
Abstract
The present study tested the hypothesis that estradiol reduces tissue infarction after middle cerebral artery occlusion (MCAO) in estradiol-deficient females by augmenting glutamic acid decarboxylase (GAD) expression and thus activity, leading to increases in γ-amino-butyric acid (GABA) tissue levels. Glutamic acid decarboxylase is the principal enzyme for GABA synthesis and has two isoforms, GAD65 and GAD67, which differ in size and cellular distribution. Rats were ovariectomized 7 to 8 days before receiving no hormone, placebo, or 25 μg estradiol via subcutaneous implant 7 to 10 days before harvesting tissue in either ischemic cohorts after 2 h of MCAO (end-ischemia) or in nonischemic cohorts. Selected cortical and striatal regions were microdissected from harvested brains. GAD65/67 mRNA levels were determined by microlysate ribonuclease protection assay. End-ischemic GABA concentrations were determined by HPLC. Steroid treatment selectively decreased ischemic cortical GAD67 mRNA levels. In most brain regions evaluated, regional GABA concentrations increased with ischemia regardless of treatment. Estradiol blocked MCAO-induced increases in GABA concentration only in dorsomedial cortex. These data suggest that estradiol repletion in ischemic rat brain selectively decreases GAD67 mRNA levels but does not alter steady-state GABA concentrations. It may be that estradiol under ischemic conditions is attenuating GABA metabolism rather than enhancing synthesis or is augmenting other aspects of GABAergic transmission such as GABA transporters and receptors.
Introduction
Estrogen has been extensively studied as a neuroprotective agent in cerebral ischemia in women, animals, and a variety of in vitro models. Most experimental data suggest that 17β-estradiol, the principal mammalian estrogen, can benefit ischemic brain and reduce cell death (for a review, see McCullough and Hum, 2003; Merchenthaler et al, 2003; Murphy et al, 2004; Yang et al, 2003). We have shown that female rats sustain less brain tissue injury after middle cerebral artery occlusion (MCAO) than male rats (Alkayed et al, 1998). We have also observed that gender-linked differences in infarct size can be abolished by ovariectomy, and exogenous estrogen is neuroprotective in both sexes (Alkayed et al, 2000; Rusa et al, 1999; Toung et al, 1998, 2004). Estrogen's proposed mechanisms of protection for ischemic brain and the cerebral vasculature are quite varied and involve both genomic and nongenomic pathways (for a review, see Bhardwaj et al, 2003; McCullough and Hurn 2003) ranging from amplification of the antiapoptotic proto-oncogene bcl-2 (Alkayed et al, 2001; Dubal et al, 1999; Singer et al, 1998) to antioxidant effects at high concentrations (Kume-Kick and Rice 1998; Mooradian 1993).
Another potential mechanism is that estrogen could amplify inhibitory, and potentially protective, γ-amino-butyric acid (GABA) tissue levels in ischemic brain regions by increasing glutamic acid decarboxylase (GAD) baseline expression. Glutamic acid decarboxylase is the principal enzyme for GABA synthesis from glutamate and is found in two isoforms, GAD65 and GAD67, which differ in size and cellular distribution. Estrogen has been reported to alter GABA conduction (Majewska, 1992); induce synaptosomal GABA release (Fleischmann et al, 1990); increase GABAA antagonist binding affinity and site number (Juptner et al, 1991; Jussofie et al, 1995; Maggi and Perez, 1986; Perez et al, 1988; Schumacher et al, 1989); increase GABA transporter 1 mRNA (Herbison et al, 1995); increase α2 and γ1 GABAa receptor subunit mRNAs (Herbison and Fenelon, 1995); and alter GAD mRNA levels (Davis et al, 1996; Grattan et al, 1996; McCarthy et al, 1995; Weiland, 1992). Previous reports indicate that estrogen receptors are expressed by hippocampal and hypothalamic GABA neurons (Su et al, 2001). The potency of estrogen on GABAergic signaling varies by brain region and is under investigation (McCarthy, 1995; Paul and Purdy, 1992; Smith, 1989). For example, estrogen in ovariectomized females increases tissue GABA levels in the hypothalamus (Luine et al, 1997).
