
Abstract
Although p53 controls cell death after various stresses, its role in neuronal death after brain ischemia is poorly understood. To address this issue, we subjected p53-deficient (p53−/− and p53+/−) mice (backcrossed for 12 generations with C57BL/6 mice) and wild-type mice (p53+/+) to transient global ischemia by the three-vessel occlusion method. Despite similar severity of ischemia, as shown by anoxic depolarization and cortical blood flow, neuronal death in the hippocampal cornus ammonis (CA)1 region was much more extensive in p53+/+ than in p53−/− mice (surviving neuronal count, 9.3% ± 3.0% versus 61.3% ± 34.0% of nonischemic p53+/+ controls, respectively, P < 0.0037). In p53+/− mice, a similar trend was also observed, though not statistically significant (43.5% of nonischemic p53+/+ controls). In p53+/+ mice, p53-like immunoreactivity in hippocampal CA1 neurons was enhanced at 12 h after ischemia, and messenger ribonucleic acid for Bax, a direct downstream target of p53, was also increased. These results indicate that p53 potentiates ischemic neuronal death in vivo and suggest that this molecule could be a therapeutic target in neuronal death after cerebral ischemia.
Introduction
Brain ischemia is a leading cause of death and disability in the aged population. Attempts to protect the brain against ischemia in the clinical setting, however, have not been successful, owing to the complex mechanisms of ischemic injury. To better understand the pathophysiology involved, animal models of transient global ischemia have been used in which the most vulnerable neurons in the cornus ammonis (CA)1 region of the hippocampus are selectively degenerated (Kirino, 1982; Pulsinelli et al, 1982; Smith et al, 1984). Even in this model, which causes uniform, delayed neuronal injury in the hippocampus, the mechanisms of ischemic neuronal injury remain largely unknown.
The apoptotic pathway modulator p53 might be a key molecule in ischemic brain injury. In response to genotoxic or oxidative stress, p53 regulates cell-cycle arrest and deoxyribonucleic acid repair, as well as cell death, by controlling many downstream molecules (Gotz and Montenarh, 1996; Levine, 1997). In several studies, cerebral ischemia led to upregulation of p53 messenger ribonucleic acid (mRNA) or protein (Li et al, 1994, 1997; McGahan et al, 1998; Tomasevic et al, 1999a). One study showed attenuation of ischemic injury in p53-deficient mice subjected to focal ischemia (Crumrine et al, 1994), but others showed exacerbation of injury (Maeda et al, 2001). These contradictory results may reflect differences in ischemic stress caused by genetic modification of the architecture of collateral flow in these mutants. In vivo evidence for a crucial role of p53 in an apoptotic pathway leading from oxidative stress to neuronal death is still lacking (Love, 2003).
To obtain such evidence, we evaluated pyramidal cell damage in the CA1 region in p53-deficient mice subjected to global cerebral ischemia according to our recently described method (Yonekura et al, 2004). Since this method yields highly reproducible and consistent CA1 neuronal injury, as compared with other models (Kitagawa et al, 1998; Wellons et al, 2000), comparison of ischemic outcomes in wild-type and p53-deficient mice could show a link between p53 and ischemic neuronal injury in vivo.
Materials and methods
Animal Preparation
All mice, aged 8–12 weeks, were purchased from Taconic (Germantown, NY, USA), and only male animals were used in this study. Mice lacking one (heterozygous or p53+/−) or both p53 alleles (homozygous or p53−/−), backcrossed for 12 generations onto the C57BL/6 background (p53-C57BL/6Ntac-Trp53tm1 N12), were used in this study (Donehower et al, 1992). As controls, mice carrying both alleles (wild-type or p53+/+) derived from the heterozygous mice were used (C57BL/6tac-Trp53tm1 N5). All animal-related procedures were conducted in accordance with the guidelines of the University of Tokyo.
Global Cerebral Ischemia
Global cerebral ischemia was induced as described (Yonekura et al, 2004). Briefly, the day before ischemia, a 3.0-mm vinyl tube (inner diameter, 1.0mm; outer diameter, 3.0 mm) was attached to the skull bilaterally with cyanoacrylate 1 mm superior and posterior to the root of the zygomatic arch for measurement of cortical microperfusion by laser Doppler flowmetry. On the day of ischemia, anesthesia was induced and maintained with 1.5% halothane in 30% O2/70% N2O, and laser Doppler microperfusion probes were inserted to the guide tube bilaterally. Atropine sulfate (0.25 mg/kg, intraperitoneally) and amikacin (20 mg/kg, intraperitoneally) were administered, and rectal and temporal muscle temperatures were maintained at 37.0°C for up to 30 mins after ischemia. A basilar artery and both common carotid arteries were occluded for 14 mins. After wound closure, the mice were returned to their cages and placed in an incubator set at 33.0°C and 50% humidity. Acetate Ringer's solution (0.5 mL) was administered subcutaneously in all mice 30 mins and 24 h after ischemia.
Physiologic Measurements
In a separate group of p53+/+ (n = 6) and p53−/− mice (n = 5) subjected to ischemia, blood pressure was measured through a PE-10 cannula inserted into the left femoral artery. Arterial blood samples were analyzed for pH, PaCO2, PaO2, and glucose before ischemia, 8 mins after induction of ischemia, and after 15mins of reperfusion.
Measurement of Direct Current Potential
In another group of p53+/+ (n = 5) and p53−/− (n = 6) mice subjected to ischemia, hippocampal direct current (DC) potential was measured through silver chloride wires and a glass micropipette electrode (tip diameter, 10–20 μm) inserted into the right hippocampus 1.9 mm posterior to the bregma, 1.2 mm lateral from the midline, and 1.2 mm below the dura (Paxinos and Franklin, 1997). A reference electrode was placed in a neck muscle (Kawahara et al, 1995).
