
Abstract
Coupling between local cerebral blood flow and local cerebral metabolic rate for glucose is involved in the pathogenesis of epilepsy-related neuronal damage in the adult brain; however, its role in the immature brain is unknown. Lithium–pilocarpine–induced status epilepticus is associated with extended damage in adult rats, mostly in the forebrain limbic areas and thalamus, whereas damage was moderate in 21-day-old rats (P21) or absent in P10 rats. The quantitative autoradiographic [14C]iodoantipyrine technique was applied to measure the consequences of lithium-pilocarpine status epilepticus on local cerebral blood flow. In adult and P21 rats, local cerebral blood flow rates increased by 50% to 400%; the highest increases were recorded in regions showing damage in adults. At P10, local cerebral blood flow rates decreased by 40% to 60% in most areas, except in some forebrain regions showing no change during status epilepticus. In areas injured when status epilepticus was induced in adults, a strong hypermetabolism (Fernandes et al., 1999) not matched by comparable local cerebral blood flow increases was present in rats of all ages, whereas in damage-resistant areas, local cerebral metabolic rate for glucose and local cerebral blood flow remained coupled in the three age groups. Thus, the level of coupling between blood flow supply and metabolism is not involved in seizure-related brain damage in the developing brain, which appears to be resistant to the consequences of such a mismatch.
Keywords
Changes in cerebral blood flow and metabolism in epilepsy have been of interest because of their involvement in the pathogenesis of neurologic damage. Indeed, during severe and prolonged seizures or status epilepticus (SE), large and sustained metabolic increases were recorded that are correlated with neuronal damage in various experimental models of severe seizures or SE (Meldrum, 1983; Ingvar, 1986; Handforth and Treiman, 1995). Nevander et al. (1985) showed that in vulnerable areas, after an initial marked metabolic increase, local cerebral metabolic rates for glucose (LCMRglc's) return to normal levels and eventually decrease below the level recorded in the control situation as seizures continue; these low levels of LCMRglc's coincide with the occurrence of neuronal damage. In addition, changes in cerebral blood flow are characterized by large increases up to 500% to 900% within seconds of the onset of seizure activity, and during this initial phase these large increases in rates of local cerebral blood flow (LCBF) are similar in amplitude to those of LCMRglc's. However, as seizures continue, LCBF values decrease to 150% to 300% of control values after 1 or 2 hours of continuous seizure activity or SE, inducing a progressively established relative hypoperfusion, while the metabolic demand remains high. This situation may reach a critical level in terms of supplying adequate amounts of substrates to the brain, leading to the hypothesis of the role of a mismatch between blood supply and energy metabolism in seizure-induced neuronal damage (Ingvar and Siesjö, 1983; Tanaka et al., 1990; Kreisman et al., 1991). Autoradiographic studies about the coupling between flow and metabolism in adult animals subjected to severe seizures or SE confirmed that neuronal damage may result from a mismatch between LCBF and LCMRglc (Ingvar and Siesjö, 1983), whereas a tight coupling exists between cerebral blood supply and metabolism in most physiologic and pharmacologic states and during the early phase of seizure activity (Sokoloff, 1981; Duncan, 1992; Kuschinsky, 1996).
Conversely, the immature brain is resistant to seizure-induced brain damage; indeed, damage is usually not seen before the third week of postnatal life in rodents (Nitecka et al., 1984; Cavalheiro et al., 1987; Sperber et al., 1991; Wasterlain and Shirasaka, 1994; Priel et al., 1996). However, large metabolic increases are recorded in various models of seizures in the developing brain (Tremblay et al., 1984; Fujikawa, 1989). Likewise, a widespread cerebral vasodilation has been shown during seizures in neonates in various species, including dogs, monkeys, and pigs (Young et al., 1984; Fujikawa et al., 1986; Park et al., 1987). The relation among cerebral metabolism, blood supply, and neuronal damage after SE or severe seizures has not been studied during brain development.
In a previous study using pentylenetetrazol to induce SE in 10-day-old (P10) and 21-day-old (P21) rats, we demonstrated a 100% to 400% metabolic increase in all brain areas studied in P10 rats, whereas blood flow increases reached only 32% to 184%, creating a mismatch between blood flow and metabolism in some areas. However, this mismatch was not associated with neuronal damage in P10 rats. In P21 rats, pentylenetetrazol seizures induced LCMRglc increases that were restricted to areas involved in the epileptic circuit, whereas other areas showed decreases or no change. At that age, the coupling between LCBF and LCMRglc was maintained and no neuronal damage was recorded (Pereira de Vasconcelos et al., 1992, 1995; Pineau et al., 1999).
To further study the relation among cerebral metabolism, blood flow, and brain lesions, we used a model of epilepsy induced by lithium and pilocarpine in which neuronal damage varies with postnatal age. Status epilepticus induced by pilocarpine with or without lithium leads to neuronal damage mostly in hippocampus, dentate gyrus hilus, piriform and entorhinal cortices, amygdala, septum, thalamus, and neocortex; this adult pattern of brain lesions develops mainly after the third week of postnatal life in the rat, whereas no such damage has been found in younger animals (Cavalheiro, 1995; Priel et al., 1996; Dubé et al., 2001). In this model, we recorded large metabolic increases during SE in adult rats. Higher LCMRglc increases occurred mostly in forebrain structures undergoing neuronal damage after SE (Fernandes et al., 1999), as previously observed in other models of epilepsy (Duncan, 1992). Large metabolic increases were also recorded during seizures in P10 and P21 rats. These increases were not correlated to any brain damage in P10 rats, and only discrete neuronal damage was recorded in P21 rats (Fernandes et al., 1999; Dubé et al., 2001).
The purpose of the present study was to measure the effects of lithium–pilocarpine SE on LCBF in the developing and adult rat brain using the [14C]iodoantipyrine quantitative autoradiographic technique (Sakurada et al., 1978), to establish if the level of coupling between cerebral blood supply and metabolism may be a possible predictive index of brain damage in immature brain.