The current literature suggests that estrogen alters GAD and GABA in the brain, although little work has been performed in the cortex and striatum under ischemic conditions. γ-Amino-butyric acid agonists have been well studied (Hagberg and Sandberg, 1998) and reduce injury when administered before and after an ischemic insult (for a review, see Schwartz-Bloom and Sah, 2001). We therefore tested the hypothesis that estradiol treatment will amplify GABA tissue levels in ischemic brain regions by increasing GAD baseline expression.
Materials and methods
Animals and Hormone Treatments
This study was conducted in accordance with the National Institutes of Health guidelines for the care and use of animals in research. The Animal Care and Use Committee of the Johns Hopkins University approved all protocols. All methods are as previously described (Alkayed et al, 1998; Toung et al, 1998). Briefly, agematched, sexually mature female Wistar rats (Hsd:WI, 200–225 g; Harlan, Indianapolis, IN, USA) were ovariectomized under halothane anesthesia (induction 4% to 5%, maintenance 1.25% to 1.5% delivered via mask in O2-enriched air) 7 to 8 days before hormone treatments. Ovariectomized females received no hormone or placebo (OVX), or 25 µg 17β-estradiol (E25) via subcutaneous implant (Innovative Research of America, Sarasota, FL, USA) 7 to 10 days before MCAO (ischemic cohorts—OVX, n = 15; E25, n = 13) or euthanasia (nonischemic cohorts—OVX, n = 6; E25, n = 6). We chose an estradiol dose that is neuroprotective in ischemic ovariectomized (Rusa et al, 1999) and reproductively senescent female (Alkayed et al, 2000) rat brain. Ovariectomized animals were randomly assigned to treatment groups.
Middle Cerebral Artery Occlusion
For MCAO, each animal was anesthetized with halothane (induction 4% to 5%, maintenance 1.25% to 1.5% delivered via mask in O2-enriched air) and a femoral artery catheter was placed for continuous monitoring of mean arterial blood pressure (MABP) and measurement of arterial blood gases. Rectal and temporalis muscle temperature was monitored with a Mon-a-therm system (Mon-a-therm, Mallinckrodt Medical, Inc., model 6510, St Louis, MO, USA) and maintained by a heating lamp. The effectiveness of vascular occlusion was determined by sustained reduction of laser-Doppler flow signal (LDF, Moor Instruments Ltd, model MBF3D, Oxford, England) obtained by probe placement 6 mm lateral and 2 mm posterior to bregma (Alkayed et al, 1998; Toung et al, 1998). Focal cerebral ischemia was accomplished using a modified intraluminal filament technique as previously described (Longa et al, 1989). Briefly, the right common carotid and external carotid arteries were exposed and ligated. The right occipital artery was cauterized, then the right pterygopalatine artery was ligated. After the baseline LDF was determined, a 4.0 nylon monofilament surgical suture with a heat-blunted tip was introduced via the right common and internal carotid arteries until the LDF signal displayed an abrupt and significant reduction, confirming ongoing ischemia. Then the suture was secured in place. LDF was measured during ischemia over 15 to 30-min intervals. Five-minute recording averages were analyzed for each time point.
Brain Preparation and Microdissection
To block postmortem GAD activity, animals were injected intraperitoneally with 1.2 mmol/L kg of 3-mercaptopro-pionic acid 2.5 mins before study end point. Rats were euthanized via decapitation under halothane anesthesia (4% to 5%) either at the end of 2 h MCAO period (end-ischemia) or 9 days after placebo/hormone implantation (nonischemic cohorts), and brains were harvested and frozen on dry ice and stored at −80°C for later microdissection. Serial 300 µm coronal sections of the brains were cut in a cryostat at −9°C, thaw-mounted onto glass microscope slides, and immediately refrozen. Using the Palkovits micropunch dissection technique (Palkovits and Brownstein, 1988), samples of brain were removed with 17-gauge sharpened stainless-steel hypodermic tubing (1067 µm internal lumen diameter) from two selected regions of cerebral cortex and two regions of caudate-putamen in each hemisphere (Figure 1). Selection of these brain regions was based on a previous regional cerebral blood flow (CBF) study (Rusa et al, 1999) to sample representative ischemic regions.
