DG) granule cells and in olfactory tubercle (Tu). During the latent period, EAAC1 expression was strongly expressed in the DG granular and molecular layers, Tu, cerebral cortex and septum, and went back to control levels in CA1, CA2 and CA3 layers. The overexpression of EAAC1 occurred mainly in structures prone to develop Fluoro-Jade-B-positive degenerating neurons. It is, however, not clear to what extent the overexpression of EAAC1 contributes to epileptogenesis and in which area it may represent a preventive or compensatory or response to injury.
Keywords
Introduction
In the adult brain, high-affinity Na+-dependent glutamate transporters control the concentration of glutamate in the synaptic cleft and terminate its actions (Danbolt, 2001). A disturbance in glutamate-mediated excitatory neurotransmission has been considered as a critical factor in the etiology of many pediatric and adult forms of epilepsy (Perry and Hansen, 1981; Sherwin et al, 1988; During and Spencer, 1993). Five subtypes of glutamate transporters— L-glutamate/L-aspartate transporter (GLAST), gliaL L-glutamate transporter (GLT-1), Cl~-dependent high-affinity glutamate transporter (EAAC1), excitatory amino acid transporter (EAAT4) and EAAT5 (Danbolt, 200l)—have been identified. A dramatic elevation of extracellular glutamate levels and lethal spontaneous seizures were reported after the loss of the glial transporters, particularly GLT-1 (Rothstein et al, 1996; Tanaka et al, 1997), which is responsible for approximately 80% of the glutamate taken up by GLAST, GLT-1 and EAAC1 in the hippocampus (Rothstein et al, 1996). However, the selective inhibition of GLT-1 appears to be insufficient to lead to glutamate accumulation and hence to N-methyl-D-aspartate receptor-induced toxicity. This suggests a critical role of the other glutamate transporters, like GLAST and/or EAAC1, in the maintenance of extracellular glutamate levels (Selkirk et al, 2005). The role of the neuronal/glial glutamate transporter EAAC1 in epilepsy is not yet clearly understood (Rothstein et al, l994; Coco et al, 1997; Conti et al, 1998; Kugler and Schmitt, 1999). Contradictory data showed that administration of the antisense oligonucleotide of EAAC1 into the rat brain produces epilepsy (Sepkuty et al, 2002) and some motor impairment (Rothstein et al, 1996), whereas EAAC1 knockout mice have reduced spontaneous locomotor activity but do not develop seizures (Peghini et al, 1997). Likewise, in surgical resections from human epileptic structures (Mathern et al, 1999; Meldrum et al, 1999; Tessler et al, 1999; Proper et al, 2002) and in animal models of epilepsy (Miller et al, 1997; Akbar et al, 1998; Ghijsen et al, 1999; Simantov et al, 1999; Doi et al 2000; Ueda and Willmore, 2000; Ueda et al, 2001; Crino et al, 2002), an increase or a decrease of EAAC1 transporter expression has been reported.
Systemic injection of pilocarpine, a non-subtype-specific partial muscarinic agonist, alone or combined with lithium in the rat, is frequently used to produce an animal model of temporal lobe epilepsy (Persinger et al, 1988; Turski et al, 1989; Dubé et al, 2001). After status epilepticus (SE), extended neuronal loss and reactive gliosis occur mainly in forebrain limbic structures, including the hippo-campus, amygdala, septum, thalamus, piriform and entorhinal cortex (Ent) (Turski et al, 1989; Motte et al, 1998; Fujikawa et al, 1999; Dubé et al, 2001). The acute phase of SE is followed by a latent period characterized by cellular reorganization, before the onset of spontaneous recurrent seizures (Turski et al, 1989; Dubé et al, 2001; Roch et al, 2002).
To study the potential contribution of EAAC1 to early epileptogenesis in the lithium-pilocarpine model, we investigated (1) the timing of changes in EAAC1 expression after SE, compared with the pattern of neuronal degeneration, (2) the regional distribution of EAAC1 and (3) the type of cells expressing these transporters.
Materials and methods
Lithium-Pilocarpine-Induced Status Epilepticus
Adult Sprague-Dawley rats (Janvier Breeding Center, Le Genest St-Isle, France) were housed in controlled standard conditions (12 h light/dark cycle, lights on at 0700 h), with food and water available ad libitum. All animal experi-mentation was performed in accordance with the rules of the European Community Council Directive of November 24, 1986 (86/609/EEC) and the French Department of Agriculture (License Number 67-97).
Rats were injected with lithium choride (intraperitoneally, 3 mEq/kg, Sigma, St Louis, MO, USA). About 20 h later, methylscopolamine bromide (subcutaneously, 1 mg/ kg, Sigma) was administered to limit the peripheral effects of the convulsant. Status epilepticus was induced by injecting pilocarpine hydrochloride (subcutaneously, 25 mg/kg, Sigma) 30 mins after methylscopolamine admin-istration. The injection of diazepam (DZP, intramuscu-larly, 2.5mg/kg, Valium, Roche, Lyon, France) 2 h after SE onset allowed to reduce the rats' anxiety, induce muscle relaxation and improve survival, but this dose did not stop the seizures. A lower dose of DZP (1 mg/kg) was administered 12 h after SE onset. For each animal, clinical signs of seizure activity were noted. Briefly, within 5 mins after pilocarpine injection, rats developed diarrhea, piloerection and other signs of cholinergic stimulation.
During the following 15 to 20 mins, rats exhibited head bobbing, scratching, chewing and exploratory behavior. Recurrent seizures started around 20 to 25 mins after pilocarpine administration. These seizures which asso-ciated episodes of head and bilateral forelimb myoclonus with rearing and falling progressed to SE at approximately 35 to 40 mins after pilocarpine injection, as described previously (Persinger et al, 1988; Dubé et al, 2001; Roch et al, 2002). For the study of the acute phase, rats (n = 3) were killed at 24 h after the onset of SE. Most often, the rats killed at 24 h after the onset of SE were still exhibiting occasional seizure activity. For the study of the latent phase, rats (n = 3) were killed at 144 h after SE. Control rats received the lithium-methylscopolamine treatment and saline instead of pilocarpine, and only a single DZP injection at 2 h after saline injection. Controls rats (n =3) were killed at 144 h.