Histologic Evaluation
Mice (n = 12/genotype) were subjected to ischemia by investigators who had no prior knowledge of the animals' genotype. After 4 days, the surviving p53+/+ (n = 7), p53+/− (n = 11), and p53−/− mice (n = 9) were anesthetized with 4% halothane and perfused transcardially with 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4. After postfixation for several hours, the brains were removed, embedded in paraffin, cut into 4-μm-thick coronal sections containing the dorsal hippocampi, and stained with cresyl violet. Intact neurons in the CA1 region were counted in masked fashion, and the average neuronal density calculated from both hemispheres (cells/mm) was used to evaluate the severity of ischemic injury.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling Staining
Paraffin-embedded brain sections of p53+/+ and p53−/− mice (n = 2 each) subjected to ischemia were processed for histologic evaluation of neuronal injury in the CA1 region. After deparaffinization, the sections were treated with proteinase K, and fragmented deoxyribonucleic acid (DNA) was visualized by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL; Roche Diagnostics, Mannheim, Germany).
Immunohistochemistry
At 6 (p53+/+, n = 2) and 12 h (p53+/+ and p53−/−, n = 2 each) after ischemia, the mice were anesthetized with 4% halothane and perfused transcardially with ice-cold 4% paraformaldehyde in 0.01 mol/L phosphate-buffered saline, pH 7.4. After fixation, the brains were cryoprotected with sucrose and embedded in ornithine carbomyltransferase compound (Tissue-Tek, Torrance, CA, USA). The same procedures were performed in nonischemic p53+/+ controls (n = 2).
The primary antibody was rabbit polyclonal antibody against wild-type p53 (1:1000; NCL-p53-CM5p, Novocastra Laboratories, Newcastle, UK; Botchkarev et al, 2001). The secondary antibody was affinity-purified biotinylated anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA, USA). Staining was visualized with a combination of colorimetric substrates for peroxidase (ImmunoPure Metal Enhanced DAB Substrate Kit; Pierce, Rockford, IL, USA). Some sections were counterstained with methyl green before mounting.
Quantitative Assessment of Messenger Ribonucleic Acid for p53 and Bax
Bilateral whole hippocampi were dissected at 4°C after decapitation under 4% halothane anesthesia (nonischemic controls, 12, and 24 h after ischemia; n = 5–6/group). The hippocampi were homogenized, and total RNA was isolated with RNeasy mini kits (Qiagen, Germany). Complementary DNA (cDNA) was synthesized from total RNA with ReverTra Ace-α (Toyobo, Osaka, Japan). Quantitative real-time polymerase chain reaction (PCR) was performed with the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). A reaction tube contained forward primer (0.2–1 μL, 10 nmol/mL), reverse primer (0.2–1 μL, 10 nmol/mL), cDNA (1 μL), distilled water (7–9 μL), and SYBR Green Master Mix (10 μL; Applied Biosystems) with a total volume of 20 μL. The appropriate primer concentration was used for each primer. The PCR program consisted of 5 mins at 95°C and then 40 cycles of 30 secs at 95°C, 30 secs at 55°C, and 1 min at 72°C. The following primers were used. β-actin: forward, GCTCTT TTCCAGCCTT CCTT; reverse, TGATCCACATCTGCTGGAAG. Bax: forward, TCTAGATGCAGAGGATGAT TGC TGAC; reverse, GAATTCACTCCAGCCAC AAAGATGGT. For p53, primers that encompass the neo cassette were used as described previously by Donehower et al (1992) to confirm the absence of normal mRNA: primer 4b, GGGACAGCCA AGTCTGTTATGTGC, and primer 6, CTGTCTTCCAGATA CTCGGGATAC. However, a significant amount of primer dimers was also amplified when these primers were used when subjected to quantitative PCR. We therefore designed a new set of primers which purely amplifies the downstream portion of the neo cassette for quantitative PCR: forward, TATCTAGATGGAAGACTCCAGTGGGAAC, and reverse, GTGAATTCCTGTAGCATGGGCATCCTTT. To confirm appropriate amplification, the size and sequence of PCR products were verified by agarose gel separation and analysis with an ABI PRISM 310 Genetic Analyzer (Applied Biosystems). To allow quantitative comparisons among multiple samples, p53 and Bax mRNA levels were normalized according to expression levels of internal control (β-actin) of each sample.
Statistical Analysis
All values are expressed as mean ± s.d. The cell counts (neuronal density) in the CA1 region, cerebral blood flow (CBF), p53 mRNA data were compared by one-way analysis of variance (ANOVA) followed by Bonferroni's post-hoc comparison (Dr SPSS II for Windows, 11.0.1J, SPSS Japan Inc., Tokyo, Japan); significance was set at P < 0.05. Bax mRNA levels were similarly compared using two-way ANOVA. The other physiologic parameters were compared with an unpaired, two-tailed t-test.
Results
Physiologic Variables
Blood pressure and blood gases in p53+/+ and p53−/− mice were mostly within the physiologic range before, during, and after ischemia (Table 1). There were no significant differences between p53+/+ and p53−/− mice.