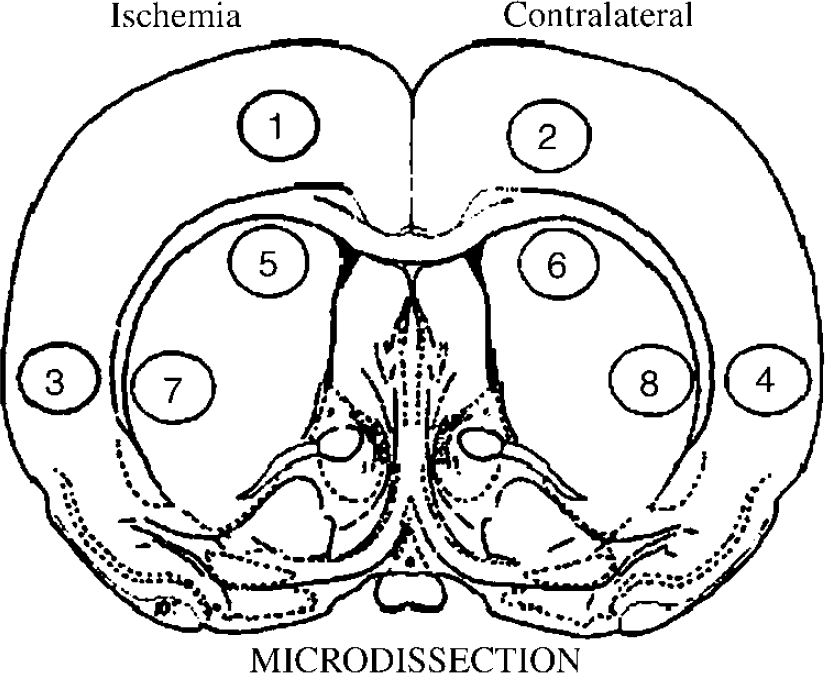
Rats were euthanized via decapitation under halothane anesthesia either at the end of 2 h of middle cerebral artery occlusion (MCAO) or at 9 days after placebo/hormone implantation. Brains were harvested and frozen on dry ice and stored at −80°C for later microdissection. Serial 300 µm coronal sections of the brains were cut in a cryostat at −9°C, thaw-mounted onto glass microscope slides, and immediately refrozen. Using a micropunch dissection technique, brain samples were removed with 17-gauge sharpened stainless-steel hypodermic tubing (1067 µm internal lumen diameter) from selected regions of the cerebral cortex and caudate-putamen in each hemisphere. Selection of these brain regions was based on previous regional cerebral blood flow (CBF) studies (Rusa et al, 1999) to sample representative ischemic regions. Dorsomedial cortex, 1 and 2; dorsolateral cortex, 3 and 4; dorsal striatum, 5 and 6; lateral striatum, 7 and 8. Odd numbers are from ipsilateral or ischemic regions, even numbers are from contralateral or nonischemic regions.
For nonischemic studies, micropunches from each sampled region were pooled from both hemispheres for each animal and placed in 4.0 N guanidine thiocyanate (GuSCN) for mRNA quantification and stored at −80°C until further analysis. For ischemic studies, micropunches from each sampled region were pooled from ipsilateral (ischemic) and contralateral (nonischemic) hemispheres for each animal. For each structure, half of the tissue was placed in 0.1 N perchloric acid (HC***IO4) for GABA determination and half in 4.0 N GuSCN for mRNA quantification. For quantification of tissue GABA content, the tissue was sonicated in 50 to 100 µL of ice-cold 0.1 N HC***IO4 and then centrifuged. The acid extract was stored at −20°C for measurement of GABA by HPLC, and the precipitate redissolved in 50 µL of 1.0 mol/L NaOH and stored at 4°C for assay of tissue protein content (Bradford, 1976). All samples for GAD65/67 mRNA analysis were stored at −80°C until further analysis.