Immunohistochemistry
Animals were deeply anesthetized with pentobarbital (1.8 g/kg, Dolethal®, Vetoquinol, Lure, France,) and perfused transcardially with ice-cold phosphate buffer (PB, 0.1 mol/L, pH 7.4), followed by ice-cold fixative (4% paraformaldehyde in 0.1 mol/L PB, pH 7.4). Brains were removed immediately after perfusion, postfixed in the same fixative one night at 4°C and impregnated with 20% sucrose diluted in PB for cryoprotection. Transverse 40 μm sections were cut from frozen blocks, collected in PB and stored at −20°C in an antifreeze solution (0.1 mol/L PB, 30% ethylene glycol, 30% glycerol).
For immunohistochemical staining, sections were pretreated with 0.3% hydrogen peroxide in PB for 15 mins at room temperature and then rinsed several times in PB and in PB saline (PBS, 0.02 mol/L, pH 7.4). Sections were serially incubated in 5% normal rabbit serum (Vector ABC kit, Vector Laboratories, Burlingame, CA, USA) in PBS for 1 h, the primary antibody EAAC1 (goat polyclonal antibodies, Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted (1/500) with 2% normal rabbit serum and 0.3% Triton in PBS for 12 h at room temperature, the secondary antibody (biotinylated rabbit anti-goat immunoglobulin G (IgG), Vector ABC kit) diluted (1/200) with 0.3% Triton in PBS for 1 h at room temperature, and then avidin-biotin complex diluted (1/200) with 0.3% Triton in PBS for 1 h at room temperature (Vectastain ABC kit, Vector). Diaminobenzidine tetrahydrochloride with hydrogen peroxide (diaminobenzidene, Biosys kit, Vector) was used as the peroxidase substrate to visualize sites of antibody binding. The sections were observed with a Nikon microscope (Diaphot 300, Hamanatsu C5 985 chilled CCD Camera) and Visilog Software (Noesis, France). The distribution of positive cells in the brain was mapped and the regional density of EAAC1 was estimated. The specificity of the antibodies against EAAC1 was controlled by incubating the slides without primary antibody, which led to lack of protein expression (data not shown). Image analysis of immunohistochemistry was performed on a PC computer using the Image J software. The optical densities were obtained from four fields taken bilaterally in each structure of interest and then averaged for three rats.
For double immunofluorescence staining of EAAC1/glial fibrillary acidic protein (GFAP) (Sigma), EAAC1/ microtubule-associated protein (MAP)-2 (Sigma), coronal free-floating sections were incubated in 5% normal horse serum in PBS for 1 h, the mixture of primary antibodies (diluted at 1/250) in 2% normal horse serum (Vector ABC kit) and 0.3% Triton in PBS for 12 h at room temperature, the secondary antibody (anti-goat IgG Cy3 conjugate and anti-rabbit or anti-mouse IgG fluorescein isothiocyanate conjugate, Sigma) diluted (1/250) with 0.3% Triton in PBS for 1 h. Sections were observed with a confocal laser-scanning microscope (BioRad MRC 1024, Hertfordshire, UK). Images were acquired using double excitation at 488 and 568 nm and were recorded through separate channels. Serial plane images were collected at a given depth or throughout the thickness of the fluorescent preparation.
Fluoro-Jade-B Staining
The sections were rinsed, mounted onto slides and dried. Slides were immersed in 100% ethanol for 3 mins, followed by 1 min in 70% ethanol and 1 min in distilled water. Slides were then transferred to a gently shaken solution of potassium permanganate for 15 mins. After rinses, sections were placed in Fluoro-Jade-B (Histo-Chem Inc., Jefferson, AR, USA) staining solution during 30 mins at room temperature. After staining, the slides were rinsed in distilled water, dried and immersed in 100% ethanol for 3 mins, in toluene for 3 mins and mounted. Sections were examined with an immunofluorescence microscope (Nikon, Diaphot 300, Hamanatsu C5 985 chilled CCD Camera) and Visilog Software (Noesis, France).
Statistical Analysis
Statistical analysis for the difference of the regional changes between control and lithium-pilocarpine rats was performed using the SPSS® 8.0 software. Values were expressed as means ± s.e.m. Data were analyzed using one-way analysis of variance (ANOVA). If the F ratios were significant, post-hoc Dunnett's multiple-comparison tests were applied to assess significance, which was set at P < 0.05.
Results
In control brains, the EAAC1 distribution pattern was in accordance with previous studies (Rothstein et al, 1994). EAAC1 was present mainly in the hippocampus (Figure 1A), olfactory tubercle (Tu) (Figure 3A), cerebral cortex (Figure 5A) and septum (Figure 6A). No detectable Fluoro-Jade-B staining was seen in control animals (Figures 1D, 5D and 6D).