Blood pressure and blood gas analysis in p53+/+ and p53−/− mice subjected to 14 mins of ischemia
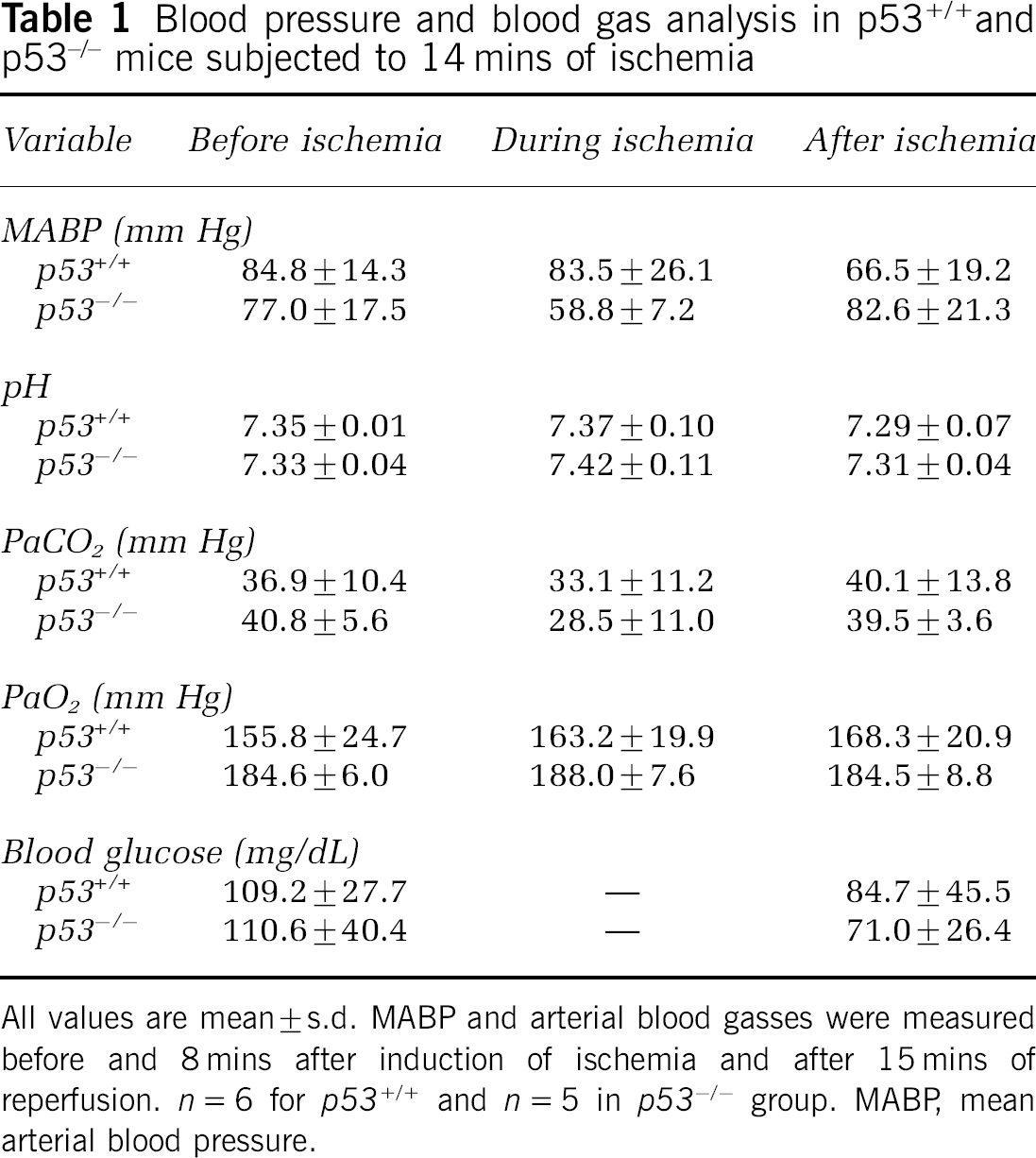
All values are mean ± s.d. MABP and arterial blood gasses were measured before and 8 mins after induction of ischemia and after 15 mins of reperfusion. n = 6 for p53+/+ and n = 5 in p53−/− group. MABP, mean arterial blood pressure.
In the mice subjected to histologic analysis, rectal and temporal muscle temperatures were strictly controlled at 37.0°C ± 0.6°C until the animals were returned to their cages. No hypothermic phase was noted in any of the mice for up to 6 h after ischemia (Table 2). In all mice, the mean rectal temperature after ischemia was within the physiologic range (36.7°C to 37.4°C). There were no significant differences among p53+/+, p53+/−, and p53−/− mice at any time point.
Temperatures and CBF profile in p53+/−, p53+/−, and p53−/− mice subjected to 14 mins of ischemia
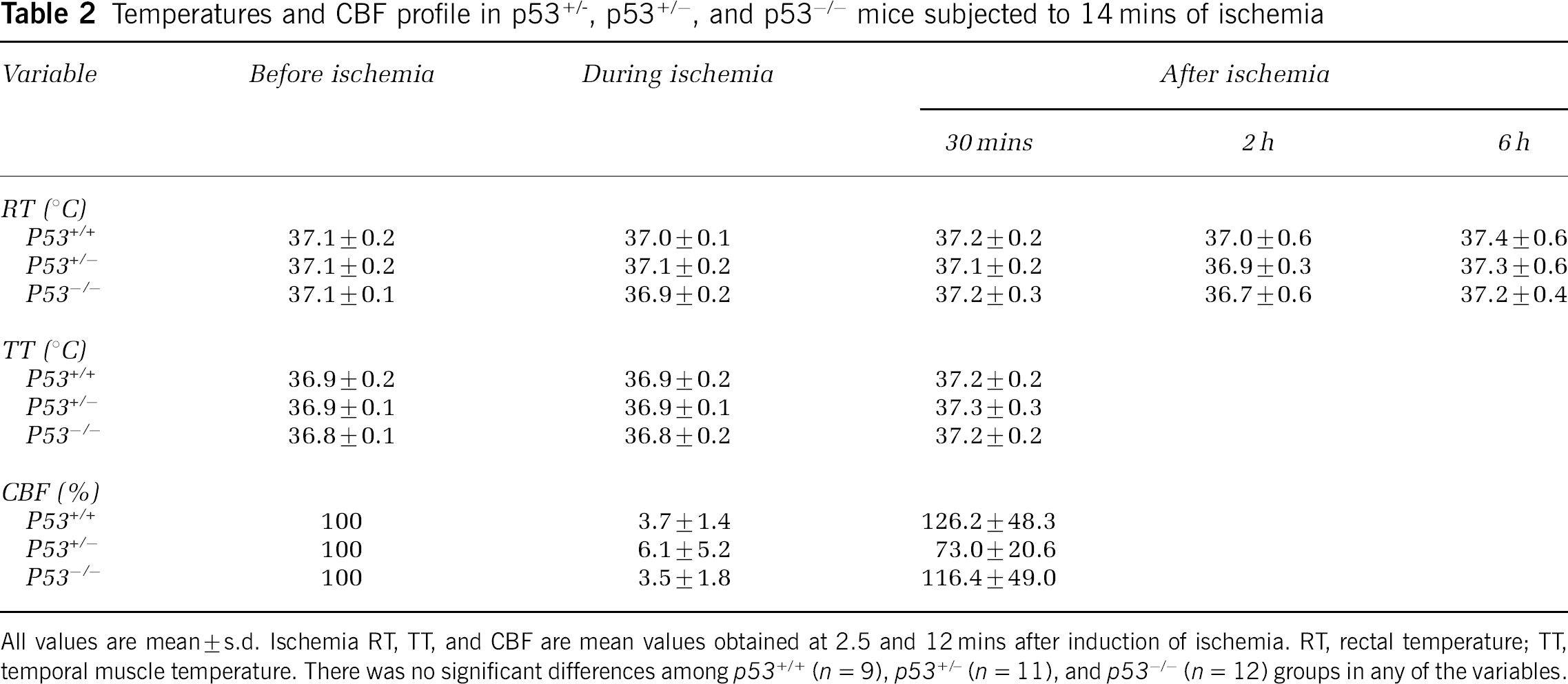
All values are mean ± s.d. Ischemia RT, TT, and CBF are mean values obtained at 2.5 and 12 mins after induction of ischemia. RT, rectal temperature; TT, temporal muscle temperature. There was no significant differences among p53+/− (n = 11), and p53−/− (n = 12) groups in any of the variables.