MATERIALS AND METHODS
Animals
One male and two female Adult Sprague-Dawley rats (Janvier Breeding Center, Le Genest-St-Isle, France) were housed together in mating groups for 5 days. Litter size was reduced to 10 offspring for homogeneity. The same breeder provided adult 3-month-old male rats. The experiments were performed on a total of 30 rats (9 P10, 11 P21, and 10 adult stage). All experiments were performed in accordance with the rules of the European Committee Council Directive of November 24, 1986 (86/609/EEC) and the French Department of Agriculture (license no. 67–7). Lithium chloride (127 mg/kg) was administered intraperitoneally 16 hours before the subcutaneous injection of pilocarpine (60, 30, and 25 mg/kg in P10, P21, and adult rats, respectively). P21 and adult rats received 1 mg/kg methyl-scopolamine subcutaneously 30 minutes before receiving pilocarpine to reduce peripheral consequences. Methylscopolamine was not administered to P10 rats because the peripheral effects of pilocarpine are limited at that age. Control animals received the same pharmacologic treatments, but were given saline instead of pilocarpine.
Measurement of local cerebral blood flow and physiologic variables
A femoral artery and vein were catheterized with polyethylene tubing using light halothane anesthesia. Both catheters were threaded under the skin up to the back of the hindpaw to allow free access to the catheters in free-moving rats. The animals recovered from surgery in their home cages (suckling rats were returned to the dams) for 17 to 24 hours before the onset of experiment.
The autoradiographic measurement of LCBF was performed using the [14C]iodoantipyrine technique (Sakurada et al., 1978) as adapted to the immature rat by Nehlig et al. (1989). The measurement of LCBF was performed 70 minutes after the beginning of SE. The onset of SE was assessed according to the behavioral criteria previously described at the different ages studied (Priel et al., 1996; Fernandes et al., 1999). IAP (4-Iodo-N-[methyl-14C]iodoantipyrine, spec. act. 56 mCi/mmol; Amersham, Les Ulis, France) was injected through the femoral vein at a concentration of 25 μCi/mL. The period of measurement was approximately 60 seconds during which an increasing rate of the tracer was administered and timed, free-flowing arterial blood samples were collected from the arterial catheter. The rats were decapitated approximately 1 minute after the initiation of IAP infusion. Brains were quickly removed, frozen in isopentane, chilled to −25°C, and stored at −80°C. The amount of radioactive tracer per unit volume of blood in each sample was measured by liquid scintillation after tissue solubilization. Tracer concentration was calculated from the amount of 14C, the weight of the blood samples, and a specific gravity of 1.06 g/mL blood. The frozen brains were cut into 20-μm coronal sections at −20°C in a cryostat. Sections were autoradiographed on Amersham Biomax MR films along with calibrated 14C standards (Amersham). Adjacent sections were stained with thionine for histologic identification of specific nuclei. Autoradiographs were analyzed by quantitative densitometry with a computerized image-processing system (Biocom 500, Les Ulis, France): optical density was measured bilaterally on a minimum of four brain sections. Structures were anatomically defined according to the developing rat brain atlas of Sherwood and Timiras (1970) and the atlas of Paxinos and Watson (1986) for adult rats. Local cerebral blood flow values were calculated according to the Fick equation using a blood-brain partition coefficient of 0.8 (Sakurada et al., 1978). The correction for time lag and washout effect was considered during the final calculation of LCBF (Nehlig et al., 1989).
Arterial blood gases (pH, arterial oxygen and carbon dioxide tensions) were measured just before pilocarpine and 14C-IAP were injected (i.e., 70 minutes after the beginning of SE) using a blood-gas analyzer (Corning model 158; Corning Medical and Scientific, Halstead, UK). Mean arterial blood pressure was recorded with an air-damped mercury manometer, and hematocrit levels were simultaneously measured.
Statistical analysis
Local cerebral blood flow values were determined in 40 structures in three age groups each of subjects and controls. The values of LCBF and physiologic variables recorded in the lithium—pilocarpine group were compared with those of the age-matched lithium-saline group by means of a Scheffé test for multiple comparisons.
RESULTS
Behavior
Behavioral changes induced by the administration of lithium and pilocarpine were time and age dependent (Priel et al., 1996; Fernandes et al., 1999). Signs of cholinergic stimulation (e.g., diarrhea, piloerection) developed in P21 and adult rats within 5 minutes after pilocarpine injection, followed by a 15 to 20 minute phase of head bobbing, scratching, chewing, and exploratory behavior. Recurrent seizures started 20 to 25 minutes after pilocarpine injection, and were associated with head and bilateral forelimb myoclonus; rearings and fallings evolved to SE in 40 to 60 minutes after pilocarpine injection (Fernandes et al., 1999). In P10 rats, behavioral seizure signs were less visible, appeared after 15 to 20 minutes, and were mainly characterized by hyperactivity, scratching, and mastication; no rearing nor falling could be seen at that age. In P10 rats, SE was reached 40 minutes after pilocarpine injection and was characterized by intense body tremor and tonic extension of the tail (Priel et al., 1996; Fernandes et al., 1999).
Effects of lithium–pilocarpine on physiologic variables
Arterial blood gas concentrations were not affected by lithium–pilocarpine seizures at any age; however, a 28% and 42% decrease in arterial carbon dioxide tension were recorded in P10 and P21 rats, respectively (Table 1). Compared with control levels in P10 and P21 rats, arterial blood pressure remained stable 70 minutes after the onset of SE, but increased by 21% in adult animals experiencing seizures. Hematocrit levels were not altered by lithium–pilocarpine seizures in any age group (Table 1).
