
Abstract
Hepatocyte growth factor (HGF), a natural ligand for the c-met protooncogene product, exhibits mitogenic, motogenic, and morphogenic activities for regeneration of the liver, kidney, and lung. Recently, HGF was clearly shown to enhance neurite outgrowth in vitro. To determine whether HGF has a neuroprotective action against the death of neurons in vivo, we studied the effect of HGF on delayed neuronal death in the hippocampus after 5-minute transient forebrain ischemia in Mongolian gerbils. Continuous postischemic intrastriatal administration of human recombinant HGF (10 or 30 μg) for 7 days potently prevented the delayed death of hippocampal neurons under both anesthetized and awake conditions. Even when HGF infusion started 6 hours after ischemia (i.e., in a delayed manner), HGF exhibited a neuroprotective action. We conclude that HGF, a novel neurotrophic factor, has a profound neuroprotective effect against postischemic delayed neuronal death in the hippocampus, which may have implications for the development of new therapeutic strategies for ischemic neuronal damage in humans.
Hepatocyte growth factor (HGF) is a pleiotrophic cytokine that exhibits mitogenic, motogenic, and morphogenic activities toward a variety of cells (Nakamura et al., 1989; Zarnegar and Michalopoulos, 1995; Matsumoto and Nakamura, 1996). Physiologically, HGF plays an important role as an organotrophic factor responsible for vigorous regeneration of the liver, kidney, and lung (Nakamura et al., 1989; Boros and Miller, 1995; Zarnegar and Michalopoulos, 1995; Matsumoto and Nakamura, 1996). Both HGF and the c-Met/HGF receptor of membrane-spanning tyrosine kinase are expressed in various regions of the brain, and functional coupling between HGF and c-Met enhances the survival of hippocampal neurons in primary culture and induces neurite outgrowth on neuronal development in vitro (Jung et al., 1994; Honda et al., 1995; Yamagata et al., 1995; Hamanoue et al., 1996). Hepatocyte growth factor plays a role as a limb mesenchyme-derived chemoattractant for motor axons (Ebens et al., 1996), and is as potent a survival factor for motor neurons as other survival factors described to date, such as brain-derived neurotrophic factor (Oppenheim et al., 1992), ciliary neurotrophic factor (Sendtner et al., 1992), and glial cell line—derived neurotrophic factor (Henderson et al., 1994). This accumulating evidence implies a neurotrophic function of HGF; however, there have been no reports on whether HGF exhibits neurotrophic activity in vivo. In the current study, to determine whether HGF has a neuroprotective action against the death of neurons in vivo, we studied the effect of HGF on delayed neuronal death after transient forebrain ischemia in gerbils (Kirino, 1982) using continuous topical administration of HGF directly into the brain (Miyazawa et al., 1997).
METHODS
Animal preparation
Fifty-eight male Mongolian gerbils weighing 50 to 70 g were used and divided into eight experimental groups. Anesthesia was performed with an initial concentration of 3% halothane, and then maintained with 1.5% halothane in a mixture of 40% O2/60% N2O under a face mask. In experiments 1 and 2, a needle electrode was placed in the subscalpal space for EEG recording to confirm electrical cessation during ischemia. The body temperature was monitored and maintained at 37°C throughout the experiments using a feedback-controlled heating pad.
Experiment 1: Continuous intrastriatal administration of HGF under halothane anesthesia
At first, an osmotic minipump (Alzet model 2001; Palo Alto, CA, U.S.A.) containing human recombinant HGF (10 or 30 μg) or a physiologic saline solution was implanted. A cannula device connected to the subcutaneously implanted osmotic minipump was inserted into the right striatum from a point 1 mm anterior, 2 mm lateral, and 4 mm ventral to the bregma, and fixed with dental cement to the skull. To start infusion just after implantation, each osmotic minipump was incubated in physiologic saline at 37°C according to the instructions of Alzet. The HGF was intrastriatally administered for 1 week during reperfusion. Approximately 30 minutes after implantation of the pump, both carotid arteries were occluded for 5 minutes with Sugita temporary aneurysm clips. Complete forebrain ischemia was confirmed by the electrical cessation on EEG. The animals were divided into four groups as follows:
Group 1 (n = 6): Sham-operated animals. The sham operation included carotid manipulation and cannula placement in the right striatum.
