
Abstract
Hepatocyte growth factor (HGF) is an interesting candidate for acute stroke treatment as shown by continuous infusion or gene delivery protocols. However, little is known about HGF-mediated long-term effects. The present study therefore analyzed long-term effects of an acute intrastriatal HGF treatment (5 μg) after a 45-minute stroke, with regard to brain injury and neurologic recovery. Hepatocyte growth factor induced long-term neuroprotection as assessed by infarct volume and neuronal cell death analysis for as long as 4 weeks after stroke, which was associated with sustained neurologic recovery as evidenced by corner-turn and tight-rope tests. Analyzing underlying mechanisms of HGF-induced sustained neuroprotection, enhanced cell proliferation followed by increased neuronal differentiation of neural precursor cells (NPCs) was observed in the ischemic striatum of HGF-treated mice, which persisted for up to 4 weeks. In line with this, HGF promoted neurosphere formation as well as proliferation of NPC and decreased caspase-3-dependent hypoxic injury in vitro. Preservation of blood—brain barrier integrity 24 hours after stroke was furthermore noticed in animals receiving HGF, which was associated with the inhibition of matrix metalloproteases (MMP)-2 and MMP-9 at 4 and 24 hours, respectively. We suggest that sustained recruitment of proliferating cells together with improved neurovascular remodeling provides an explanation for HGF-induced long-term neuroprotection.
Keywords
Introduction
Hepatocyte growth factor (HGF) exhibits unique features that make it a promising agent for stroke treatment. In human patients, blood HGF levels in the acute stroke phase correlate closely with clinical recovery in the postacute stroke phase, more closely than other growth factors (Ozaki et al, 2007), suggesting that HGF induces favorable responses in the brain tissue that facilitate brain remodeling. Several studies showed that HGF and its corresponding receptor c-Met, which are present in the mature brain (Achim et al, 1997; Honda et al, 1995), exert mitogenic, morphogenic, angiogenic, and antiapoptotic effects on various kinds of tissues and cells (Matsumoto and Nakamura, 1996; Nakamura et al, 1989). In these studies, reasons for the beneficial effects in the brain remained unclear.
Therapeutic approaches using HGF in experimental stroke research have so far focused on structural tissue preservation in the acute stroke phase (Date et al, 2006; Miyazawa et al, 1998; Niimura et al, 2006a, 2006c; Shang et al, 2010; Shimamura et al, 2004). These studies have mostly been performed on rats using either continuous infusion or gene delivery strategies in models of focal or global cerebral ischemia. Tissue reorganization in the subacute stroke phase similarly as long-term functional outcome after the discontinuation of treatment have not been examined in these studies. As such, the question remains open, why HGF is able to induce persisting neurologic recovery.
In view of their pleiotropic effects on cell proliferation, differentiation, and survival, growth factors have the potential to influence tissue remodeling in a profound way far beyond the time point of delivery (Greenberg and Jin, 2006; Hermann and Zechariah, 2009). Notably, the adult brain possesses its own source of resting progenitor cells, that is, neural precursor cells (NPCs) in the subventricular zone (SVZ) (Ma et al, 2009; Zhao et al, 2008). Upon ischemia, SVZ-derived NPCs proliferate and migrate toward the developing brain lesion (Arvidsson et al, 2002; Yamashita et al, 2006). Whereas many of the newborn cells undergo degeneration in the postacute stroke phase (Dayer et al, 2003; Yoo et al, 2008), the survival of NPCs can greatly be enhanced by growth factors or antiapoptotic proteins (Doeppner et al, 2009; Wada et al, 2003; Wang et al, 2004).
In view of the promising features of HGF in human stroke patients, we were interested to see whether HGF, which initially had been characterized as organotrophic growth factor outside the brain (Matsumoto and Nakamura, 1996), may induce sustained neuroprotection when delivered in the acute phase of a stroke. Furthermore, the question whether or not tissue survival translates to functional neurologic recovery was addressed. We therefore conducted the present experiments, in which we administered HGF intrastriatally over a time interval of 3 days, analyzing subsequent processes of tissue remodeling, with special focus on the endogenous proliferation and differentiation of NPCs.
Materials and methods
Animals
Experimental procedures were performed according to the European Institutes of Health guidelines for the care and use of laboratory animals and approved by local authorities. Animals were housed under a regular circadian rhythm regimen with free access to food and water. In total, 119 (including sham operations) adult male C57/BL6N mice weighing 23 to 28 g were used. If not stated otherwise, each experimental group consisted of eight to nine animals. Mortality rates for animals of experimental groups including survival periods for up to 7 days were zero for each experimental condition. For animals surviving 4 weeks after induction of stroke, we observed survival rates of 88.9% (vehicle treated) and 87.5% (HGF treated). All experiments were performed in a blinded manner with regard to the kind of treatment chosen for each animal. For analysis of postischemic cell proliferation, animals were intraperitoneally injected with 5-bromo-2-deoxyuridine (BrdU; Sigma, Steinheim-Taufkirchen, Germany) on days 4 to 6 (7-day survival) or on days 8 to 18 (28-day survival), with a daily dose of 50 mg/kg body weight.
Induction of Focal Cerebral Ischemia
Induction of focal cerebral ischemia was performed as previously described (Doeppner et al, 2009). Briefly, animals were anesthetized (0.8% to 1.5% isoflurane, 30% O2, remainder N2O) and rectal temperature was maintained employing a feedback-controlled heating system. Regional cerebral blood flow was assessed using a laser Doppler flowmeter connected to a probe that was attached to the skull overlying the core region of the middle cerebral artery territory. Cerebral ischemia was induced by transient occlusion of the middle cerebral artery using a silicon-coated filament (180 μm diameter; Doccol, Redlands, CA, USA), which has been removed after 45 minutes to allow reperfusion of the middle cerebral artery. Laser Doppler flowmeter recordings continued for 15 minutes after thread removal to monitor appropriate reperfusion (>80% of initial regional cerebral blood flow). Sham-operated animals underwent the same experimental procedure except for insertion of silicon-coated monofilaments.
Stereotactic Intracerebral Injections
Stereotactic surgeries were performed during reperfusion under constant anesthesia as described above. The mice were placed in a stereotactic apparatus (Kopf Instruments, Düsseldorf, Germany) and the skull was exposed followed by drilling a hole at 0.4 mm anterior and 1.8 mm lateral (left ischemic hemisphere) from bregma. From these coordinates, intrastriatal injections were made 3.5 mm ventral to the bregma using a 10-μL Hamilton syringe (Switzerland). Control mice received 5 μL of 0.1 mol/L phosphate-buffered saline (PBS) per animal and day as vehicle, whereas treated animals obtained HGF at doses of 1, 5, or 10 μg on occasion of each injection, diluted in 5 μL 0.1 mol/L PBS. At the end of injection, the syringe was kept in place for additional 5 minutes before removal. For experiments with survival periods of 7 or 28 days, all animals received additional injections on days 1 to 3 using the same protocol as described above. For dose-finding studies, HGF was kindly provided by Genentech (San Francisco, CA, USA), whereas for the remaining experiments, HGF was purchased from R&D Systems (Wiesbaden-Nordenstadt, Germany). In the latter experiments, an HGF dose of 5 μg per animal and day was used.