Effects of lithium–pilocarpine SE on physiologic variables
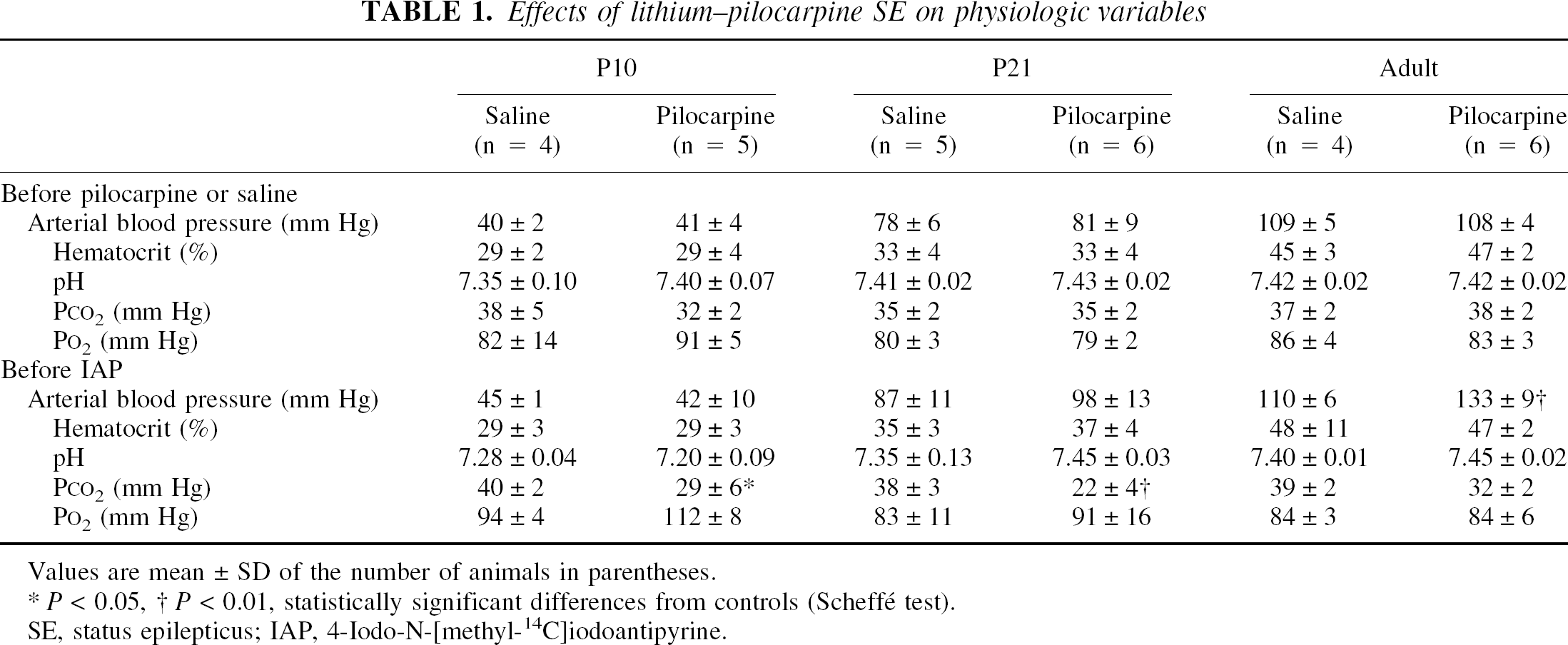
Values are mean ± SD of the number of animals in parentheses.
P < 0.05
P < 0.01, statistically significant differences from controls (Scheffé test).
SE, status epilepticus; IAP, 4-Iodo-N-[methyl-14C]iodoantipyrine.
Effects of lithium–pilocarpine status epilepticus on local cerebral blood flow
In control animals of all ages, LCBF rates measured in the present study were in the same range as those reported previously (Nehlig et al., 1989; Pereira de Vasconcelos et al., 1995).
In P10 rats, LCBF values decreased by 40 to 60% in most structures, with significant changes recorded in 17 of 33 structures studied. Although these changes occurred in most systems, no changes were observed in forebrain limbic areas (Table 2). No significant blood flow changes were recorded in limbic cortices (e.g., entorhinal, piriform, and perirhinal cortices), in other limbic areas (e.g., hippocampus, amygdala), and in the caudate nucleus.
Effects of lithium–pilocarpine seizures on LCBF
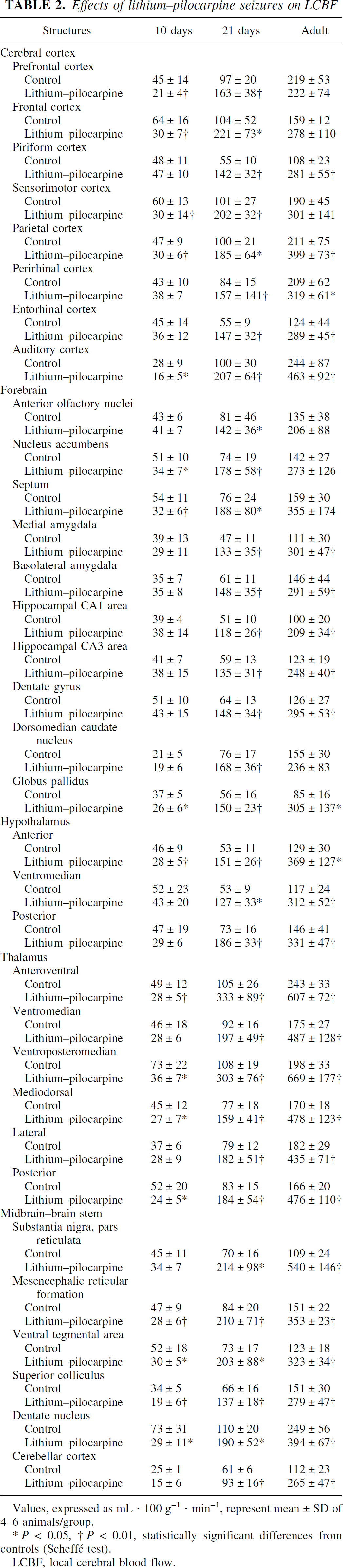
Values, expressed as mL · 100 g−1 · min−1, represent mean ± SD of 4–6 animals/group.