Group 2 (n = 9): Ischemia and implantation of a minipump containing physiologic saline solution
Group 3 (n = 9): Ischemia and implantation of a minipump containing 10 μg HGF
Group 4 (n = 9): Ischemia and implantation of a minipump containing 30 μg HGF
Experiment 2: Delayed start of continuous intrastriatal administration of hepatocyte growth factor
Animals were subjected to 5-minute transient forebrain ischemia similar to that in experiment 1. After recovery from the anesthesia, the animals were returned to their cages and given free access to water. Six hours after the beginning of recirculation, an osmotic minipump including 30 μg HGF was implanted under halothane reanesthesia. The osmotic minipump had been incubated in physiologic saline at 37°C before the experiment. Thus, the administration of HGF started 6 hours after ischemia and continued for 7 days (group 5, n = 9). Group 2 was used as a control group.
Experiment 3: Continuous intrastriatal administration of hepatocyte growth factor under awake conditions
To exclude the possibility of a secondary neuroprotective effect of halothane per se (Edgehouse and Dorman, 1987) and postischemic secondary hypothermia (Corbett et al., 1990; Kuroiwa et al., 1990), animals were subjected to ischemia under awake conditions. Anesthesia was initially performed and an osmotic minipump was implanted without incubation. To accomplish awake forebrain ischemia, the first thread was looped around both common carotid arteries, while the second thread was ligated on the loop. The ends of the first thread were led out through the nuchal muscle and back skin, and then left until the start of ischemia. After closure of the wounds halothane inhalation was discontinued, followed by at least 2 hours until complete recovery from the anesthesia. The animals were subjected to 5-minute forebrain ischemia under awake conditions, without any pain, by occlusion of both common carotid arteries by pulling the ends of the first thread. The loss of the righting reflex was taken as evidence of forebrain ischemia under awake conditions. The ends of the second thread were pulled to release the carotid occlusion. In this experiment, since the osmotic minipumps were not incubated at 37°C, the infusion usually started 3 to 4 hours after implantation (i.e., 1 to 2 hours after ischemia). The animals were divided into two groups as follows:
Group 6 (n = 7): Awake ischemia and implantation of a minipump containing physiologic saline solution
Group 7 (n = 7): Awake ischemia and implantation of a minipump containing 30 μg HGF
Experiment 4: Pathologic observation of the striatum after administration of hepatocyte growth factor
In the animals in group 8 (n = 2), 30 μg HGF was infused for 7 days into the striatum without ischemia; the brains were investigated pathologically to determine whether HGF had a toxic effect on glia and neurons of the striatum. Coronal sections of the two brains, passing through the trajectory of the cannula in the striatum, were stained with hematoxylin and eosin for pathologic observation.
Evaluation
Seven days after ischemia, the animals were transcardially perfused with 4% paraformaldehyde in 0.01 mol/L phosphate-buffered saline. Infusion of the agent into the striatum was confirmed by the implanted pump being empty when the animals were killed. The brains were removed, immersed in the same fixative for 24 hours, and then embedded in paraffin. Three-micrometer paraffin sections were stained with cresylviolet. The number of intact neurons in the center of the hippocampal CA1 sector was determined, averaged, and expressed as both the neuronal density per 1 mm linear length. The investigator performing the cell counting was blind with regard to the treatment of each section. Statistical significance was analyzed using one-factor analysis of variance, followed by Scheffe's F test. Data are expressed as means ± SD (Fig. 1).