Cerebral Blood Flow
After three-vessel occlusion, cortical CBF was immediately reduced and remained constant during the ischemic period in all mice (Figure 1). In all groups, the mean CBF during ischemia was < 10% of the preischemic control value (Table 2). In the early postischemic period, some mice showed insufficient recovery of CBF, resulting in relatively larger s.d.'s. The mean values, however, were not statistically significant.
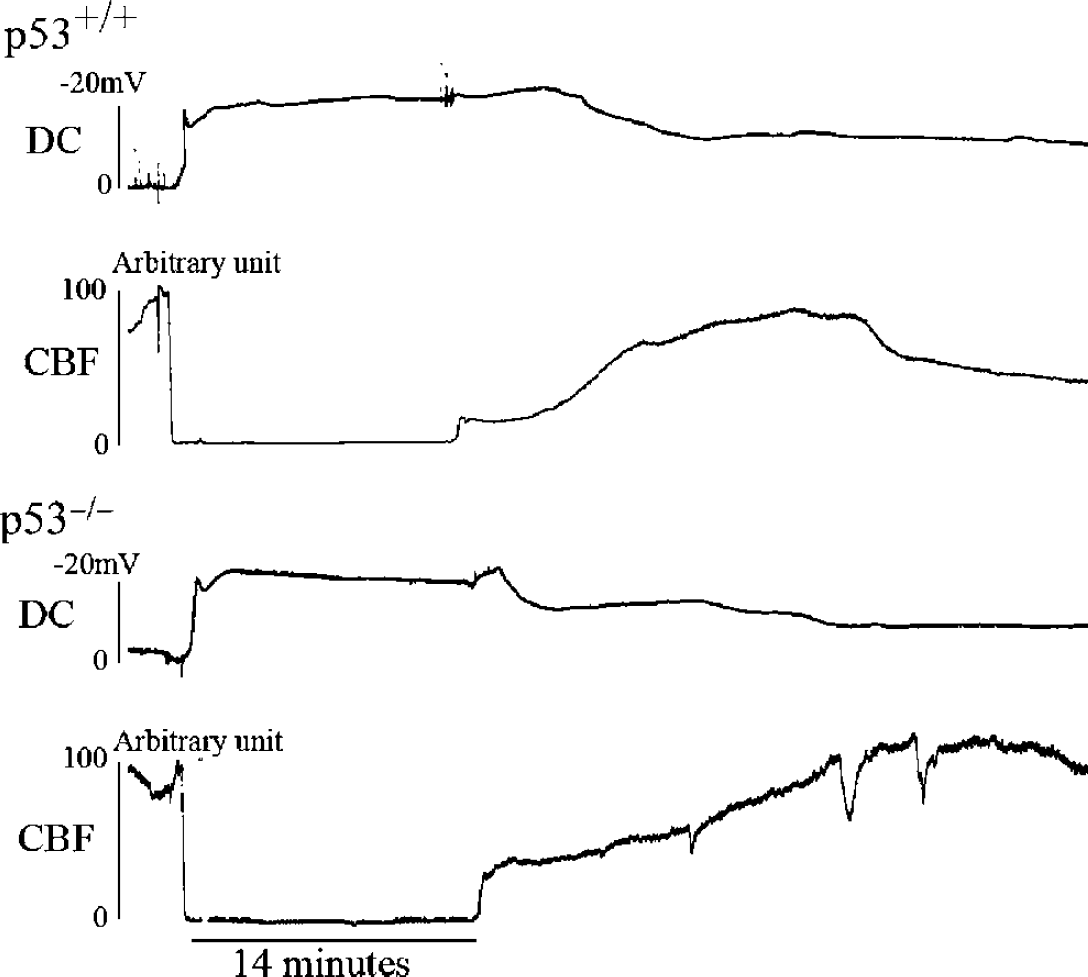
Direct current potential and CBF in p53+/+ and p53−/− mice. In both genotypes, CBF was reduced to < 10% of the control value immediately after ischemia and remained constant during the 14-min ischemic period. A negative shift of DC potential was observed at 49.5 ± 14.4 secs in p53+/+ mice (n = 5) and at 63.2 ± 7.9 secs in p53−/− mice (n = 6); this difference was not statistically significant (P > 0.05; unpaired two-tailed t-test).
Anoxic Depolarization
After three-vessel occlusion, an abrupt negative shift of DC potential was noted with a delay of 54.6 ± 16.9 secs in the p53+/+ mice and 66.5 ± 11.5 secs in the p53−/− mice (Figure 1); this difference was not statistically significant (P = 0.17). Direct current potential showed a further negative shift, followed by slow recovery during recirculation. Anoxic depolarization time, when measured at the halfmaximal value, was 31.1 ± 6.6mins in p53+/+ and 23.0 ± 10.1 mins in p53−/− mice: this difference was not statistically significant either (P = 0.16).