P < 0.05
P < 0.01, statistically significant differences from controls (Scheffé test).
LCBF, local cerebral blood flow.
In P21 rats exposed to lithium–pilocarpine SE, significant increases in LCBF values (50% to 200% compared with control values) were recorded in all structures studied (Table 2). The highest increases (> 150%) occurred in the piriform and entorhinal cortices and forebrain areas, mainly in the septum, amygdala, hippocampus, basal ganglia, thalamus, and hypothalamus. More moderate LCBF increases (50% to 100%) were recorded in some cortical and posterior areas of lithium–pilocarpine–treated rats when compared with controls.
In adult rats, LCBF significantly increased over control values by 50% to 400% in all but nine structures. The highest increases (> 250%) were recorded in the piriform and entorhinal cortices, amygdala, hypothalamus, thalamus, and basal ganglia. In adults, increases in LCBF rates during SE were of the same amplitude as those recorded in P21 rats, except that no significant changes in LCBF were seen in the dorsal anterior cortices (prefrontal and cingulate).
Relation between LCBF and LCMRglc during lithium–pilocarpine status epilepticus
During prolonged seizures or SE, a mismatch between local blood supply and metabolism has been shown to participate in brain damage in adult animals (Meldrum, 1983; Ingvar and Siesjo, 1983). But the relation between these two parameters of functional activity and brain damage is not clear in immature animals. Thus, in the present model of SE, we studied the coupling between LCMRglc and LCBF using LCMRglc values that we obtained previously in a separate group of rats during the second hour of lithium–pilocarpine SE (Fernandes et al., 1999).
In areas lesioned after lithium–pilocarpine SE in the adult brain (i.e., mostly limbic forebrain and thalamic areas), LCMRglc's reached 400% to 850% of control levels during lithium–pilocarpine SE, whereas LCBF values increased more moderately, reaching only 150% to 300% of control levels (Figs. 1A and 3). In P21 rats, the LCBF and metabolic responses to SE were similar to those in adult rats, with major increases in LCMRglc (450% to 800%) not compensated by similar blood flow increases (100% to 200%) (Figs. 1B and 3). In P10 rats, SE induced a severe hypermetabolism (up to 600% of basal levels), whereas LCBF rates were unchanged compared with controls. This resulted in a severe mismatch between LCMRglc's and LCBF rates in all areas in which damage develops after SE in adult rats (Figs. 1C and 3) (Priel et al., 1996; Dubé et al., 2001).
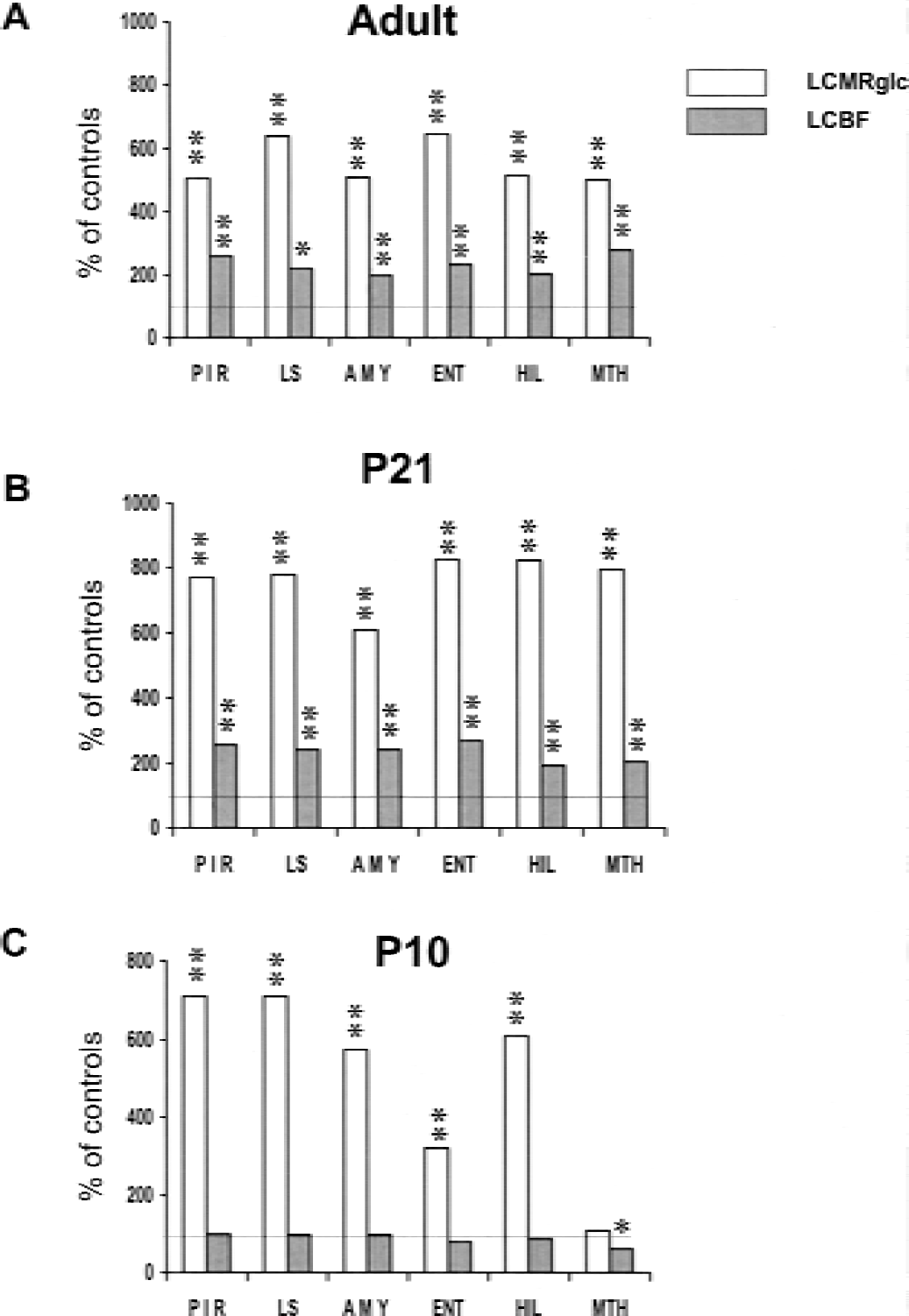
(A–C) Effects of lithium–pilocarpine–induced status epilepticus on local cerebral blood flow and local cerebral metabolic rate for glucose (LCMRglc) in vulnerable structures. Values are expressed as percentages of control rates (the present study, Fernandes et al., 1999). PIR, piriform cortex; LS, lateral septum; AMY, amygdala; HIL, hilus of dentate gyrus; MTHAL, mediodorsal thalamus. *P < 0.05, **P < 0.001, significantly different from controls.