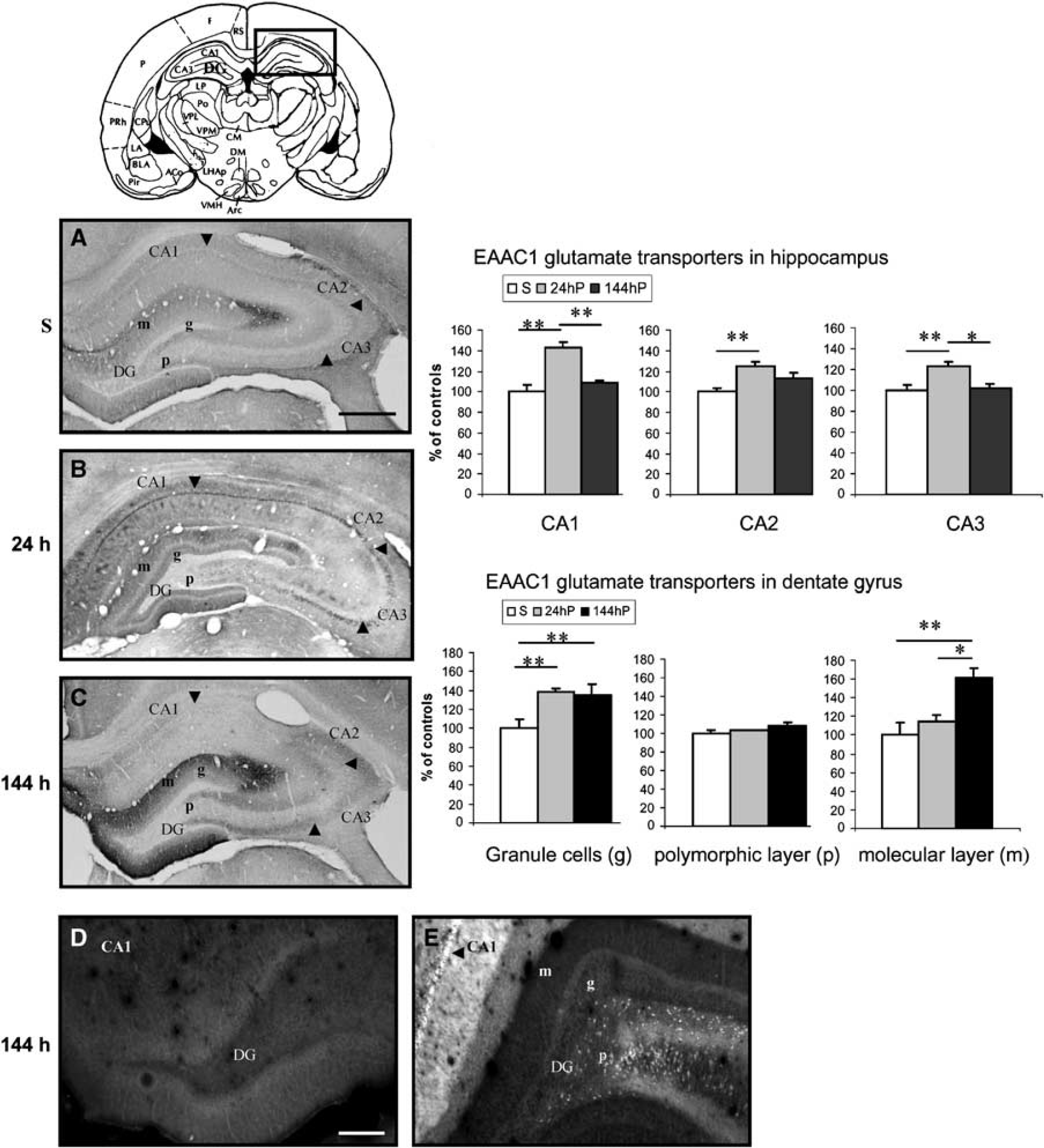
Composite drawing of representative coronal sections taken at the level of hippocampus (Paxinos and Watson, 1986) showing the expression of EAAC1 immunoreactivity at 24 and 144 h after SE. (
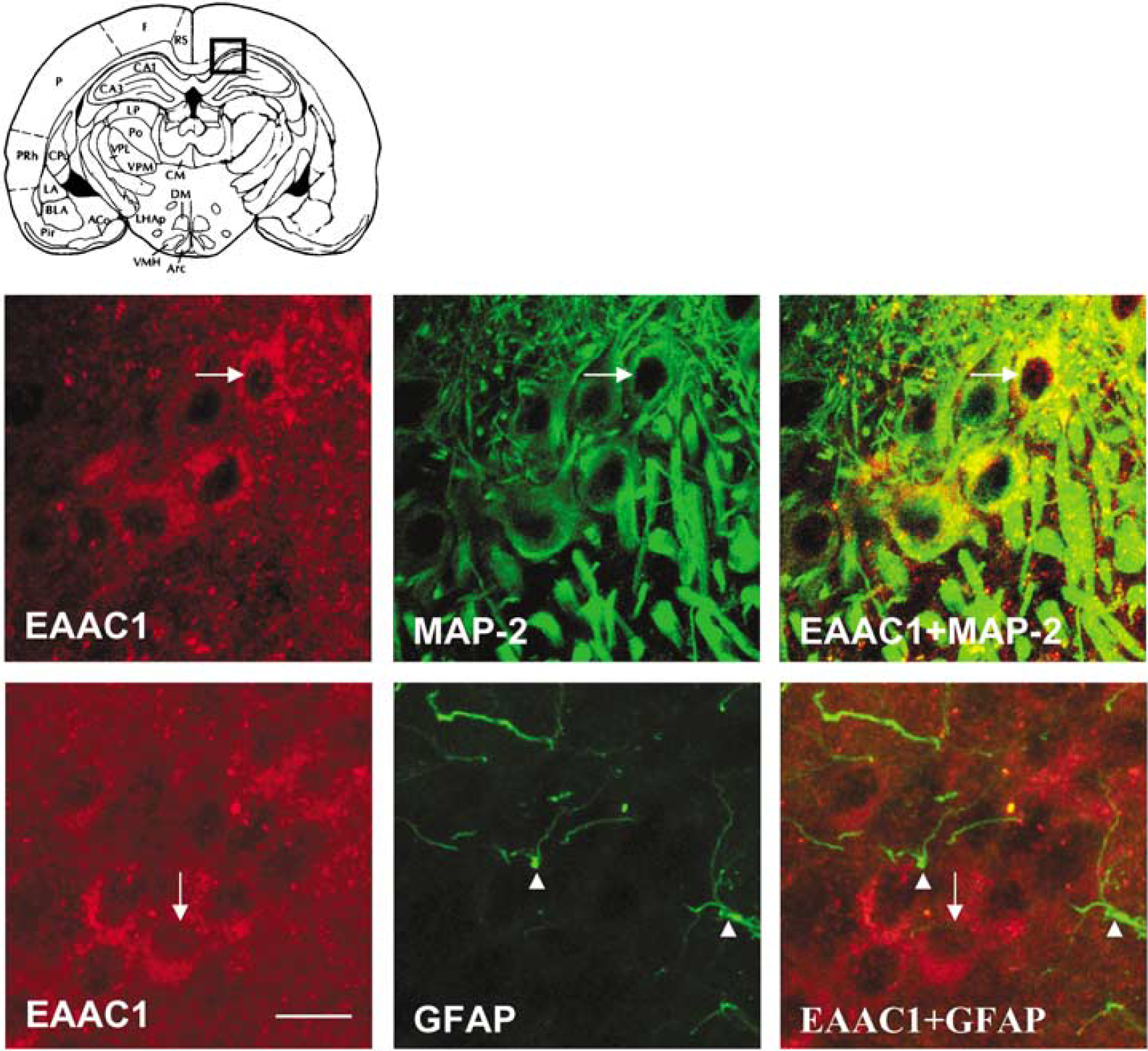
Composite drawing of representative coronal sections taken at the levels of hippocampus (Paxinos and Watson, 1986) and showing the phenotype of EAAC1 expression in the CA1 hippocampal pyramidal cell layer at 24 h after SE. Double immunofluorescence labelling with anti-EAAC1 (red) and anti-GFAP (green) (scale bar: 21.5 mm) or anti-MAP-2 (scale bar: 21.5 mm) shows the expression of EAAC1 in neurons (arrow) and not in astrocytes (arrow), respectively.