Surgical Success and Survival Rates
Surgical success and survival rates are critical for the assessment of histologic outcome after ischemia in this small rodent model. The surgical success rate was 91.7% (n = 33), and the overall survival rate at 4 days was 81.8% (n = 27). Within groups, the survival rate after successful surgery was 77.8% (7/9) in p53+/+ mice, 91.7% (11/12) in p53+/− mice, and 75% (9/12) in p53−/− mice.
Neuronal Injury in the Hippocampal CA1 Sector
Neuronal injury in the CA1 region was observed 4 days after ischemia (Figure 2). In the p53+/+ mice, most of the pyramidal neurons exhibited pyknotic, shrunken nuclei. In contrast, few degenerated neurons were observed in p53+/− (data not shown) and p53−/− mice, indicating attenuation of neuronal injury in the mutant mice. This observation was also confirmed by the absence of DNA fragmentation in p53−/− mice, as detected by TUNEL staining (Figure 3). Quantitative assessment showed that CA1 neuronal density was significantly higher in p53−/− mice (149.6 ± 82.9 cells/mm) than in p53+/+ mice (22.8 ± 7.4 cells/mm, P < 0.005, Figure 4), 61.3% and 9.3% of nonischemic controls (243.9 ± 4.4 cells/ mm), respectively. In p53+/− mice, a similar trend was observed, though not statistically significant (neuronal count, 106.2 ± 95.7 cells/mm, 43.5% of controls, P > 0.05).
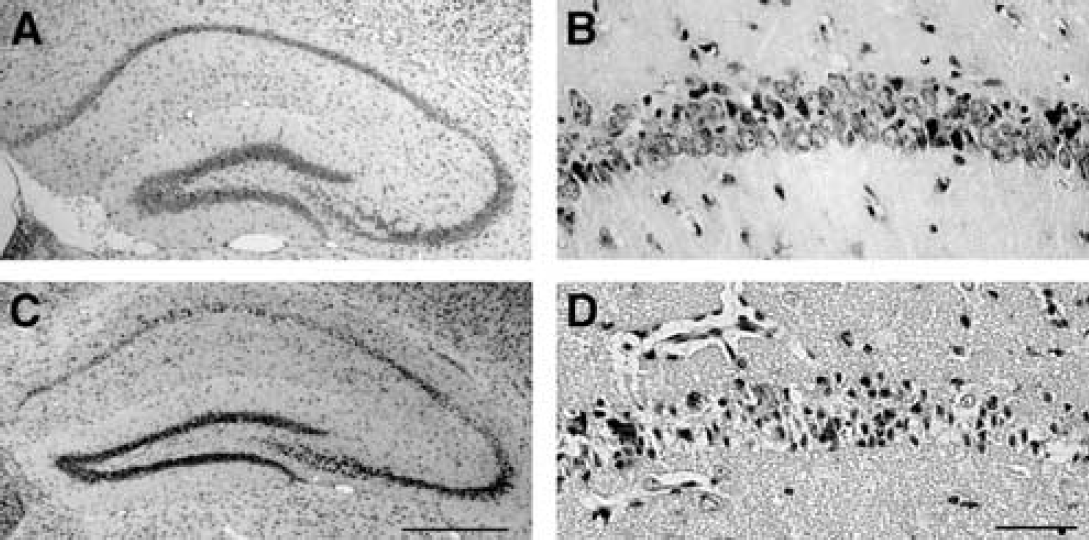
Histologic evaluation of the hippocampal CA1 region with cresyl violet staining in p53+/+ and p53−/− mice. At 4 days after ischemia, CA1 neurons were less degenerated in p53−/− mice (
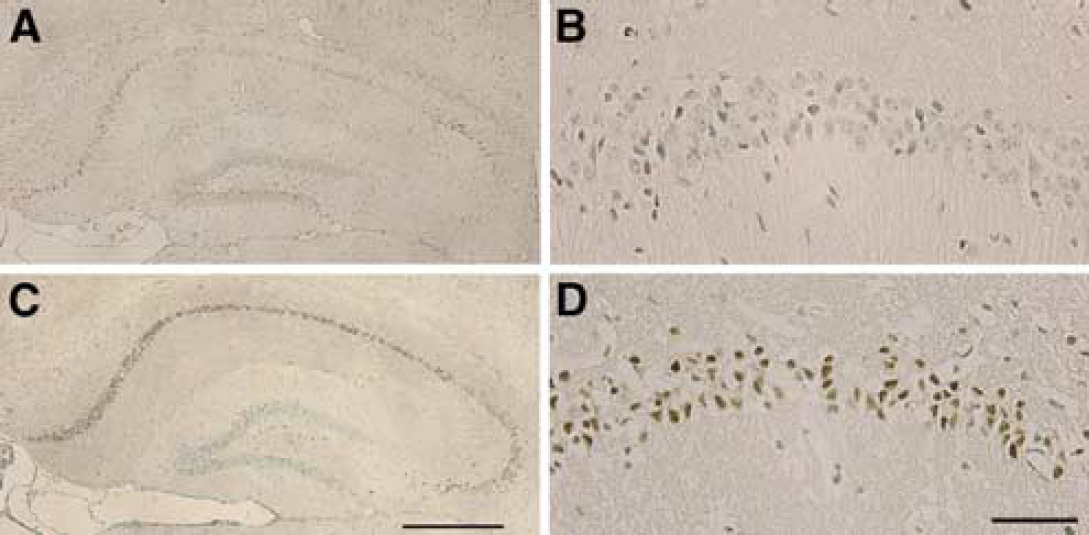
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining of the hippocampal CA1 region in p53+/+ and p53−/− mice. Deoxyribonucleic acid fragmentation was observed in most CA1 neurons in p53+/+ mice at 4 days after ischemia (C, D). In p53−/− mice, however, only a few CA1 neurons displayed TUNEL-positive staining (
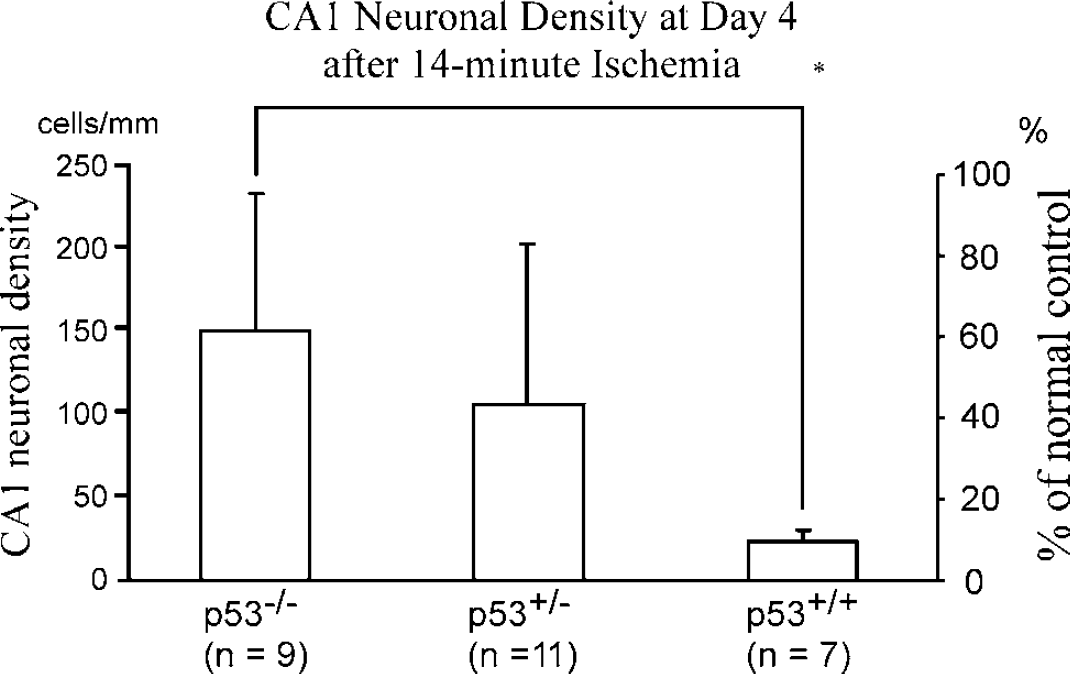