In areas resistant to lithium–pilocarpine–induced damage (mainly the brainstem and midbrain), the adjustment between metabolic demand and blood supply was maintained in all age groups despite a slight hyperperfusion in some areas of the adult brain and a moderate hypoperfusion in P10 rats (Fig. 2).
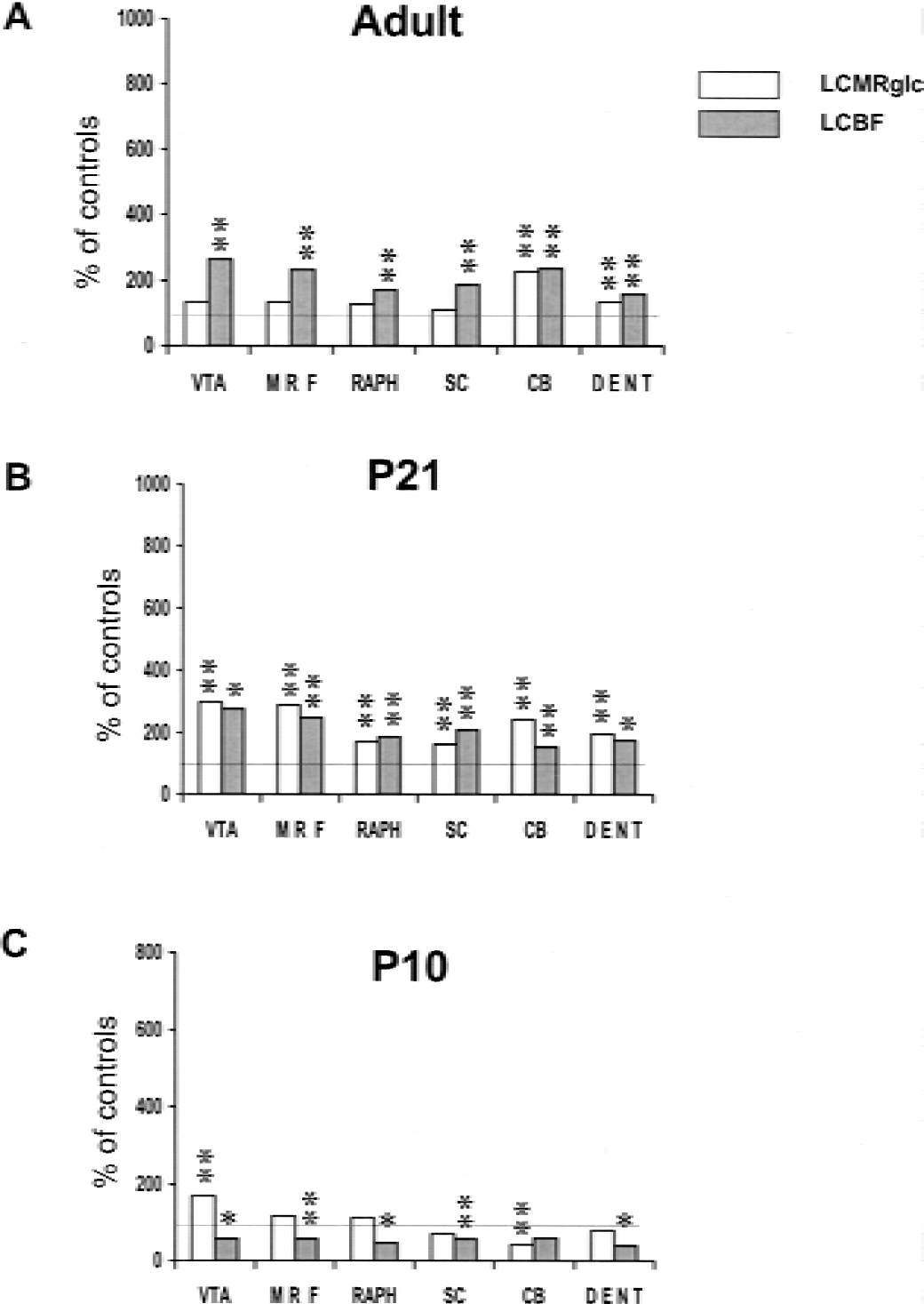
(A–C) Effects of lithium–pilocarpine–induced status epilepticus on local cerebral blood flow and local cerebral metabolic rate for glucose (LCMRglc) in resistant structures. Values are expressed as percentages of control rates (the present study, Fernandes et al., 1999). VTA, ventral tegmental area; MRF, mesencephalic reticular formation; MRAPH; medial raphe nucleus; SCOL, superior colliculus; CBCX, cerebellar cortex; DENT, dentate nucleus. P < 0.05, **P < 0.001, significantly different from controls.