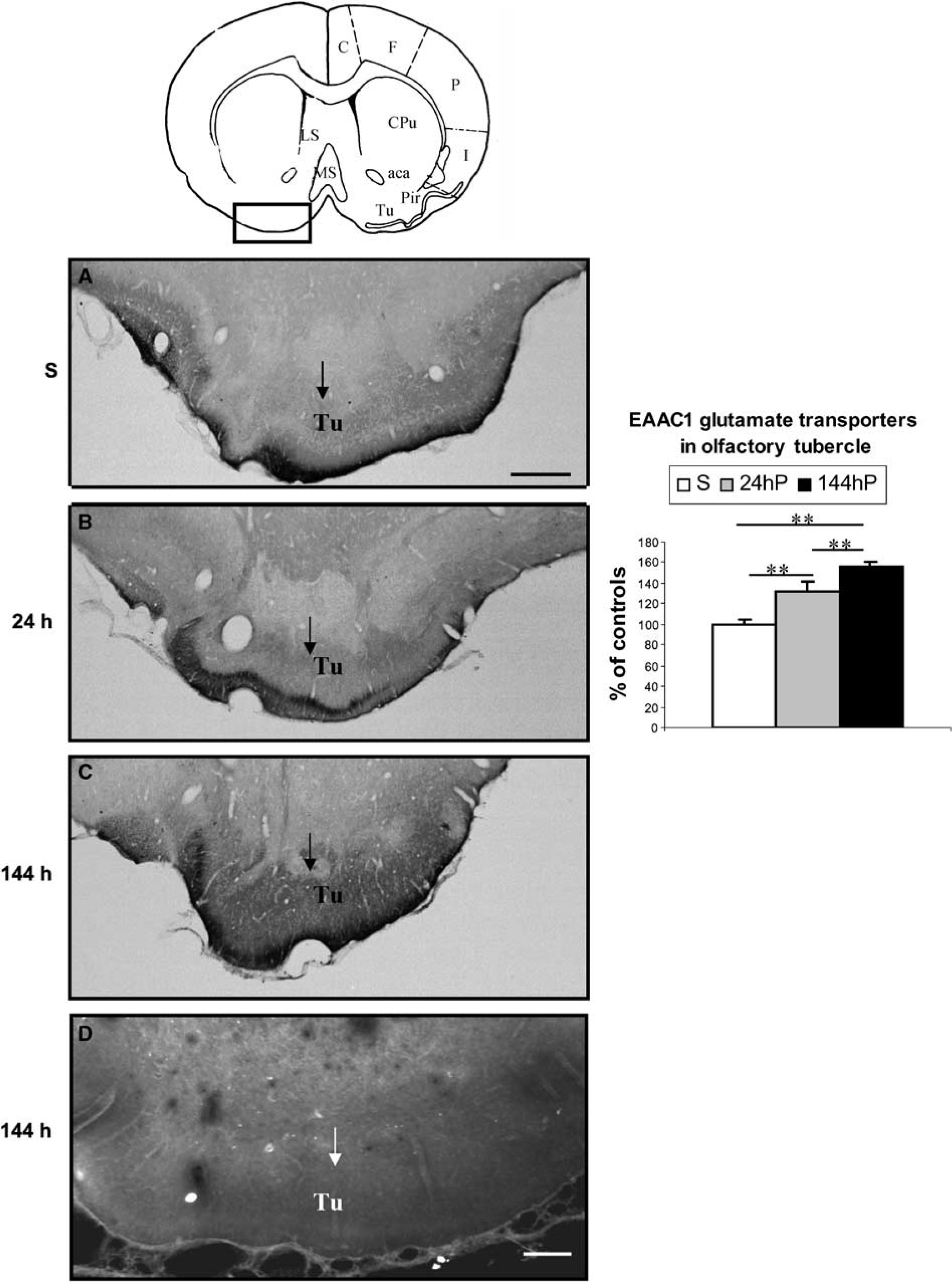
Composite drawing of representative coronal sections taken at the levels of Tu (Paxinos and Watson, 1986) showing the expression of EAAC1 immunoreactivity at 24 and 144 h after SE (
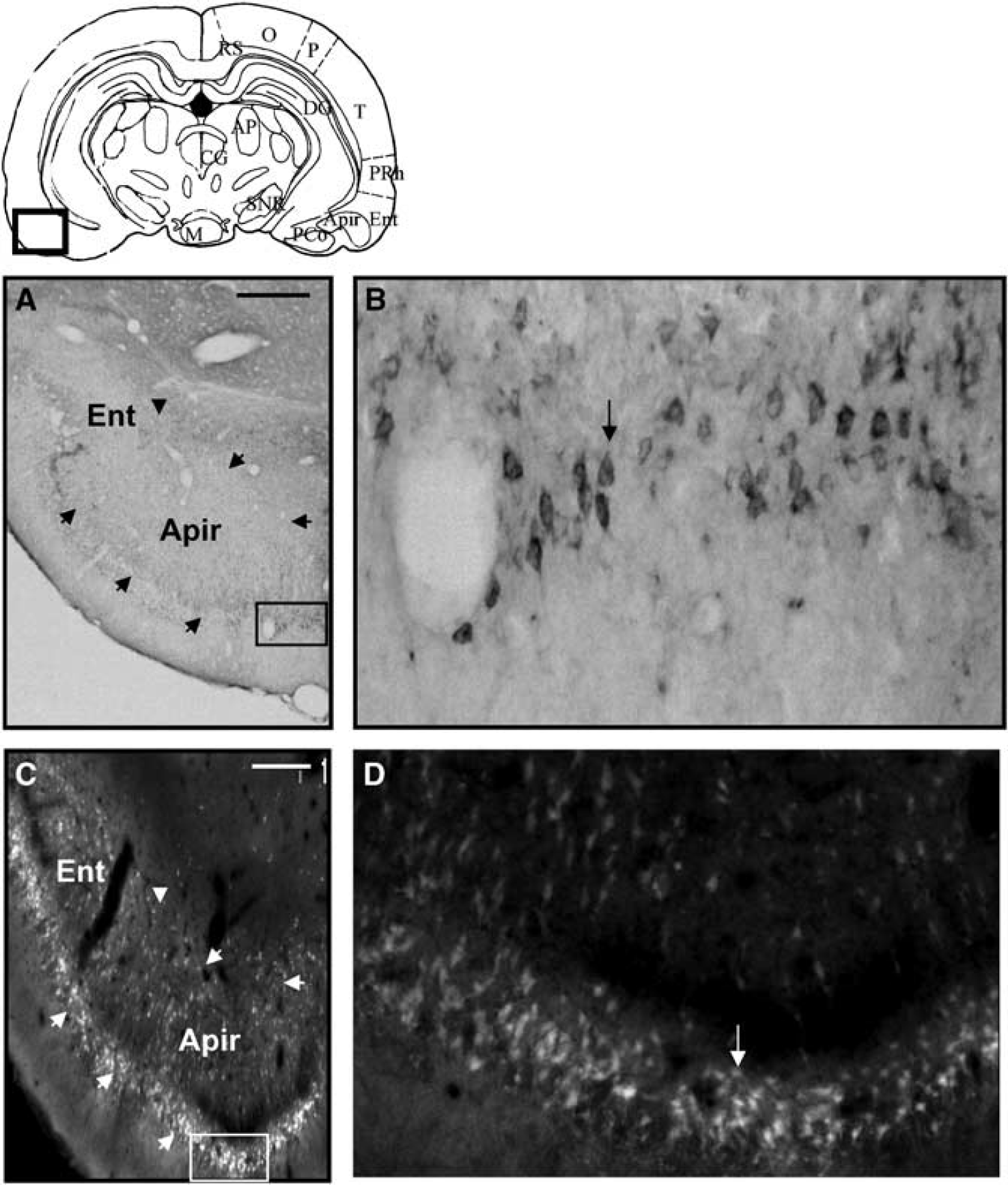
Composite drawing of representative coronal sections taken at the levels of Ent (Paxinos and Watson, 1986), showing the expression of EAAC1 immunoreactivity (
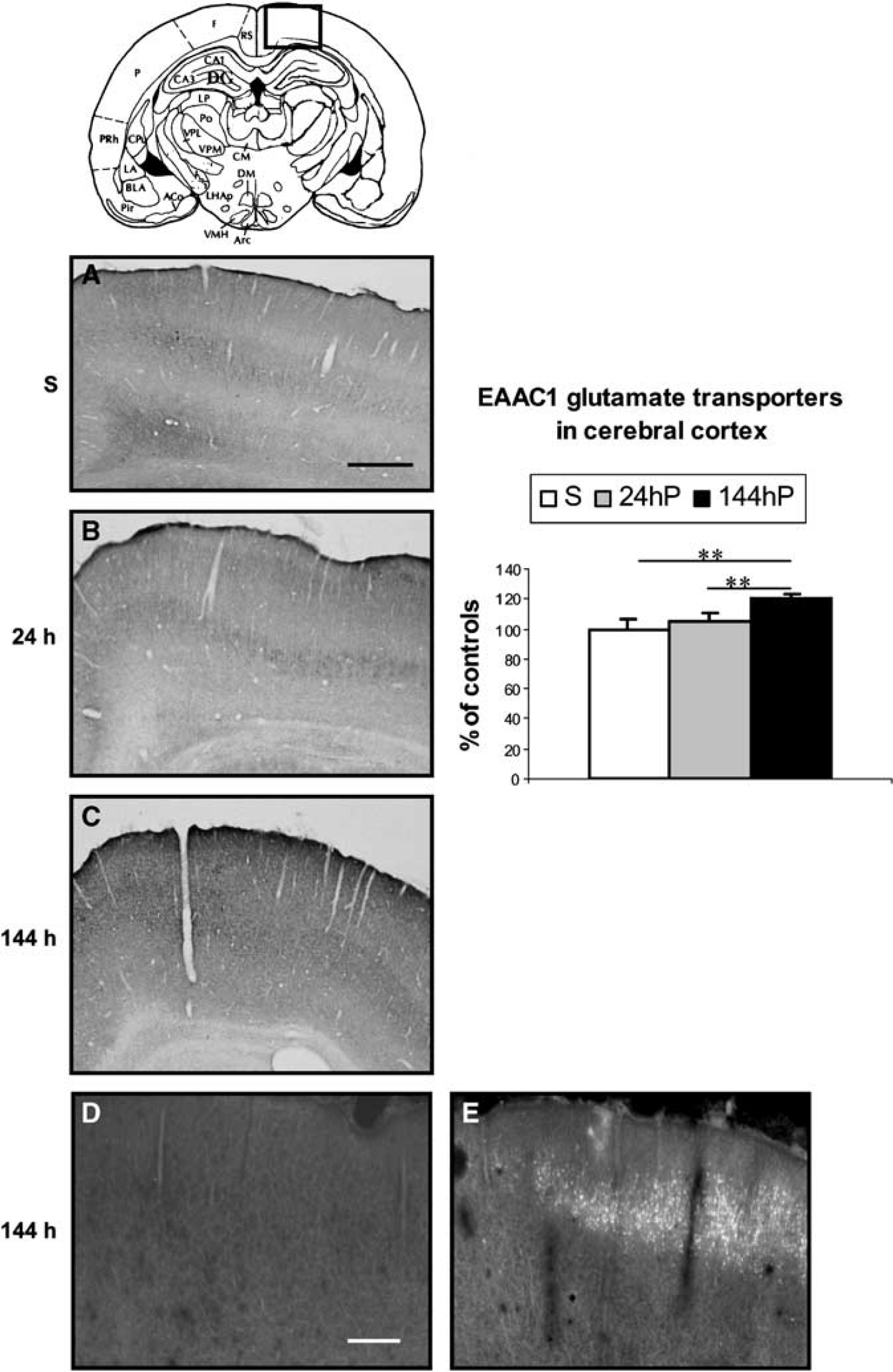
Composite drawing of representative coronal sections taken at the level of neocortex (Paxinos and Watson, 1986), showing the expression of EAAC1 immunoreactivity at 24 and 144 h after SE (
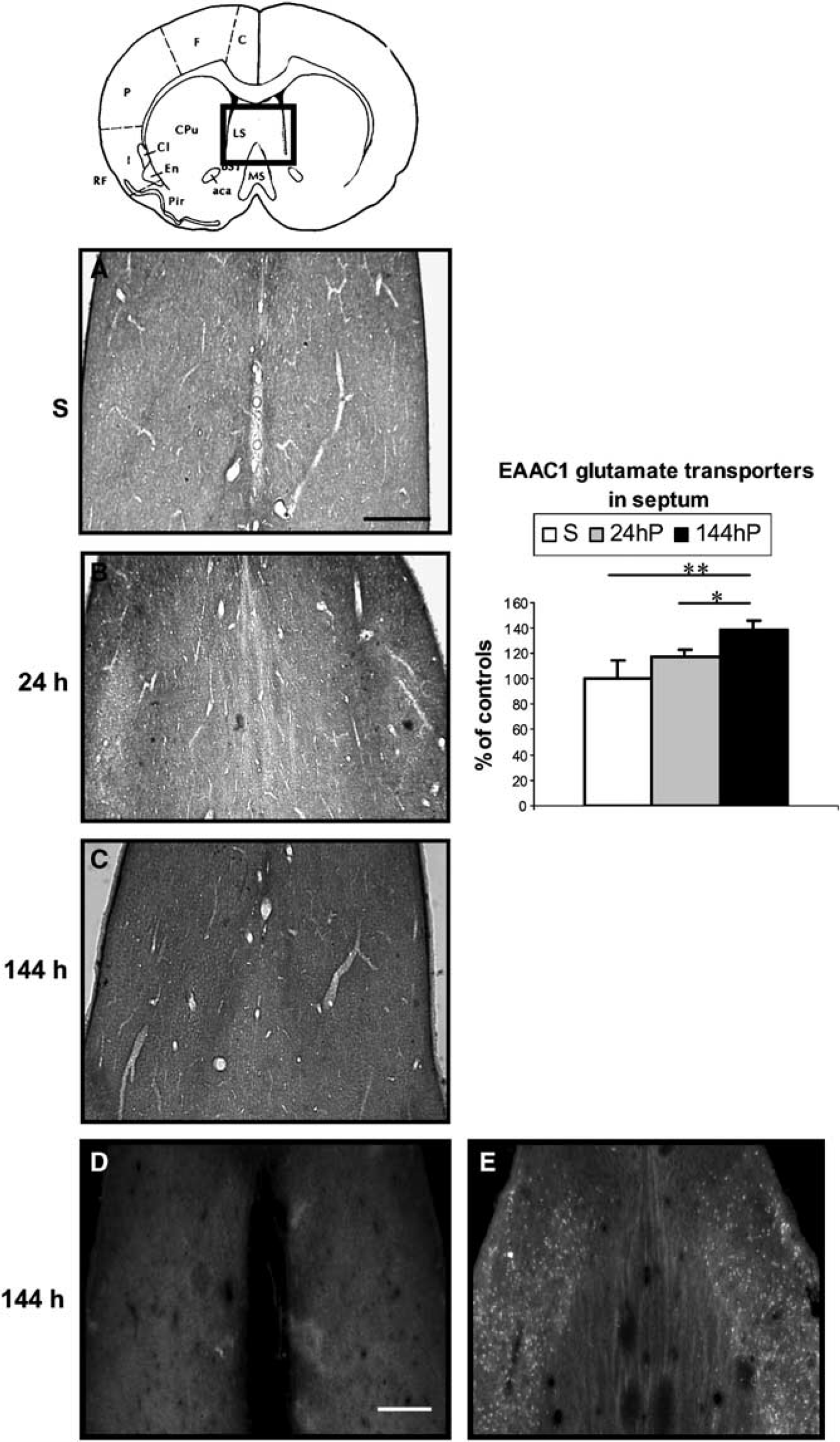