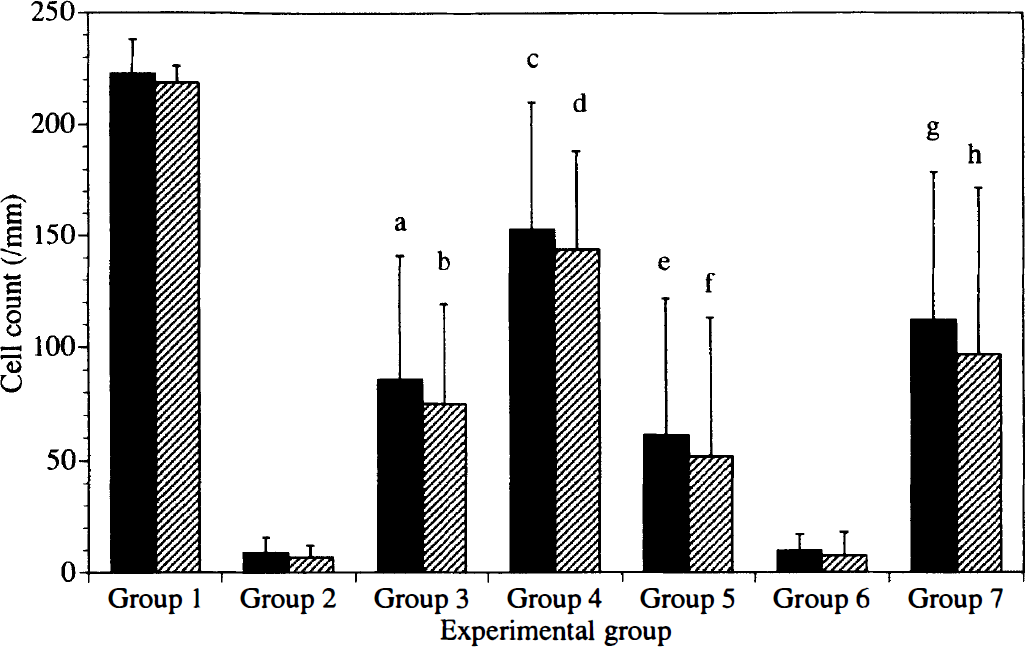
Graphic representation of the number of viable neurons in the center of a bilateral hippocampal CA1 sector. The number of viable neurons was determined, averaged, and expressed as the neuronal density per 1 mm linear length, the data being expressed as mean ± SD. Black bars, right side; hatched bars, left side. Group 1 (n = 6); Sham-operated animals. Group 2 (n = 9): ischemia and implantation of a minipump containing PSS. Group 3 (n = 9): ischemia and implantation of a minipump containing 10 μg HGF. Group 4 (n = 9): ischemia and implantation of a minipump containing 30 μg HGF. Group 5 (n = 9): delayed start of HGF infusion 6 hours after ischemia. Group 6 (n = 7): awake ischmeia and implantation of a minipump containing PSS. Group 7 (n = 7): awake ischemia and implantation of a minipump containing 30 μg HGF. Statistical differences were evaluated by one factor analysis of variance followed by Scheffe's F-test. Groups 3, 4, and 5 were compared with group 2 on each side. Group 7 was compared with group 6 on each side. a, b, c, and d: P < 0.001 versus group 2; e and f: P < 0.05 versus group 2; g and h: P < 0.01 versus group 6.
RESULTS AND DISCUSSION
The current results constitute the first hard evidence that HGF can potently protect hippocampal neurons from ischemic insult in vivo. At first, gerbils were subjected to transient forebrain ischemia under halothane anesthesia, and then human recombinant HGF or saline was infused using an osmotic minipump. The absolute values per 1 mm of intact neurons in a bilateral CA1 sector of the hippocampus after 7 days of reperfusion are given in Fig. 1. In control animals infused with saline (group 2), only 3% to 4% of the neurons survived compared with sham-operated animals (group 1) after forebrain ischemia. In contrast, continuous intrastriatal administration of HGF for 7 days during reperfusion prevented the delayed neuronal death in a dose-dependent manner (groups 3 and 4). In animals infused with 30 μg HGF (group 4), approximately two thirds of the neurons survived compared with sham-operated animals, with the number of surviving neurons being approximately 20-fold higher than in saline-infused animals.