Immunohistochemistry and Terminal Transferase dUTP Nick End Labeling
Animals were intraperitoneally injected with chloralhydrate (420 mg/kg body weight) and transcardiacally perfused with 4% paraformaldehyde on day 7 or day 28 after stroke. Brains were removed, postfixed in paraformaldehyde, embedded in paraffin, and coronal sections were prepared. Sections were deparaffinized, boiled in 0.2% citrate buffer, and incubated with blocking solution. For analysis of BrdU+ cells and Ki-67+ cells, sections were exposed to blocking solution and subsequently stained with a rat monoclonal anti-BrdU antibody (1:50; 18 hours, 4°C; Serotec, Düsseldorf, Germany) or a rabbit monoclonal anti-Ki-67 antibody (1:100; 18 hours, 4°C; Abcam, Cambridge, UK), followed by incubation with a goat antirat Alexa 594 (1:500, 1 hour; Molecular Probes, Darmstadt, Germany) or a donkey anti-rabbit Alexa Fluor 568 (1:400, 1 hour; Molecular Probes) secondary antibody. For differentiation analysis of BrdU+ cells, sections were incubated with a mouse monoclonal anti-BrdU antibody (1:500; Roche, Basel, Switzerland), a goat polyclonal antidoublecortin antibody (1:50; Santa Cruz Biotechnology, Heidelberg, Germany), a rat polyclonal anti-glial fibrillary acidic protein antibody (1:500; Zymed, Darmstadt, Germany), or a rat biotin-conjugated antiisolectin B4 (IB4) antibody (1:100; Vector, Peterborough, UK) for 18 hours at 4°C. For further microglia analysis, colocalization of IB4 with major histocompatibility complex (MHC)-II was performed using a mouse monoclonal anti-MHC-II antibody (1:200; Santa Cruz Biotechnology). For double staining against BrdU and neuronal nuclei (NeuN) or 2',3'-cyclic nucleotide 3'-phosphodiesterase, a rat monoclonal anti-BrdU antibody (see above) and a mouse monoclonal anti-NeuN antibody (1:200, 18hours, 4°C; Chemicon, Schwalbach, Germany) or a mouse monoclonal anti-2',3'-cyclic nucleotide 3'-phosphodiesterase antibody (1:400; Chemicon) were used. After washing, sections were incubated for 1 hour at room temperature with the following secondary antibodies: goat anti-mouse Cy-3 (1:400; Dianova, Hamburg, Germany) or goat anti-rat Alexa 594 (1:400; Dianova) for MHC-II and BrdU staining, goat anti-rat Alexa 488 (1:250; Molecular Probes) or donkey anti-goat Alexa 488 (1:250; Molecular Probes) for glial fibrillary acidic protein or doublecortin staining, goat anti-mouse Cy-3 (1:100; Jackson Immuno-Research, Newmarket, UK) for 2',3'-cyclic nucleotide 3'-phosphodiesterase staining, and goat anti-mouse Alexa 488 (Molecular Probes) for NeuN (1:400) staining. Terminal transferase dUTP nick end labeling (TUNEL) staining was performed incubating sections with proteinase K (7 minutes, 37°C), followed by exposure to the TdT enzyme reaction according to the manufacturer's manual (Roche). After several washing steps and exposure to TUNEL blocking solution (20 minutes, room temperature), sections were stained with a streptavidin-Alexa594-conjugated secondary antibody (2 hours, room temperature; Molecular Probes). Quantitative analyses were performed in four regions of interest within the ischemic striatum located 0.14 mm anterior, 2.5 to 3.25 mm ventral, and 1.5 to 2.25 mm lateral from bregma. For each regions of interest, three sections per animal were evaluated, for which mean values were computed.
Infarct Volumetry and Determination of Edema Formation
Infarct volume analysis was performed on days 1 and 7 after stroke. Brains were removed and cut into four slices of 2 mm each. Slices were stained with 2,3,5-triphenyltetrazolium chloride (2%), and computer-based analysis of infarct volumes was performed using the software Image J by subtracting the area of the nonlesioned ipsilateral hemisphere from that of the contralateral side. Infarct volume sizes were calculated by integration of the lesioned areas. Postischemic brain edema was measured also using Image J software as the increase of ipsilateral hemispheric volume in comparison to the contralateral hemisphere as described by Wacker et al (2009).
Analysis of Motor Coordination Deficits
For analysis of poststroke motor coordination deficits, animals were evaluated using the corner-turn and tight-rope tests. One day before induction of stroke, all animals were trained before the beginning of the actual tests on days 7, 14, 21, and 28 after ischemia. Each test was performed twice per time point, for which means were calculated. For the tight-rope test, animals were placed on a 60-cm long tight rope grasping the string with their forepaws. Healthy animals grasp the string with four legs and tail and move to reach a side pole, whereas mice suffering from ischemia-induced motor deficits cannot lift their hind legs and eventually fall off onto the cage bedding placed underneath. Maximum test time was 60 seconds and the results were scored from 0 (min) to 20 (max) according to a validated score (Gerlai et al, 2000) depending on the time animals spent on the rope and whether or not they reached the platform. Scores were used for statistical analysis.
For the corner-turn test, animals were placed in a corner consisting of two vertical boards attached at one side with an angle of 30°. Each mouse was tested for the side chosen over 10 trials per day, that is right or left rearing in the corner. Whereas healthy animals leave the corner without side preference, mice suffering from stroke preferentially turn to the left, nonimpaired body side when leaving the corner. The laterality index was calculated according to the following formulae: (number of left turns—number of right turns)/10.
Blood—Brain Barrier Permeability
For this study, animals that were treated with vehicle, HGF (see above), or the matrix metalloprotease (MMP) inhibitor BB-1101 (0.75 mg per animal) at the beginning of reperfusion were used. Mice received intravenous injections of 2% Evans Blue (2 mL/kg) 22 hours after the stroke, as previously described (Chiba et al, 2008). Two hours later, animals were killed by transcardiac perfusion with 0.1 mol/L PBS. Brains were removed and separated into hemispheres. Left hemispheres were weighed, homogenized in 2 mL of 50% trichloroacetic acid, and centrifuged at 10,000 r.p.m. for 20 minutes. The extracted Evans Blue dye was then further diluted with ethanol, and the fluorescence signal was measured with a luminescence spectrophotometer (excitation at 620 nm, emission at 680 nm). An external standard (62.5 to 500 ng/mL) was used for the calculation of Evans Blue contents. Evans Blue contents of left hemisphere tissue was calculated from four animals as (μg) Evans Blue per (g) tissue.
Zymography of Matrix Metalloproteases
Left hemispheres from ischemic and nonischemic animals were homogenized in cold lysis buffer (basic buffer) containing 50 mmol/L Tris-HCl (pH 7.6), 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3, and 1% Triton X-100. Homogenates were centrifuged at 4°C at 12,000 g for 5 minutes and supernatants were thereafter incubated with a 1:10 volume of gelatin sepharose 4B for 1 hour at 4°C with constant shaking. After centrifugation and resuspension of pellets in elution buffer (basic buffer containing 10% dimethyl sulfoxide (DMSO) and 20% volume of lysis buffer), purified samples were analyzed by zymography. Protein concentrations were determined by the bicinchoninic acid method (BCA kit, Thermo Scientific, Bonn, Germany).