Composite drawing of representative coronal sections taken at the level of the septum (Paxinos and Watson, 1986), showing the expression of EAAC1 immunoreactivity at 24 and 144 h after SE (
Acute Period
During the acute period (24 h after SE), EAAC1 immunostaining increased in cornus ammonis (CA)1, CA2 and CA3 (fields of the hippocampus) pyramidal cell layers of the hippocampus (Figure 1B) by 43%, 25% and 23%, respectively, and in dentate granule cells by 39% (P < 0.01) compared with control rats (Figure 1A). EAAC1 immunoreactivity in CA1 was colocalized with MAP-2, but not with GFAP (Figure 2), showing that the induction of the expression of the EAAC1 protein by SE was exclusively neuronal. The distribution of EAAC1 in the hippocampus was similar to that of Fluoro-Jade-B-stained neurons, with the exception of the polymorphic layer of the dentate gyrus (DG), in which EAAC1 was not upregulated while Fluoro-Jade-B staining was increased (data not shown). In addition, EAAC1 immunoreactivity was also in-creased by 31% (P < 0.01) in the Tu (Figure 3B) compared with control rats (Figure 3A). However, there was no Fluoro-Jade-B expression in Tu (data not shown). In amygdala, entorhinal and piriform cortices, edema was present 24h after lithium-pilocarpine injection (Figure 4A). Some EAAC1 immunopositive cells were present at the periphery of the edema, but their low number did not allow quantification (Figure 4B). Fluoro-Jade-B-positive neurons were located within and at the periphery of the edema (Figures 4C and 4D).