With regard to the clinical application of neurotrophic factors, they must be neuroprotective when administered after, not before or during, cerebral ischemia. In most studies on the neuroprotective effects of neurotrophic factors on a brain ischemia model, however, the neurotrophic factors usually were administered before the initiation of ischemia (Shigeno et al., 1991; Beck et al., 1994; Tsukahara et al., 1994). We therefore examined whether HGF has a neuroprotective action, even when HGF infusion started 6 hours after forebrain ischemia. About a quarter of the neurons survived in animals subjected to delayed HGF infusion (group 5), which was approximately sevenfold higher than in control saline-infused animals. Thus, the delayed infusion of HGF significantly prevented the delayed neuronal death of CA1 pyramidal neurons after forebrain ischemia. The significant neuroprotection by HGF infused in a delayed manner is encouraging for the practical application of HGF for the treatment of patients with ischemic neuronal injury.
During the verification of neuroprotective agents, the neuroprotective effects of halothane per se (Edgehouse and Dorman, 1987) and postanesthetic secondary hypothermia (Corbett et al., 1990; Kuroiwa et al., 1990) were found to frequently modify experimental results. To exclude the possibility of such secondary neuroprotective effects, animals were subjected to ischemia under awake conditions. At 2 hours after discontinuation of anesthesia, the animals were subjected to 5-minute forebrain ischemia under awake conditions. The HGF infusion substantially began by 1 to 2 hours after ischemia. In the ischemic animals under awake conditions, 30 μg HGF significantly prevented the delayed neuronal death even after awake ischemia (group 7) compared with the data for the control group (group 6), thereby indicating that the neuroprotective effect of HGF did not result from postanesthetic secondary hypothermia or halothane per se. Furthermore, the fact that ischemia probably was more severe under awake conditions than under halothane anesthesia (Imon et al., 1991) strongly supports the neuroprotective effect of HGF.
Since hippocampal CA1 neurons in rats express c-Met mRNA and HGF prolongs the survival of hippocampal neurons in primary culture (Honda et al., 1995), we presume that the neuroprotective action of HGF on CA1 neurons is mediated by c-Met. Yamagata and others (1995) demonstrated selective HGF mRNA expression in the microglia of rat brain, and c-Met mRNA expression in neurons as well as in astrocytes and microglia. This also implies the possibility of interactions between neurons, astrocytes, and microglia through HGF and its receptor. In our study, the influence of long-lasting postischemic mild hypothermia from HGF per se could not be completely excluded (Dietrich et al., 1993; Colbourne and Corbett, 1994), and we do not have data on the effect of HGF at the chronic stage (1 to 2 months after ischemia). Since previous studies clearly demonstrate a neurotrophic effect of HGF on neurons in primary culture under strict control through the medium temperature (Honda et al., 1995), we believe that the neurotrophic effect of HGF after forebrain ischemia persists longer, rather than that HGF delays the progression of “delayed neuronal death,” as seen in hypothermic animals (Dietrich et al., 1995).
It is crucial to determine the appropriate amount of each neurotrophic factor for the neuroprotective action according to either the type of ischemic model or the mode of administration. Since there was no degenerative change of neurons or glia around the trajectory of the cannula, except for the traumatic injury caused by the cannula penetration, a toxic effect of HGF per se could be excluded (experiment 4, data not shown). Although the exact cellular mechanism of the neuroprotective activity of HGF is not known, our results should facilitate further studies on different modes of HGF administration as well as on the effective time window for the neuroprotective action of HGF toward ischemic neuronal injury.
Footnotes
Acknowledgments
The authors thank Miss Namiko Nomura and Miss Akiko Yano for their excellent technical assistance.