Separation of MMP-2 and MMP-9 as pro-form and active form was performed using Novex Zymogram Gels (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Briefly, samples were incubated in nonreducing sample buffer (0.4 mol/L Tris, pH 6.8, 5% sodium dodecyl sulfate, 20% glycerol, 0.05% bromophenol blue) for 10 minutes at room temperature. Thereafter, the samples were loaded onto 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels containing 0.1% gelatin. After electrophoresis, the samples were incubated with 2.5% Triton X-100 twice for 20 minutes, equilibrated with developing buffer (Novex), and incubated for over 18 hours at 37°C. Gels were stained with Coomassie blue for 30 minutes and destained in washing solution (30% methanol, 10% acetic acid). White bands on a dark background indicated zones of digestion corresponding to the presence of pro-MMPs and activated MMPs on the basis of their molecular weight. As standards, 0.1 ng of human pro-MMP-9 and human pro-MMP-2 (Merck Biosciences, Darmstadt, Germany) and 0.01 ng of activated MMP-9 and activated MMP-2 (Merck Biosciences) were used. Thereafter, gels were scanned and densitometrically analyzed.
Preparation and Cultivation of Subventricular Zone-Derived Neural Precursor Cells
Subventricular zone-derived NPCs were prepared from 11-to 12-week-old C57/BL6N mice according to a previously described protocol (Doeppner et al, 2010). Subventricular zones were microdissected under stereomicroscopic control and minced into small pieces, mechanically triturated, and dissociated to single-cell suspension. Cells were cultured in serum-free basic medium (DMEM-F12; PAA, Pasching, Austria), supplemented with epidermal growth factor (2 μg/mL), basic fibroblast growth factor (2 μg/mL), and penicillin-streptomycin (Invitrogen, Darmstadt, Germany). Cells were incubated under standard cell culture conditions and growth factors were added every 2 to 3 days. Cells were passaged every 7 to 10 days and used for experiments after passage 3 or 4.
Oxygen—Glucose Deprivation
For each experiment, 100,000 NPCs were used (n = 4). Before induction of oxygen—glucose deprivation (OGD), cell culture medium was substituted by a crystalloid solution (‘Thomajodin’ plus 1 mM mannitol; DeltaPharm, Dortmund, Germany), and the cells were incubated in a hypoxic chamber (1% O2, 5% CO2, and 94% N2) for 45 minutes and reincubated in normal glucose containing cell culture medium for 24 hours. Control cells were incubated with 0.1 mol/L PBS 1 hour before OGD, whereas other cells were treated with recombinant human HGF at a final concentration of 20 ng/mL. The number of dead cells was counted using a LIVE/DEAD-Viability-Cytotoxicity-Assay kit (Lonza, Basel, Switzerland).
Caspase-3 Assay
Caspase-3 activity was measured in cultivated NPCs (n = 4 per condition) at 1, 2, 4, and 8 hours after OGD using the fluorophore substrate Ac-DEVD (aspartic acid-glutamic acid-valine-aspartic acid)-7-amino-4-methylcoumarin (Biomol, Hamburg, Germany). Cells were incubated either with HGF (20 ng/mL) or with the caspase-3 inhibitor z-DEVD.fmk (50 μmol/L, solved in 0.2% DMSO; Biomol) at the beginning of OGD. Controls were incubated with either 0.1 mol/L PBS or 0.2% DMSO in 0.1 mol/L PBS. At the time points given, NPCs were complemented with lysis buffer that contained 25 mmol/L HEPES (pH 7.5), 1 mM EDTA, 10 mM 1,4-dithiothreitol (Roche), 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (Sigma-Aldrich), 10% sucrose, 0.1% Triton X-100, with 10 μg/mL of pepstatin A, aprotinin, and leupeptin each plus 1 mM PMSF (all Sigma-Aldrich) as protease inhibitors. Thereafter, the cells were centrifuged and supernatants were used for caspase-3 activity measurement. For each measurement, 90 μL of samples were incubated with 10 μL of substrate (50 μmol/L final concentration) and fluorescence (355 to 460 nm) was detected at 2-minute intervals for 20 minutes. The increase in fluorescence was linear between 2 and 16 minutes. Caspase-3 activity was recalculated from the slope (fluorescence units per time) and is given as picomoles of substrate cleaved per milligram of protein per minute based on a standard curve for 7-amino-4-methylcoumarin (Biomol). Protein concentration in the supernatant was determined using the Bradford assay.
Statistics
All data are given as means ± s.d. For comparison between two groups, the Student's t-test was used, whereas for a multigroup comparison, a one-way analysis of variance followed by the Tukey's post hoc test was performed. A P value of < 0.05 was considered to be statistically significant.
Results
Hepatocyte Growth Factor Induces Neuroprotection in a Dose-Dependent Manner
Although HGF-induced neuroprotection has been described before, these studies were mostly performed on rats and included either continuous release or overexpression of HGF. We therefore first determined whether or not a single injection of HGF is sufficient to induce neuroprotection after transient focal cerebral ischemia in mice (Figure 1A). When 1 μg of HGF was injected into the ischemic striatum during early reperfusion, no effect on infarct size was observed 24 hours after stroke. On the contrary, HGF at dosages of 5 or 10 μg, respectively, yielded significant neuroprotection after stroke. In line with this, ipsilateral edema formation was significantly reduced in animals treated with 5 or 10 μg of HGF, but not in animals treated with 1 μg of HGF (Figure 1B). Hepatocyte growth factor-induced neuroprotection was not associated with changes in regional cerebral blood flow (Figure 1C); animals from all groups exhibited similarly high reperfusion rates. As no significant difference with regard to infarct volume and edema formation between animals treated with 5 or 10 μg of HGF was observed, further in vivo experiments were performed using doses of 5 μg only.