Quantitative evaluation of CA1 neuronal injury in p53+/+, p53+/−, and p53−/− mice. Neuronal density in the CA1 region was significantly greater in p53−/− mice than in p53+/+ mice. A similar trend was also observed in p53+/− mice, though not statistically significant. All values are mean ± s.d. *P < 0.005. Scale on the right side is expressed as % of nonischemic wild-type mice.
Postischemic p53-Like Immunoreactivity
In nonischemic p53+/+ controls, no p53-like immunoreactive cells were observed (Figure 5A); however, p53-like immunoreactivity with faint staining was observed in the CA1 region 6 h after ischemia (Figure 5B), which was enhanced in both intensity and extent at 12 h (Figure 5C). This immunoreactivity is highly specific for p53 because it was not observed in p53−/− mice even after ischemia (Figure 5D).
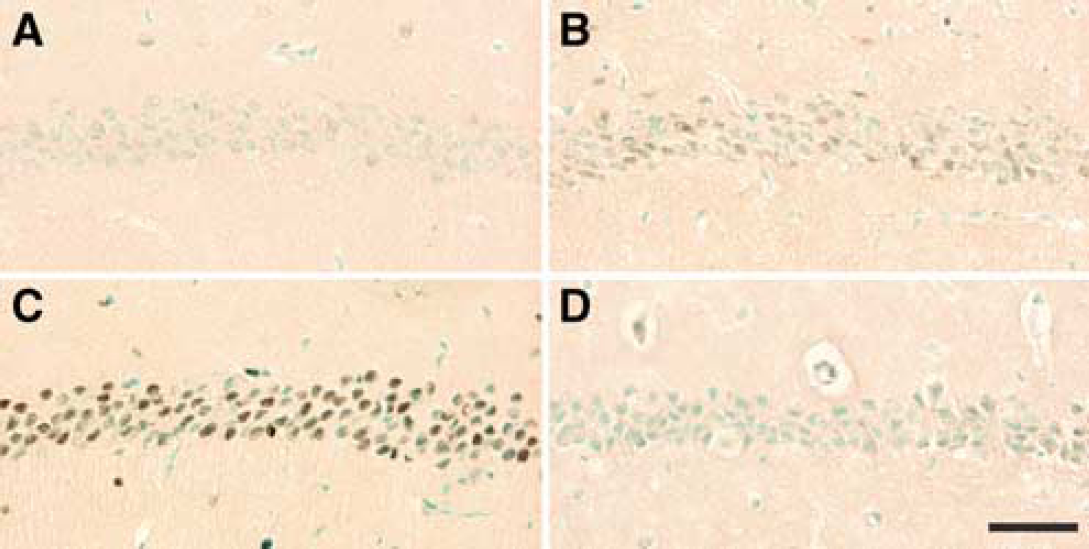
Immunostaining for p53 in the hippocampal CA1 region in p53+/+ and p53−/− mice. p53-like immunoreactive cells were not observed in CA1 sector of nonischemic p53+/+ mice (
Postischemic p53 and Bax Messenger Ribonucleic Acid Levels
After reverse transcription of mRNA in the hippocampal samples, the size and sequences (data not shown) of the target endproducts were confirmed (Figure 6A). As expected in p53−/− mice, normal p53 transcripts were not amplified in nonischemic controls, as well as after ischemia up to 24 h (Figure 6B). In contrast, quantitative real-time PCR in p53+/+ mice revealed that p53 transcripts were significantly increased at both 12 (1.7 ± 0.4-fold, P < 0.05) and 24 h (1.8 ± 0.5-fold, P < 0.01) after ischemia (oneway ANOVA, n = 6 at each time point, Figure 6C). Bax mRNA levels were also increased, by 1.6 ± 0.1-fold at 12 h and 1.8 ± 0.5-fold at 24h, after ischemia compared with that in nonischemic p53+/+ mice (Figure 6D). Statistical analysis showed significant main effects of genotype (P < 0.01) and time (P < 0.01), as well as interaction of both factors (P < 0.05), indicating that Bax mRNA increases significantly in p53+/+ mice after ischemia. Post-hoc analysis revealed a significant overall increase at 12 h (P < 0.01), which persists up to 24 h genotype-dependently (P < 0.05).