DISCUSSION
Choice of the timing for radioactive-tracer injection
In the present model, SE develops within 1 hour after pilocarpine administration and lasts for 4 to 12 hours in P10, P21, and adult rats. To measure cerebral metabolic activity during the acute period of SE, [14C]2-deoxyglucose was injected 50 minutes after the onset of SE (Fernandes et al., 1999) and LCMRglc's measured for the next 45 minutes, as required for quantification (Sokoloff et al., 1977). Because of the differences in the duration of measurements between LCMRglc and LCBF experiments (45 minutes and 1 minute, respectively), and because the final value of LCMRglc calculated according to the operational equation of the 2-deoxyglucose method mostly represents the cerebral metabolism during the first 10 to 20 minutes after tracer injection (Sokoloff et al., 1977), the measurement of LCBF rates in the present study was performed at 70 minutes after the onset of lithium–pilocarpine SE.
Characteristics of the blood flow response during status epilepticus
In the mature and immature brain, the LCBF response during lithium–pilocarpine SE appears relatively homogeneous, as previously shown in various SE models in developing (Pereira de Vasconcelos et al., 1995) and adult animals (Ingvar, 1986; Tanaka et al., 1990). Indeed, in the present study, LCBF rates increased by 100% to 200% in most areas of the adult and P21 rat. Fujikawa (1989) previously reported similar increases in LCBF in adult rats subjected to seizures induced by repetitive subconvulsive doses of pilocarpine. These relatively moderate vasodilations do not match cerebral metabolic rates, which largely increased compared with control values during the second hour of lithium–pilocarpine SE (fourfold to sevenfold in most areas, Fernandes et al., 1999). This mismatch between blood flow and metabolism may originate in the timing of the measurements after the onset of SE. Indeed, after 1 to 2 hours of SE in adult rats, a decrease in the magnitude of the increase in LCBF rates has been shown while metabolic rates remain high, leading to a relative hypoperfusion (Horton et al., 1980; Ingvar and Siesjö, 1983; Kreisman et al., 1991).
Conversely, in P10 rats, LCBF responses to lithium– pilocarpine seizures remained homogeneous, but the vascular changes were characterized by decreases not seen in the other age groups in this model. These decreases ranged from 30% to 60% and occurred in mainly in the brainstem and midbrain, but also in the thalamus, hypothalamus, and dorsal limbic cortices. At that age, only the limbic areas directly involved in the circuit of lithium– pilocarpine seizures (e.g., hippocampus, amygdala, entorhinal and piriform cortices) showed LCBF rates within the normal range. However, in another model of SE induced by pentylenetetrazol, LCBF increases ranging from 30% to 185% were recorded in all structures in P10 rats (Pereira de Vasconcelos et al., 1995). Thus, depending on the convulsant, the P10 rat brain is able to show LCBF increases during SE (Pereira de Vasconcelos et al., 1995), as previously shown in newborns of different species, such as dogs, monkeys, and pigs (Young et al., 1984; Fujikawa et al., 1986; Park et al., 1987). Moreover, in 7- to 18-day-old marmoset monkeys, bicuculline-induced seizures lead to a redistribution of LCBF in favor of the brainstem pontomedullary regions. This region-specific circulatory response is lost in older animals aged 4 to 8 weeks (Fujikawa et al., 1986). Thus, LCBF response to seizures appears to depend on developmental age, but also on the epilepsy model and the species.
In the present study, normal or decreased values of LCBF were recorded in P10 rats during lithium–pilocarpine SE. This age-specific response may have several origins. First, it may be because the increase in cerebral blood flow linked to seizure activity may occur more rapidly and not last as long as in older animals, and may relate in turn to the severity or duration of the seizures. Indeed, in P10 rats, because of the incomplete maturation of the limbic circuitry (Tremblay et al., 1984), the behavioral response to lithium–pilocarpine SE is not as strong as in older animals, but has a clear electroencephalograph expression, with continuous spiking activity in the hippocampus spreading to the cortex (Cavalheiro et al., 1987; Hirsch et al., 1992, personal observations). Likewise, the duration of SE is reduced in P10 rats compared with older animals (Fernandes et al., 1999). Second, the lack of cerebral blood flow response to SE might be due to the immaturity of the cholinergic innervation because seizures were induced by the muscarinic agonist, pilocarpine. Acetylcholine has been shown to regulate cerebral blood flow, particularly via intracranial cholinergic nerve fibers, both in basal conditions and during various stimuli including neuronal activation (Scremin and Jenden, 1996). In the rat brain, monitoring of the cholinergic system becomes possible around gestational age 14, but the complete development of cholinergic neurons and receptors occurs only around P16 to P21 in the rat (Rotter et al., 1979; Schlumpf et al., 1991; Tice et al., 1996). This delayed maturation might partly explain the absence of cerebral blood flow increase during seizures in P10 rats compared with older animals.
Relation between seizure-induced metabolic and circulatory changes and neuronal damage
In the adult brain, LCBF rates and LCMRglc's increase to a similar degree during the early phase of seizure activity. Then, a clear mismatch between these two variables progressively develops as seizures continue and a pronounced hypermetabolism and a relative hypoperfusion are recorded in brain areas in which damage will develop (Ingvar and Siesjö, 1983). By contrast, the immature brain of all species is resistant to seizure-induced brain damage (Priel et al., 1996; Holmes et al., 1998; Sankar et al., 1998). Thus, our objective was to determine whether the level of coupling between LCBF rates and LCMRglcs might participate in the relative protection of the immature brain against SE-induced damage.
In the adult rat, our results confirm previous data showing the high sensitivity of the mature brain to sustained hypermetabolism, concomitant with relative hypoperfusion. This mismatch is predictive of brain damage after SE or prolonged seizures (Siesjö et al., 1983; Tanaka et al., 1990). Indeed, in the present study, we observed a close relation between areas showing a pronounced mismatch between blood supply and metabolic demand (Figs. 1A and 3) and extensive damage after lithium–pilocarpine SE, such as the hippocampus, amygdala, limbic cortices or thalamus (Dubé et al., 2001). In these structures, LCMRglcs increased by 3.5-fold to 6.5-fold whereas LCBF rates increased only by 1.3-fold to 2.5-fold compared with control levels. However, brain structures in which no damage develops (mostly the brainstem and hypothalamus) showed a good adjustment between LCBF rates and LCMRglc's, with increases ranging from 1.1-fold to 3.7-fold compared with control values for both variables (Fig. 2A).