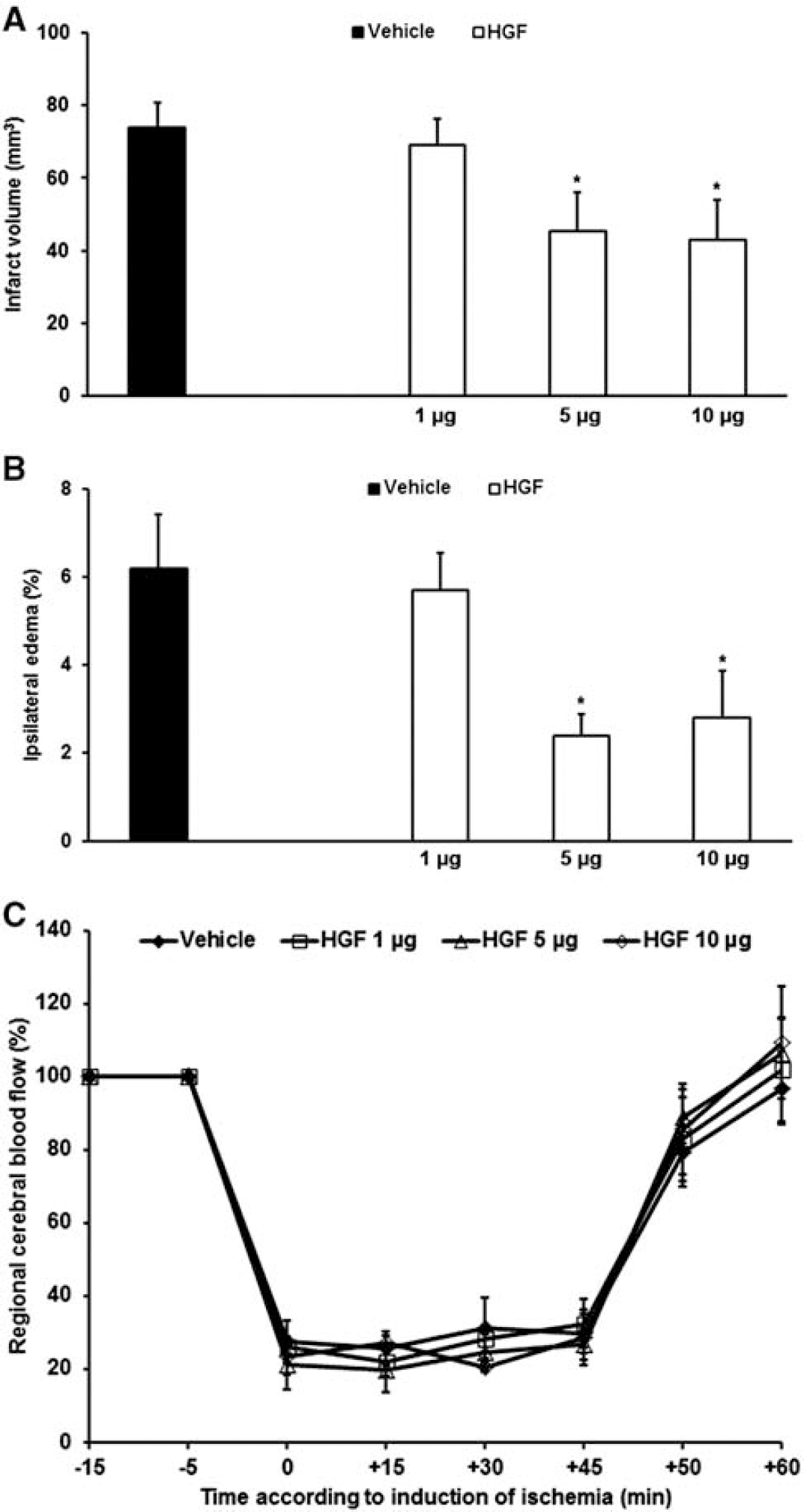
Hepatocyte growth factor (HGF) reduces postischemic injury and edema formation in a dose-dependent manner. Hepatocyte growth factor was injected at different dosages into the left ischemic striatum at the beginning of reperfusion followed by infarct analysis 24 hours after stroke (
Hepatocyte Growth Factor-Induced Neuroprotection Persists in the Postacute Stroke Phase, and Is Associated with Ameliorated Motor Coordination Deficits
As acute reduction of infarct size and edema formation (Figure 1) does not imply sustained neuroprotection or beneficial functional outcome, ischemic injury was further analyzed on both day 7 and day 28 after the stroke (Figure 2). Treatment with HGF resulted in persistent neuroprotection as assessed by means of both TUNEL (day 7) and NeuN (day 28) staining (Figure 2A). We observed 407.6 ± 54.8 TUNEL+ and 701.8 ± 62.2 NeuN+ cells per mm2 within the periinfarct area of HGF-treated animals compared with 649.0 ± 80.3 TUNEL+ and 533.3 ± 50.9 NeuN+ cells per mm2 in control animals. In line with the sustained neuroprotection, animals treated with HGF showed reduced postischemic functional deficits as assessed by both the corner-turn and the tight-rope tests (Figures 2B and 2C). Although animals from both vehicle and HGF groups gradually improved over time, animals treated with HGF always performed better than vehicle-treated animals, thereby almost approximating test performance of healthy sham-operated animals.
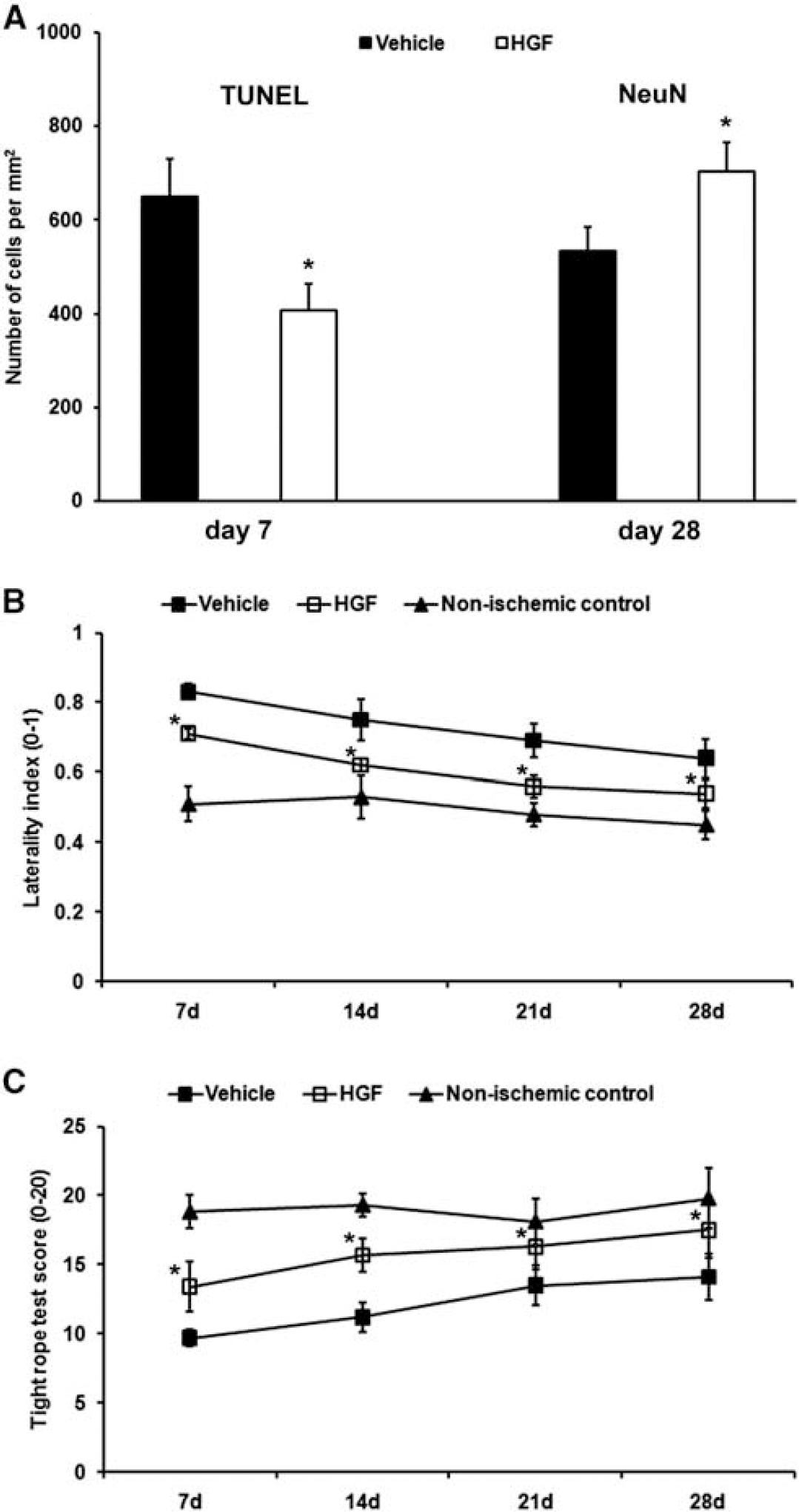
Sustained neuroprotection and amelioration of functional neurologic deficits induced by hepatocyte growth factor (HGF). Treatment with HGF significantly reduced the number of terminal transferase dUTP nick end labeling (TUNEL+) cells on day 7 after stroke (
Hepatocyte Growth Factor Stabilizes Blood—Brain Barrier Integrity via Inactivation of Matrix Metalloproteases
As development of acute infarct injury involves enhanced blood—brain barrier (BBB) permeability, we analyzed HGF-mediated effects on Evans Blue extravasation 24 hours after induction of stroke. Whereas control animals showed distinct Evans Blue extravasation, animals treated with HGF displayed significantly reduced extravasation of the dye (Figure 3A). Similarly, treatment with BB-1101 (0.75 mg per animal), a broad inhibitor of MMPs that are critically involved in BBB breakdown, also resulted in reduced Evans Blue extravasation. Whereas HGF treatment lead to subacute neuroprotection as suggested by decreased infarct volumes on day 7, animals treated with BB-1101 did not benefit from this therapy. As such, infarct volume was not influenced by BB-1101 (Figure 3B). Matrix metalloproteases gelatin zymography (Figures 3C-3E) revealed activation of MMP-2 in vehicle-treated control animals 4 hours after the stroke, which was not found in sham-operated animals. Both HGF and BB-1101 treatments reduced MMP-2 activation without changing levels of inactive pro-MMP-2. Activation of MMP-2 was, however, only transient, decreasing below levels of detection within 24 hours after stroke. On the other hand, active MMP-9 was only detected as late as 24 hours after stroke. Matrix metalloprotease-9 but not pro-MMP-9 activity was again reduced by HGF and BB-1101.