Latent Period
During the latent period (144 h after SE), the immunoreactivity of EAAC1 was unchanged in hippocampal CA1, CA2 and CA3 pyramidal cell layers (Figure 1C) compared with control rats (Figure 1A). The EAAC1 expression decreased by
34% (P < 0.01) in CA1 and CA3 and by 21%(P < 0.05) compared with the acute period. Fluoro-Jade-B-positive neurons were still present in CA1, CA2 and CA3 pyramidal cell layers (Figure 1E). The expression of the transporter was increased in dentate granule and molecular layers by 35% and 61%, respectively (P < 0.01) compared with control rats. EAAC1 immunoreactivity in the molecular layer increased by 46% (P < 0.05) compared with the acute period. As this time point, there was no Fluoro-Jade-B expression in the dentate granular and molecular layers, whereas Fluoro-Jade-B-positive neurons were present in the polymorphic layer (Figure 1E). In the Tu (Figure 3C), cerebral cortex (Figure 5C) and septum (Figure 6C), EAAC1 expression was increased by 56%, 21% and 39%, respectively (P < 0.01), over control levels (Figures 3A, 5A and 6A) and by 25%, 16% (P < 0.01), and 22% (P < 0.05), respectively, compared with the acute period. With the exception of the Tu, Fluoro-Jade-B neuronal labelling paralleled the expression of EAAC1 in the cerebral cortex (Figure 5E) and septum (Figure 6E).
Discussion
Prolonged seizures induced by lithium-pilocarpine are triggered by the binding of the muscarinic receptor agonist, pilocarpine, to its receptors. This first phase is rapidly followed by secondary glutamatergic generalization (Smolders et al, 1997). Moreover, the distribution of neuronal death coincides with that of glutamatergic receptors reflecting mostly excitotoxic mechanisms (Olney et al, 1986).
In the present study, the expression of EAAC1 was mainly increased during the acute and latent phase. This enhanced expression was occurring in various types of structures and situations. First, in the Tu, the increased expression of EAAC1 correlates with the cholinergic onset of lithium-pilocarpine seizures, since the latter structure contains a high density of muscarinic receptors (Rotter et al, 1979). This is in accordance with the observation that isolated focal seizures induced by moderate doses of pilocarpine lead to Fos expression in the Tu (Barone et al, 1993). Second, EAAC1 expression was upregulated in structures prone to develop Fluoro-Jade-B-positive degenerating neurons like CA1, CA2 and CA3 pyramidal cell layers of the hippocampus, septum and cerebral cortex (Motte et al, 1998; Fujikawa et al, 1999; Covolan and Mello, 2000; Dubé et al, 2001; Roch et al, 2002, Voutsinos-Porche et al, 2004). Third, in a few structures such as the granular and molecular layers of the DG, enhanced EAAC1 expression was not paralleled by Fluoro-Jade-B staining and neuronal death. Finally, in structures prone to develop very rapid damage, like amygdala nuclei, piriform and Ent, and the polymorphic layer of the DG (Motte et al, 1998; Andre et al, 2001; Roch et al, 2002), no increase in EAAC1 expression could be detected. In amygdala and parahippocampal cortices that developed marked edema, EAAC1-positive cells were present mostly at the periphery of the edema and a few neurons remaining within the edema were positive for EAAC1.
EAAC1 expression is naturally high in hippocampus, cerebral cortex, septum and amygdala (Rothstein et al, 1994; Conti et al, 1998; Wang et al, 1998). In accordance with this regional distribution, EAAC1 transporter expression increased in pyramidal neurons of CA1, CA2, CA3 areas, DG granule cells, cerebral cortex and septum during the acute and/or latent period of lithium-pilocarpine-induced SE. Likewise, EAAC1 expression is increased in hippocampal tissue of patients with temporal lobe epilepsy (Mathern et al, 1999; Proper et al, 2002). Our data confirm that the primary cholinergic stimulation followed by the secondary glutamatergic generalization activates EAAC1 expression mainly in regions involved in the circuit of seizures and epileptogenesis (Lothman, 1994). Moreover, the present data agree with previous reports that seizures induced by kainate, pilocarpine and kindling elicit the expression of EAAC1 mainly in regions involved in the circuit of the seizures (Miller et al, 1997; Ghijsen et al, 1999; Mathern et al, 1999; Ueda and Willmore, 2000; Crino et al, 2002; Furuta et al, 2003).