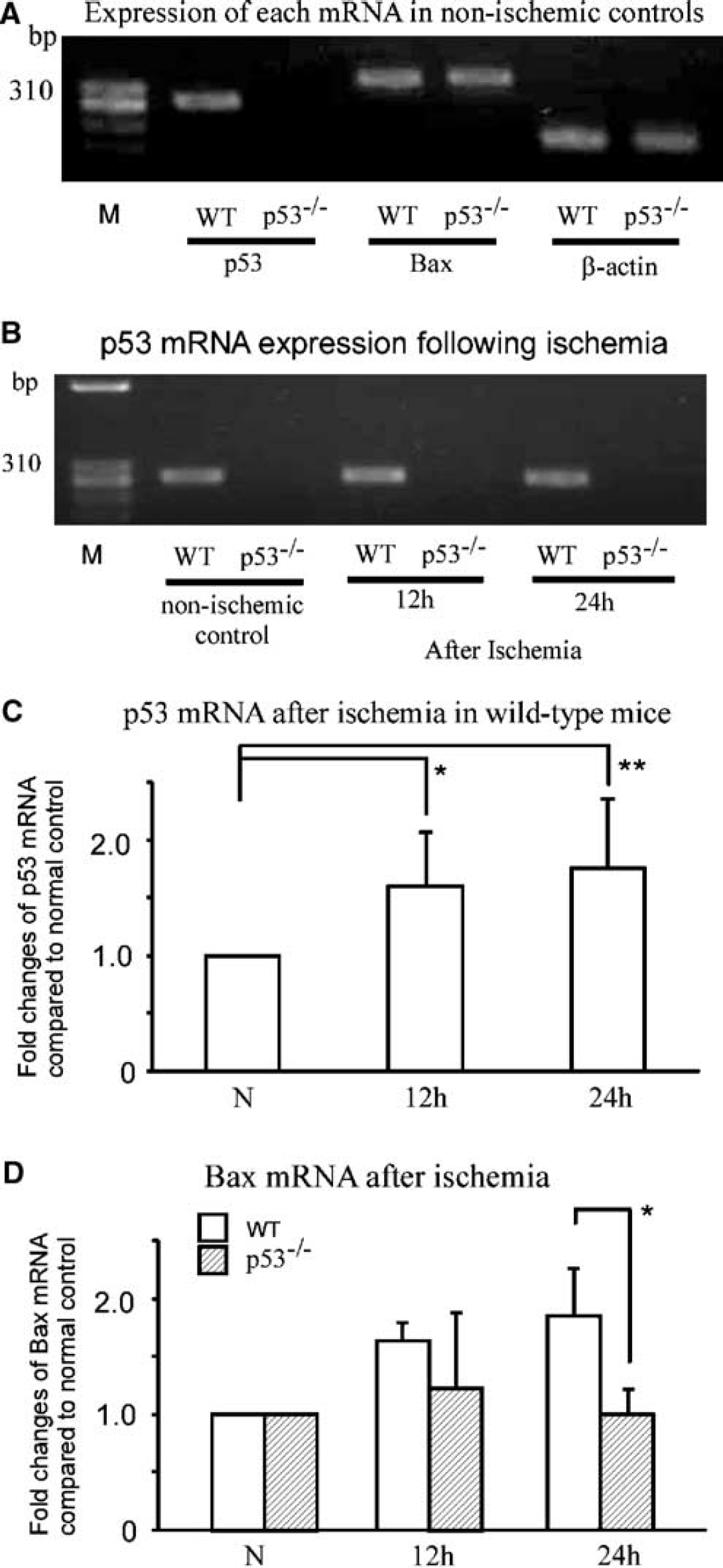
(
Discussion
This study shows that neuronal injury in the CA1 region of the hippocampus after global ischemia was attenuated considerably in p53−/− mice compared with that in p53+/+ mice. Since no difference in ischemic stress was noted, as revealed by CBF and anoxic depolarization, these results suggest that p53 potentiates the ischemic neuronal death.
Generally, gene-engineered mice are generated from multiple genetic backgrounds (Gerlai, 1996). The original chimera used to generate the mutant mice in this study was made from C57BL/6 and SV129 strains, which differ in their vulnerability to ischemia (Yonekura et al, 2004). However, all mutant mice used were backcrossed with C57BL/6 mice for 12 generations from the original chimera, theoretically eliminating 99% of the SV129 background (Gerlai, 1996). Thus, it is unlikely that our results were affected by genetic differences other than the absence of one or both p53 alleles in the mutant mice.
Since genotype can affect the cerebral vascular architecture in knockout mice (Maeda et al, 1999), it was important to rule out differences in ischemic stress. Evaluation of cortical CBF and DC potential during ischemia showed near-complete ischemia in both p53+/+ and p53−/− mice (Figure 1 and Table 1), as indicated by severe reduction of CBF and early depolarization time similar to cardiac arrest model in both groups (Kawahara et al, 2002). Though the s.d.'s were large, length of depolarization time was not different between the groups, further indicating similar ischemic impact in both groups. During the early postischemic period, recovery of CBF was attenuated in some mice (Table 2), which resulted in relatively large s.d.'s, implying that CBF recovery is variable, probably due to severe ischemic impact of this model. However, CBF does not seem to affect the histologic outcome, since there was no statistically significant difference among these groups. Therefore, we assume that the CA1 ischemic neuronal injury in the p53−/− mice was attenuated by the null mutation in the target gene.