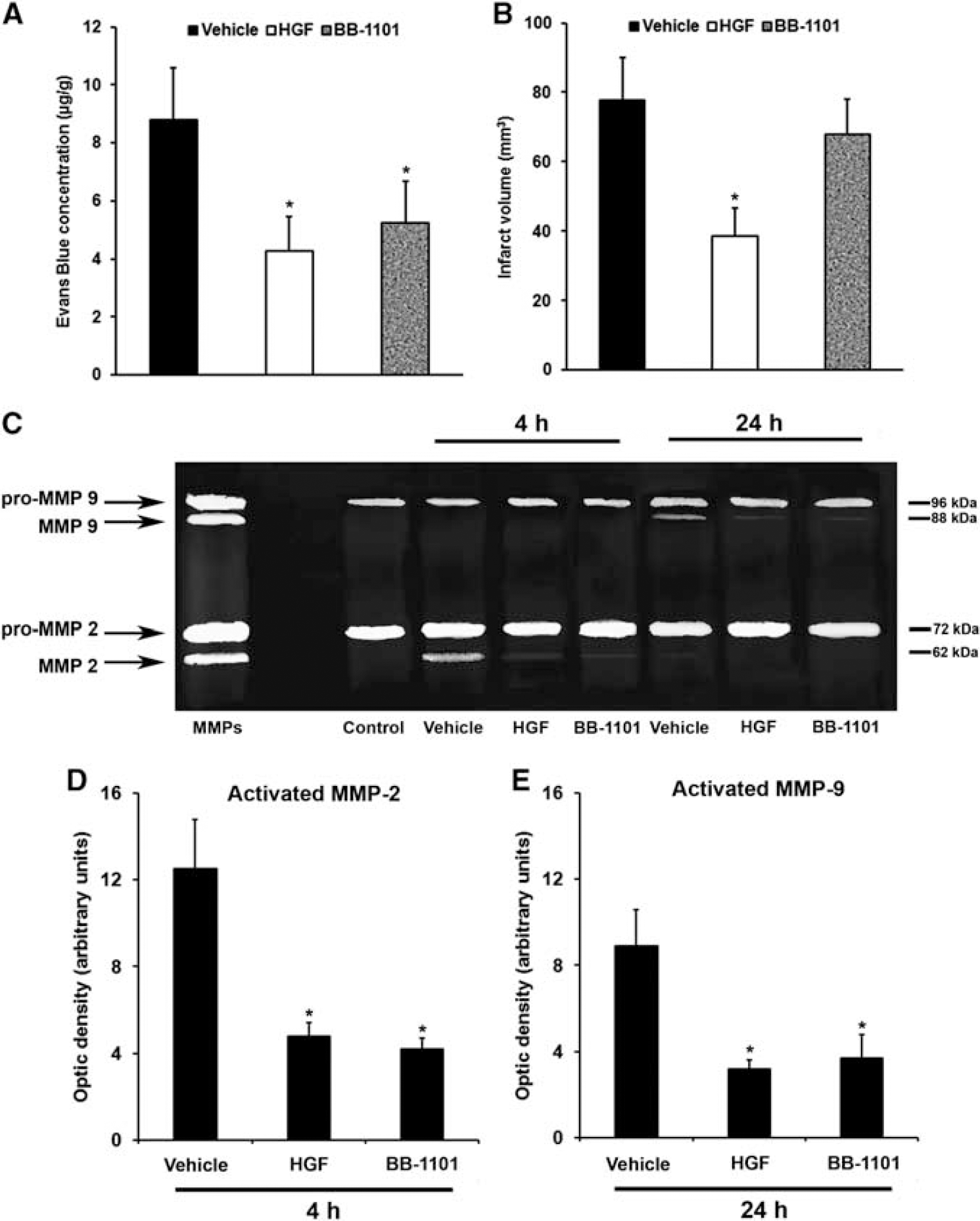
Hepatocyte growth factor (HGF) reduces blood—brain barrier (BBB) permeability and inhibits matrix metalloproteases (MMP). Blood—brain barrier permeability (
Hepatocyte Growth Factor Enhances Postischemic Cell Proliferation
To further elucidate the mechanisms underlying the sustained neuroprotection and recovery induced by HGF, cell proliferation was analyzed using the thymidine analog BrdU on day 7 and day 28 after stroke (Figure 4A). Although cell proliferation declined over time within the ischemic striatum of animals treated with either HGF or vehicle, the number of BrdU+ cells in animals treated with HGF was always higher than in vehicle-treated controls. As BrdU labeling does not exclusively reflect current cell proliferation, analysis of Ki-67+ cells was additionally performed at the time points given (Figure 4B). Similarly, HGF treatment resulted in enhanced numbers of Ki-67+ cells for up to 4 weeks after induction of stroke, demonstrating that HGF enhances postischemic endogenous cell proliferation.