The situation is different in the P21 rat. A this age, a pronounced mismatch between metabolic demand and blood flow supply also occurs in all forebrain areas during SE, as in adults (Figs. 1B and 3); however, neuronal injury was recorded only in a limited number of regions (Dubé et al., 2001). Thus, the consequences of the mismatch between blood flow and metabolism appear to be variable at that age. Some structures, such as the entorhinal cortex, lateral thalamus and hilus of the dentate gyrus (the only injured regions, Dubé et al., 2001) behave like those in the adult rat and display the typical high sensitivity to hypermetabolism–low LCBF-induced brain damage. However, other structures, such as the anterior thalamus, amygdala, and most cortices that are injured in the adult brain subjected to SE, are protected from the deleterious consequences of hypermetabolism–i.e., low LCBF, as in the P10 rat brain. Indeed, in P10 rats, lithium–pilocarpine SE induced a marked mismatch between LCBF and LCMRglc in forebrain regions (Figs. 1C and 3), though no injury developed (Dubé et al., 2001). Conversely, in structures resistant to SE-induced damage in adults (mainly the brain stem and midbrain areas) a good adjustment between LCBF and LCMRglc was recorded in P10 rats and at the other ages studied (Fig. 2).
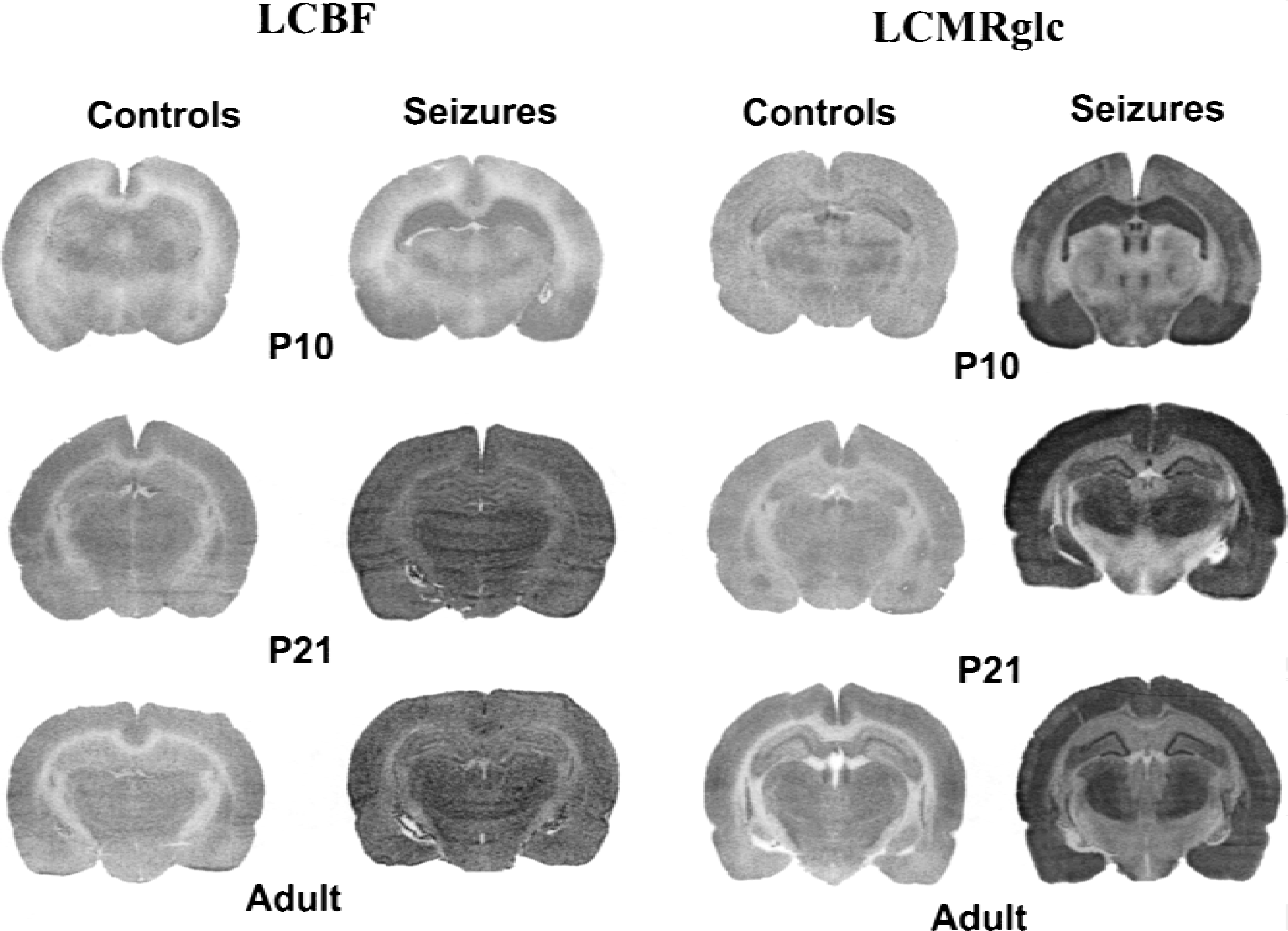
[14C]2-Deoxyglucose and [14C]iodoantipyrine autoradiograms of rat brain sections taken at the level of the dorsal hippocampus and median thalamus in P10, P21, and adult rats during control conditions (lithium-saline) and during lithium–pilocarpine status epilepticus. Radioactive tracers were injected 50 and 70 minutes after the beginning of status epilepticus for metabolic and blood flow studies, respectively. Note the relatively lower and homogeneous tracer distribution in blood flow images compared with higher rates and heterogeneous distribution of the radioactive tracer in metabolic images in the three age groups.