Glutamate transporters are considered to be crucial for preventing accumulation of neurotoxic levels of extracellular glutamate (Danbolt, 2001). Sepkuty et al (2002) reported that rats treated with EAAC1 antisense protein develop epilepsy and limbic hyperexcitability, and that this hyperexcitability may be partly attributable to a reduction in new GABA synthesis. Thus, increased EAAC1 levels observed during the acute and latent period in the lithium-pilocarpine model might be an important compensatory change that both enhances clearance of glutamate from the synaptic cleft and may serve to enhance inhibitory synaptic transmission by increasing GABA production. This upregulation of EAAC1 could be sufficient to prevent neuronal death in the Tu, and granular and molecular layers of the DG, where no degenerating cells or axon terminals were detected 24 h after the injection of lithium-pilocarpine, in accordance with previous studies (Turski et al, 1989; Persinger et al, 1988; Motte et al, 1998; Andre et al, 2001; Roch et al, 2002). In all these structures, the upregulation of EAAC1 persisting up to 144 h after the onset of SE could be a protective mechanism against the potentially deleterious consequences of seizure-enhanced glutamatergic neurotransmission.
A second situation is encountered in CA1-CA3 pyramidal cell layers of hippocampus, septum and neocortex. In these regions, the upregulation of the EAAC1 transporter coexists with degenerating neurons, indicating that the increased expression of the transporter was not sufficient to prevent neuronal death. This situation was even more pronounced in the polymorphic layer of the DG, amygdala and parahippocampal cortices, where neuronal death is rapid (Andre et al, 2001; Roch et al, 2002). In the first region, no upregulation of the glutamate transporter could be detected and in the latter two ones, almost no expression of EAAC1 was present within the edema, where most likely the majority of neurons has already disappeared by 24 h after SE, as reported previously (Roch et al, 2002). However, in these regions, overexpression of the transporter could be detected at the periphery of the edema. In that case, perinuclear deposits of EAAC1 protein were found mainly in neurons with a reduction of Golgi staining at 24 h after SE. Perinuclear EAAC1 immunoreactivity corresponds to the translocation to the Golgi complex (Furuta et al, 2003). EAAC1 internalization might be important for the rapid regulation of high-affinity glutamate uptake, preventing the excessive glutamate uptake during epileptic seizures. Alternately, increases in EAAC1 expression can also be deleterious, promote neuronal death and hence epileptogenesis by increasing extracellular glutamate levels by reversed glutamate transport. Glutamate transporters have been reported to reverse readily if the driving forces such as ion gradients and membrane potential are compromised (Nicholls and Attwell, 1990). For example, transporter-mediated glutamate homeostasis fails dramatically in ischemia; instead of removing extracellular glutamate to protect CA1 pyramidal neurons, transporters act as a site of efflux of glutamate, triggering neuronal death. This reversible glutamate transport is mainly associated with EAAC1 (Rossi et al, 2000). Whether this reversal of EAAC1 transporter function occurs in the present model of SE is not known, but if it is the case, it could contribute to the very rapid neuronal degeneration found in the polymorphic layer of the DG, amygdala, piriform and entorhinal cortices (Andre et al, 2001; Roch et al, 2002 and the present study).
EAAC1 is strongly expressed in the granular and molecular cell layers of the DG, whereas no Fluoro-Jade-B staining is seen in cells or axon terminals. These layers contain the terminations of the glutamatergic perforant path (up to 95% of the fibers) (Ottersen and Storm-Mathisen, 1989), which origi-nates in the Ent (Hjorth-Simonsen and Jeune, 1972). The lack of degeneration in the granular and molecular cell layers of the DG may reflect the strong decrease of most glutamatergic terminals resulting from widespread seizure-induced neurodegeneration within the Ent (Motte et al, 1998; Dubé et al, 2001; Roch et al, 2002 and the present study).
Together with the neuronal transporter glutamate EAAC1, a failure of the two glial glutamate transporters, GLT-1 and GLAST, could also be involved in neurodegeneration triggered by seizure-induced excitotoxicity. Indeed, the uptake of glutamate by astrocytes is the main pathway for clearing glutamate from the synaptic cleft. This pathway is part of the well-described glutamate-glutamine cycle allowing the uptake of glutamate from the synaptic cleft, its conversion into glutamine in the astrocytes and the shuttling of glutamate skeletons back to neurons (Berl and Clarke, 1983; Sonnewald et al, 2005). By 24 h after SE, preliminary studies showed that the expression of GLAST and GLT-1 was not changed in the same brain regions as those where the expression of EAAC1 was increased (Voutsinos, unpublished data). These data are in agreement with previous studies reporting that, 16-24 h after the onset of SE, the expression of GLAST and GLT-1 was slightly reduced or unchanged, but never increased (Simantov et al, 1999; Doi et al, 2000). Thus, the neurodegeneration induced by prolonged lithium-pilocarpine seizures seems to depend on a lack of adaptation of all glutamate transporters, both neuronal and glial.
Mossy fiber sprouting is a characteristic of human (Sutula et al, 1989) and animal models of temporal lobe epilepsy (Mello et al, 1993; Mathern et al, 1998b). During the latent or reorganization phase, cell loss in the polymorphic layer of the DG leads to the loss of their synaptic contacts by mossy cells. These cells send collateral fibers that cross the granule cell layer to connect to their own dendrites in the inner molecular layer. This remodelling generates new circuits underlying the occurrence of chronic seizures (Mathern et al, 1998a). Although sprouting has been mostly described in the supragranular layer (De Lanerolle et al, 1989; Houser, 1990), it has also been reported in the infrapyramidal layer (Babb et al, 1991, 1992). The overexpression of EAAC1, recorded at 144 h after SE, mainly at the level of the DG granule cells and molecular layer and in the pyramidal cell layers of CA1 and CA3, could participate in this plasticity, in the same way as enhanced EAAC1 expression was reported to be associated with axotomy-induced nerve regeneration (Kiryu et al, 1995).
In conclusion, our data suggest that selective increases in EAAC1 glutamate transport occur in association with epileptogenesis. Their density and localization in specific brain regions may have critical implication in neuronal death and reorganization. A pivotal question for further studies is whether these changes contribute to epileptogenesis or are a compensatory response to neuronal injury.