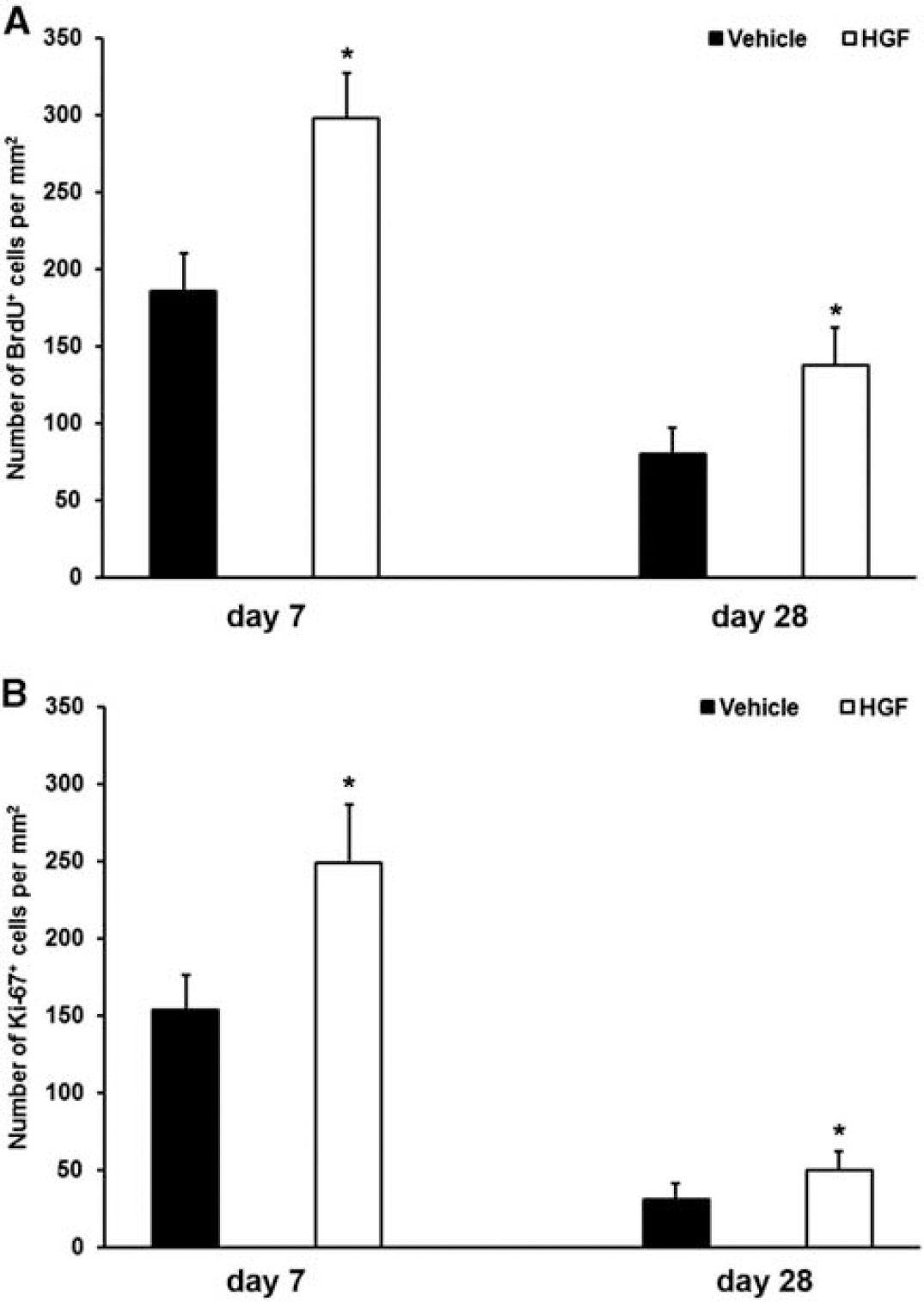
Hepatocyte growth factor (HGF) promotes postischemic cell proliferation. Postischemic treatment with HGF resulted in increased cell proliferation as assessed by 5-bromo-2-deoxyuridine (BrdU) (
Endogenous Poststroke Neurogenesis Is Enhanced After Hepatocyte Growth Factor Treatment
Differentiation analysis of BrdU+ cells revealed high relative amounts of BrdU+ cells expressing the microglial marker IB4 on day 7 after stroke, suggesting that a majority of proliferating cells rather reflect microglia than newborn neural cells (Figure 5A). There was, however, no significant difference between animals treated with either HGF or vehicle. With regard to the state of activation of IB4+ microglial cells, double staining against IB4, and MHC-II on day 7 revealed no difference between vehicle and HGF group. We observed 93.1% ± 12.5% in the vehicle and 89.6% ± 7.5% of colocalizations in the HGF group, representing a high state of activation of microglial cells. Regarding neural differentiation of BrdU+ cells, animals treated with HGF showed significantly higher numbers of BrdU+ cells expressing the immature neural marker nestin than controls, albeit cell numbers declined over time in both experimental groups (Figure 5B). On the contrary, the number of BrdU+ cells expressing glial fibrillary acidic protein gradually increased over time in animals treated with both HGF or vehicle (Figure 5C). There was, however, no significant difference between these two groups. Likewise, oligodendroglial differentiation of BrdU+ cells was not affected by HGF (Figure 5D). Interestingly, long-term neuronal differentiation was enhanced in animals treated with HGF compared with vehicle-treated mice (Figures 5E–5H). Hepatocyte growth factor treatment resulted in increased numbers of BrdU+ cells expressing either doublecortin or NeuN at day 28 after stroke when compared with control animals. Whereas no mature neuronal differentiation of newborn proliferating cells was observed on day 7 (data not shown), the number of BrdU+ cells expressing NeuN was still low on day 28 in both experimental groups.