Therefore, at the three ages studied, a clear difference appears between forebrain and midbrain–brain stem structures. Forebrain regions belong to the circuit of initiation and propagation of lithium–pilocarpine SE and are activated by the prolonged seizures, as reflected by the large metabolic increases. Conversely, in midbrain-brainstem regions not directly involved in the circuit of the seizures, metabolic increases remain moderate (Fernandes et al., 1999). These region-specific changes are not apparent in the blood flow response, which is homogeneous throughout the brain; as a result, the mismatch between flow and metabolism is observed at all ages only in forebrain regions. Neuronal damage will develop in all of these areas in adult animals; however, damage will develop in only a few areas in P21 rats, and no damage will develop in P10 rats (Dubé et al., 2001).
In accordance with our data in the P10 rat, newborn marmoset monkeys are completely protected against bicuculline SE-induced brain damage. However, when they are subjected to SE, the following two groups of brain structures can be identified: (1) structures showing a clear mismatch between blood flow and metabolism in which damage develops when SE is induced in adults, and (2) structures showing comparable LCBF and LCMRglc increases that are resistant to seizure-induced damage in adults (Söderfeldt et al., 1990; Wasterlain and Shirasaka, 1994). Thus the immature brain is able to undergo large metabolic increases accompanied by pronounced mismatches between blood flow and metabolism without any evidence of brain damage.
The P21 rat might represent an intermediate age in the maturation processes involved in the metabolic and vascular consequences of severe seizures. The increased fragility of areas such as the hilus of the dentate gyrus, lateral thalamus, and entorhinal cortex might arise from the severe hyperexcitability generated within the primary circuit involved in the epileptic activity rather than from the mismatch between blood flow and metabolism. In most other models of severe, prolonged seizures such as those induced by kainic acid, neuronal injury occurs after the third week of life and increases with maturation (Nitecka et al., 1984; Stafstrom et al., 1992), as in the present study. Most of the damage induced by seizure activity is generated by glutamatergic neurotransmission-driven excitotoxicity. At the moment, changes in the expression of glutamate receptors cannot explain why seizure-induced neuronal injury does not occur before the end of the third week of life (Sperber and Moshe, 2001). However, the degree of Ca2+ entry into the hippocampal CA1 pyramidal cell field is minimal in P1 to P3 rats, whereas it is marked in P21 to P23 rats causing subsequent injury at that age (Marks et al., 1996). In other models of SE (e.g., pentylenetetrazol), no neuronal damage occurs even in P21 rats after approximately 80 minutes of seizure activity. In that model, the lack of neuronal damage may arise from relatively moderate metabolic increases compared with those recorded in the lithium–pilocarpine model and the maintenance of coupling between LCMRglc and LCBF rates (Pereira de Vasconcelos et al., 1992, 1995).
Does the coupling between cerebral blood flow and metabolism during status epilepticus play a critical role in the genesis of neuronal damage in the immature brain?
The goal of this study was to determine whether the resistance of the immature brain to seizure-induced brain damage could result from the maintenance of a tight coupling between flow and metabolism. The present data do not validate this hypothesis because in the immature brain, no neuronal damage is seen regardless of whether blood flow and metabolism are locally coupled. Within protective factors of the developing brain, metabolic factors such as low basal LCMRglc's or the use of ketone bodies for energy metabolism might participate in the resistance of the immature brain to seizure-induced brain damage (Nehlig and Pereira de Vasconcelos, 1993). Moreover, the developing brain, particularly at P10, is mostly dependent on anaerobic glycolysis because oxidative metabolism enzymes mature mostly after P21 (Nehlig and Pereira de Vasconcelos, 1993). Thus, because of the low energetic yield of glycolysis compared with oxidative pathway, any activation at P10 will largely increase glucose breakdown, as seen during lithium–pilocarpine seizures (Fig. 1C) (Fernandes et al., 1999). However, the predominant anaerobic pathway for glucose breakdown and the low basal level of metabolism decrease the capacity of the brain to generate free radicals and oxidative stress compared with the adult brain. Indeed, free radicals generated particularly by the mitochondria, but also by the arachidonic acid metabolism or activated leukocytes, may participate to seizure-related neuronal damage in the adult brain (Liang et al., 2000, Sudha et al., 2001). P21 may represent an intermediate age displaying some of the characteristics of the adult brain and some of the protective mechanisms that operate at P10. The degree of maturation of limbic or cholinergic systems, and of systems such as those involved in the production of free radicals and the genesis of oxidative stress, most likely participate in the sensitivity of brain structures to seizure-induced brain damage at P21.
Whatever the postnatal age, no brain structure shows damage without uncoupling between blood flow and metabolism, but the opposite may occur in the immature brain (i.e., a mismatch between blood flow and metabolism with no brain damage after SE). In these prolonged seizure states, one important point may be whether the cerebral structure is outside or inside the epileptic circuit. Outside the epileptic circuit, the coupling between LCBF and LCMRglc is well preserved at all ages. However, most structures inside the epileptic circuit show uncoupling between metabolism and blood supply, regardless of the postnatal age. This uncoupling might reflect the strong hyperexcitability generated by sustained epileptic activity, which can evolve to excitotoxicity and neuronal death, depending on the degree of maturity.
Therefore, large metabolic increases take place within the structures of the epileptic circuit at all ages. Depending on brain maturity, these increases may or not lead to neuronal injury, mainly as a result of the excitotoxicity generated within neurons during prolonged seizure activity. Conversely, the circulatory response recorded at this already advanced stage of seizure activity can no longer match metabolic demand in the regions of the epileptic circuit, while it is still adjusted to metabolic demand in the regions outside the circuit. Thus, it appears that in the immature brain, the level of coupling between cerebral metabolism and blood flow during the acute phase of SE is not predictable of brain damage.
Footnotes
Acknowledgments:
The authors thank S. Boyet for excellent technical assistance.