If this assumption is correct, the overall activity of p53 should be enhanced in p53+/+ mice after ischemia, as reflected by an increased protein level by post-translational change into the active form. Indeed, p53-like immunoreactivity was detected 12 h after ischemia in the most vulnerable CA1 neurons that were destined to die (Figure 5C). This observation could reflect an alteration in the affinity of the epitope structure for the antibody after ischemia. Although oxidative stress can potentially modulate proteins through free radical reaction, this has not been reported for p53. Alternatively, ischemie stress may modify the protein into several active forms by phosphorylation, leading to a conformational change (Fuchs et al, 1995; Oda et al, 2000b; Koumenis et al, 2001; Latonen et al, 2001). Using antibodies against p53 phosphorylated al serines 18, 23, 37, 312, and 392 (kind gift from Di Taya), we could not identify these forms (data not shown). Another possibility is increased protein content. In agreement with our results, hippocampal p53 mRNA was upregulated after global ischemia in a rat model (Tomasevic et al, 1999a, b ). However, persistent translational inhibition is a well-known phenomenon after transient ischemia in irreversibly injured neurons (Paschen, 2003; MacManus et al 2004), suggesting that de novo synthesis of p53 protein from mRNA is unlikely, though not completely excluded. However, p53 is a short-lived protein, and its cellular level is regulated by degradation through the ubiquitin–proteasome pathway, which is severely impaired after global ischemia in the hippocampus, as we reported previously (Asai et al, 2002). Thus, it seems reasonable to assume that global ischemia increases the p53 protein level in vulnerable hippocampal neurons presumably due to impaired degradation pathway.
To induce cell death, p53 must be modified post-translationally. One such modification is phosphorylation. Since we could not identify the phosphorylated forms of p53, we assessed its transcriptional activity indirectly by measuring mRNA for Bax, a direct target of p53 (Miyashita and Reed, 1995). Quantitative real-time PCR studies showed that Bax mRNA level was significantly increased in p53+/+ mice after ischemia compared with that in p53−/− mice. These results suggest that the overall p53 activity in the hippocampus is elevated before neuronal death.
The potential role of p53 in ischemia has been extensively studied. In vitro, p53 levels increase after hypoxia, depolarization, and kainite or glutamate exposure, and the resultant neuronal cell death was attenuated by inhibiting p53 with antisense oligonucleotides (Banasiak and Haddad, 1998; Jordan et al, 2003) and by p53 deficiency. Although these findings indicate a causal relationship between p53 and neuronal cell death (Xiang et al, 1996), in vivo studies in ischemia are still controversial. Although one study reported no increase of p53 mRNA and protein after permanent focal ischemia up to 6 h (van Lookeren Campagne and Gill, 1998), other studies showed persistent increase of p53 protein in vulnerable CA1 neurons after global ischemia (McGahan et al, 1998; Tomasevic et al, 1999a, b ), as well as in dying neurons in the ischemic core after focal ischemia (Li et al, 1994, 1997), when studied after 6 h. However, these studies also showed a transient increase of p53 in ischemia-resistant CA3 and hypothermia- or pre-conditioning-protected CA1 neurons after global ischemia (McGahan et al, 1998; Tomasevic et al 1999a, b ), indicating that increased p53 expression is not necessarily associated with ischemic neuronal death. To directly establish a link between p53 and ischemic cell death, p53 mutant mice were subjected to permanent focal ischemia, where p53 deletion led to the reduction of infarct volume, but not in a gene dose-dependent way (Crumrine et al, 1994). A similar investigation employing rather mild transient focal ischemia for 1 h, however, disclosed aggravation of metabolic derangements and infarct volume in homozygous mutants, suggesting that p53 may have a protective role in ischemia (Maeda et al, 2001). Since ischemia is also a genotoxic stress (Liu et al, 1996), these rather conflicting observations are in line with the notion that p53 has a dual role after genotoxic stress as a ‘guardian of the genome’, such that it halts cell cycle to allow for DNA repair when the damage is mild, while it activates apoptotic pathway to eliminate cells when the damage is severe (Lane, 1992). In this regard, a role of p53 in ischemia would be context- and severity-dependent. In the present study, ischemic neuronal injury in CA1 was significantly attenuated in p53 mutants in a gene dose-dependent way. Although the mRNA and protein could not be quantitated after ischemia in the mutants, the phenotype (tumorigenesis and radiation-induced apoptosis) apparently correlates with the three different genotype (Donehower et al, 1992; Lowe et al, 1993), suggesting a corresponding amount of functional p53 after ischemia. We therefore assume that, in CA1 neurons after severe global ischemia, p53 mainly aggravates neuronal death. Consistent with this notion, the novel p53 inhibitor pifithrin-α was recently shown to have protective effect in CA1 ischemic neuronal death (Culmsee et al, 2003; Leker et al, 2004).
Although p53 directly regulates a variety of proapoptotic genes (Fei et al, 2002; Vousden and Lu, 2002), a major pathway leading to ischemic neuronal death has not been identified. Among proapoptotic genes, Bax has been shown to increase at mRNA and protein levels after ischemia in vulnerable CA1 region (Krajewski et al, 1995; Chen et al, 1996). In our study, the increase of Bax mRNA was also shown by quantitative PCR, but modest compared with that reported previously by in situ hybridization study in CA1 (Chen et al, 1996). This difference might be related to sample preparation in the current study, where whole hippocampi were used instead of CA1 region. Nonetheless, our result is consistent with previous studies and suggests a potential role of Bax in ischemic neuronal death. Other proapoptotic genes regulated by p53, such as Noxa and Puma (Oda et al, 2000a; Yu and Zhang, 2003; Cregan et al, 2004), might also be involved. Further studies are needed to determine the precise mechanisms of p53-dependent ischemic neuronal death.
In conclusion, a p53-dependent pathway appears to be a major and critical modulator of ischemic neuronal death in the CA1 region after global ischemia. Thus, p53 might be a therapeutic target for ischemic neuronal injury. Further understanding of this p53-dependent pathway may contribute to the development of novel treatments for stroke.
Footnotes
Acknowledgements
The authors thank Yoichi Taya for the kind gift of phosphorylated p53 antibodies and Ms Reiko Matsuura for technical assistance.