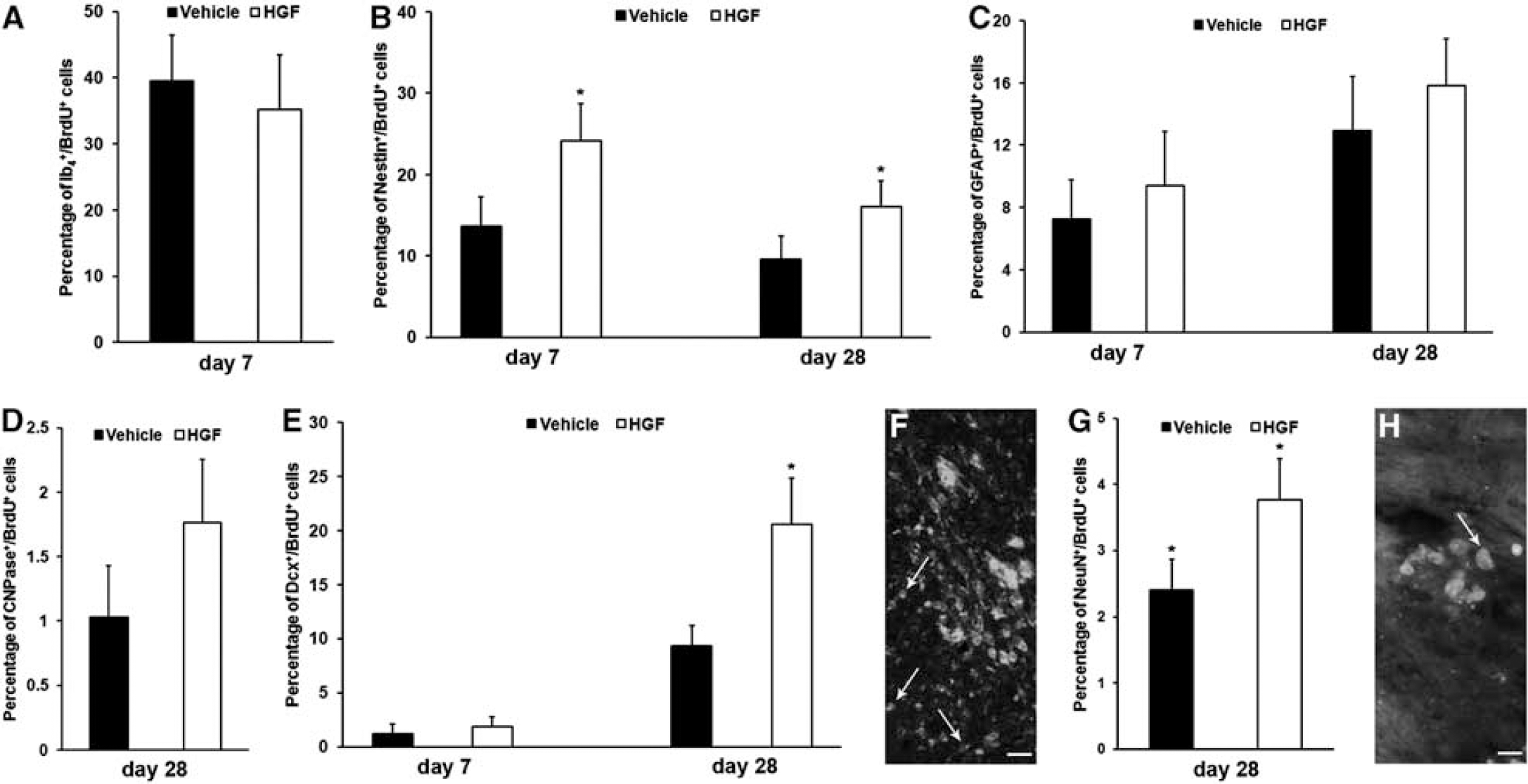
Hepatocyte growth factor (HGF) promotes neural differentiation. Fate of differentiation of 5-bromo-2-deoxyuridine (BrdU+) proliferating cells was analyzed using the microglial marker isolectin B4 (IB4) (
Hepatocyte Growth Factor Stimulates Neural Precursor Cell Proliferation In Vitro and Protects Neural Precursor Cells Against Hypoxic Injury
As enhanced cell proliferation (Figure 4) can be either due to stimulation of cell division itself or due to the protection of proliferating cells, we analyzed NPC proliferation under physiological conditions in vitro. When NPCs were cultivated under standard cell culture conditions, neurosphere formation was observed in NPCs treated with either vehicle or HGF (Figures 6A and 6B). However, incubation of NPCs with HGF resulted in significantly higher numbers of neurospheres as compared with control NPCs (Figure 6C). Likewise, treatment of NPCs with HGF resulted in significantly enhanced numbers of NPCs incorporating BrdU (Figure 6D), suggesting that HGF stimulates proliferation of SVZ-derived NPCs in vitro. Apart from HGF-mediated stimulation of NPC proliferation, HGF also preserved cultivated NPC from injury when exposed to 45 minutes of OGD followed by subsequent reexposure to oxygen and glucose under standard cell culture conditions (Figure 6E). Whereas control cells developed prominent and distinct cell injury, NPCs incubated with HGF either 4 hours before OGD or at the beginning of OGD showed significantly enhanced survival (Figure 6E). Furthermore, HGF-induced neuroprotection of NPCs in vitro was associated with attenuated caspase-3 activation. As such, significantly reduced caspase-3 activity was observed for up to 8 hours after OGD in cells that were treated with HGF. As a matter of fact, caspase-3 activities in HGF-treated cells were as low as caspase-3 activities from NPCs treated with z-DEVD.fmk, a well-known inhibitor of caspase-3. These data suggest that HGF promoted the proliferation and survival of NPCs via a mechanism involving caspase-3.
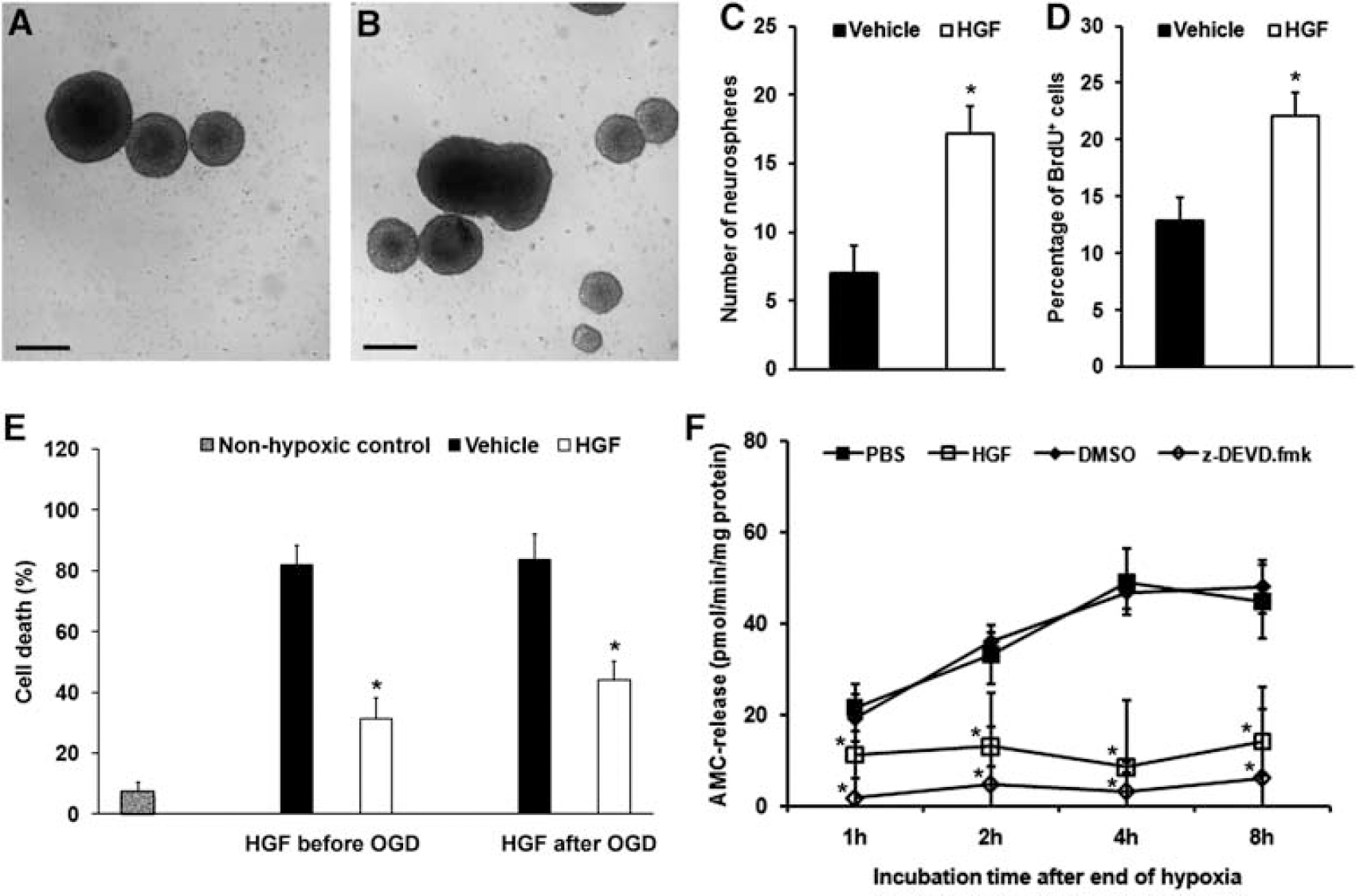
Hepatocyte growth factor (HGF) increases proliferation of neural precursor cells (NPCs) and protects against hypoxic injury in a caspase-3-dependent way in vitro. Neural precursor cells cultured in serum-free basic medium were treated with vehicle (= 0.1 mol/L phosphate-buffered saline (PBS)) (
Discussion
The present study reports that an acute HGF delivery in the first 3 days after stroke induces long-term neuroprotection associated with enhanced motor coordination recovery. Enhanced proliferation and differentiation of SVZ-derived NPCs were noticed in HGF-treated mice, which based on in vitro studies were a consequence of both enhanced cell division and increased resistance against hypoxic injury. Blood—brain barrier integrity was also enhanced and MMP-activation inhibited, pointing toward a profound remodeling of the brain that clearly outlasted the time window of HGF delivery. Our data exemplify that HGF induces potent restorative changes in ischemic brain tissue. This might explain the previously reported association of HGF values measured in the blood of stroke patients early after stroke with postacute recovery.
Although HGF-induced neuroprotection has previously been described in rats using continuous infusion or gene delivery strategies in models of focal or global cerebral ischemia (Date et al, 2006; Niimura et al, 2006b; Shang et al, 2010; Shimamura et al, 2006), we for the first time report that HGF is also protective in mice. Different to earlier studies that focused on neuroprotection in the acute injury phase, we for the first time evaluated effects of HGF after the discontinuation of treatment, showing that the promotion of neuronal survival is sustained over as long as 4 weeks, and that it goes along with functional neurologic recovery. In dose-finding studies, we showed that doses of 5 to 10 μg per animal and day are needed to induce neuroprotection, whereas a dose of 1 μg per day was ineffective. Sustained neuroprotection has previously also been shown for other growth factors with pleiotropic action, namely for vascular endothelial growth factor (Sun et al, 2003; Wang et al, 2006) and granulocyte-colony stimulating factor (Shyu et al, 2004). Compared with the huge number of studies analyzing effects of various growth factors on tissue survival in rodent models of ischemic stroke, the number of studies analyzing long-term structural and functional effects in the postacute stroke phase is still small. The lack of studies with clinically relevant structural and functional end points may represent one reason for the failure of clinical neuroprotection trials in the past.
Growth factors have multiple effects on cellular growth, proliferation, differentiation, and survival, which may help stimulate neurologic recovery and promote remodeling, once a stroke has occurred (Greenberg and Jin, 2006). This explains why growth factors may induce beneficial effects that outlast the termination of treatment. Indeed, HGF induced a long-lasting proliferation response of endogenous NPCs, which persisted until the end of the experiments at 28 days after stroke. Although HGF has been shown to enhance neuronal differentiation of embryonic stem cells in vitro before (Kato et al, 2004; Kokuzawa et al, 2003), HGF-induced effects on postischemic proliferation of NPCs have not been studied until now. The propagated proliferation of NPCs represents a reasonable explanation of the sustained action of HGF. Interestingly, both vascular endothelial growth factor (Sun et al, 2003; Wang et al, 2006) and granulocyte-colony stimulating factor (Shyu et al, 2004) promote neurogenesis. In our study, the majority of NPCs maintained at a largely undifferentiated state, expressing markers of immature neural cells. Nonetheless, the rate of differentiation into mostly immature neurons, astroglia, and oligodendroglia was also increased. It is well conceivable that these cells act in a bystander manner (Bacigaluppi et al, 2008), orchestrating restorative responses of the ischemic brain tissue.
Hepatocyte growth factor-mediated stimulation of postischemic endogenous cell proliferation and neurogenesis can result from both neuroprotection of newborn cells due to antiapoptotic properties of HGF or due to induction of cell proliferation itself. We therefore analyzed the effects of HGF treatment on SVZ-derived NPCs in vitro. Interestingly, HGF did not only induce neurosphere formation and cell proliferation under physiological conditions, but also resulted in acute protection of NPCs against hypoxic injury in a caspase-3-associated mechanism. Like cerebral ischemia (Mehta et al, 2007), hypoxic injury comprises a set of different mechanisms and cellular pathways such as activation of p38 or JNK. Hepatocyte growth factor-mediated inhibition of caspase-3 activation in cultured NPCs can therefore only be regarded as one possible mechanism by which HGF induces neuroprotection against hypoxic injury. Although few studies reported an HGF-mediated induction of cell proliferation and neuronal differentiation of embryonic-derived stem cells in vitro before (Kato et al, 2004; Kokuzawa et al, 2003), only one recent study observed an enhanced proliferation of cultured SVZ-derived NPCs after HGF treatment (Nicoleau et al, 2009). Characteristics of SVZ-derived NPCs, however, also depend on the species and the age of animals (Baker et al, 2005; Van Kampen et al, 2004), and the aforementioned study analyzed HGF-mediated effects on NPCs derived from rats or mice that were either newborn or adult. Taken into account that cell proliferation of SVZ-derived NPCs from mice gradually declines over time with aging animals (own unpublished observation), these results cannot be transferred to our study. According to our in vitro studies, stimulation of postischemic neurogenesis in vivo by HGF might be a consequence of both induction of NPC proliferation and protection of newborn cells within an inflammatory and proapoptotic postischemic milieu.
Among different pathophysiological mechanisms that lead to acute ischemic injury, breakdown of the BBB is one key factor. In our study, HGF treatment resulted in reduced extravasation of Blue Evans and reduced brain edema formation compared with vehicle control animals. Although HGF-mediated poststroke modulation of the BBB and the expression of tight junction proteins has been described before after continuous HGF infusion in rats (Date et al, 2004, 2006), effects after single bolus injections have not yet been studied. One has to keep in mind, however, that a 45-minute stroke in mice as chosen for the present study might only induce mild BBB breakdown in comparison to extended ischemia durations. As activation of MMP is critically involved in BBB breakdown, we further analyzed the activation of both MMP-2 and MMP-9 within 24 hours after stroke onset. We found acute activation of MMP-2 and MMP-9 in control animals after 4 or 24 hours, respectively, which is in line with previous reports (Candelario-Jalil et al, 2009). Interestingly, inhibition of MMP activation by HGF was as effective as treatment with BB-1101, a well-known unspecific MMP inhibitor. Whereas both HGF and BB-1101 significantly stabilized BBB integrity, only HGF yielded neuroprotection. These results confirm previous studies, indicating that MMP inhibition does not influence ischemic injury in the long run, despite an early attenuation of brain damage (Rosenberg et al, 1998; Sood et al, 2008).
In conclusion, the present study shows that acute HGF treatment induces long-term neuroprotection lasting beyond the discontinuation of treatment, translating into functional neurologic recovery in the postacute stroke phase. As histopathological correlate of its perpetuated action, an enhanced proliferation and differentiation of SVZ-derived NPCs was noticed, which again outlasted the duration of HGF treatment and therefore may have orchestrated the remodeling of the brain tissue. Further insights are urgently needed into denominators of successful and nonsuccessful brain recovery, so that therapies may be tailored to improve functional restitution.
Footnotes
The authors declare no conflict of interest.