γ-Amino-Butyric Acid Measurements>
γ-Amino-butyric acid concentrations in microdissected brain tissue were determined by isocratic reverse-phase HPLC with electrochemical detection after precolumn derivatization with o-phthaladehyde and β-mercaptoethanol as described previously (Grattan and Selmanoff, 1993; Grattan et al, 1996). To correct for minor variances in derivatization efficiency, homoserine (Sigma, St Louis, MO, USA) was added to each sample as an internal standard. γ-Amino-butyric acid content was determined by comparing the ratio of GABA and the internal standard in sample chromatograms with the same ratio of peak areas in chromatograms from external GABA standards.
Microlysate Ribonuclease Protection Assay
Tissue punches microdissected for the ribonuclease protection assay (RPA) were immediately placed into 20 µL of ice-cold 4.0 mol/L GuSCN and stored at −80°C until assay. Briefly, the RPA for GAD65/67 mRNAs was performed directly in the tissue microlysate after the method of Strauss and Jacobowitz (1993) as described previously (Grattan et al, 1996; Searles et al, 2000). Radiolabeled antisense riboprobes for GAD65 and GAD67 were transcribed using T7 and SP6 RNA polymerases (Promega, Madison, WI, USA), and UTP (New England Nuclear, Boston, MA, USA, 800 Ci/mmol, 10 mCi/ml). As an internal control, an antisense riboprobe specific for rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was also transcribed, using T3 RNA polymerase (375 bp, template from Ambion Inc., Austin, TX, USA). Probes were transcribed to a specific activity of ~10scpm/ µg. A mixture of the 3 riboprobes (GAD65, GAD67, and GAPDH; 4 to 10 ng each in 5 µL total volume) was added to the tissue punches contained in 20 µL of the GuSCN solution and hybridization performed overnight (12 to 24 h) at 37°C. Nonhybridized RNA was removed using RNase, and samples were then treated with proteinase K and N-lauryl-sarcosine at 37°C for 1 h. Protein and lipid components were removed by organic extractions with Tris-equilibrated phenol and the hybrids precipitated in ice-cold ethanol and isopropyl alcohol at −80°C for 1 h. The protected hybrids were electrophoresed on a 6% native Polyacrylamide gel and dried on an EasyBreeze drier (Hoefer-Pharmacia Biotech, Piscataway, NJ, USA). The dried gels were exposed to X-ray film for 3 h and the gels were exposed on a phosphoimage plate (Kodak, New Haven, CT, USA) for 12 h to perform PhosphorImage (Molecular Dynamics, Sunnyvale, CA, USA) scanning. The bands were quantified with the use of ImageQuant software (Molecular Dynamics, Sunnyvale, CA, USA). Additionally, using the autoradiograph for alignment, fulllength protected fragments (597 nt for GAD65, 780 nt for GAD67, and 353 nt for GAPDH) were excised from the gel for scintillation counting. All samples in the experiments were processed in a single assay run. Using twice blank as the sensitivity criterion, the assay detects ~20 pg (~0.1 fmol) of each transcript. Representative assay autoradiograms have been published previously (Grattan et al, 1996; Searles et al, 2000).
Statistical Analysis
All values are reported as mean ± s.d. unless otherwise indicated. All repeated-measures data, including physiologic parameters, and LDF, during MCAO were analyzed by two-way analysis of variance (ANOVA) with post hoc Newman–Keuls for multiple comparisons (Jandel Sigma-Stat Statistical Software, Version 2.0, Jandel Corporation, San Rafael, CA, USA). Differences in mean ischemic LDF, GABA concentrations, and GAD mRNA levels were determined by one-way ANOVA with post hoc Newman–Keuls for multiple comparisons (Jandel SigmaStat Statistical Software, Version 2.0, Jandel Corporation, San Rafael, CA, USA). The criterion for statistical significance was P < 0.05.
Results
Baseline, intraischemic, and early reperfusion MABP and blood gases were equivalent among ischemic treatment groups within the GAD and GABA cohorts and were combined (Table 1). Rectal and temporalis muscle temperatures were monitored and controlled and were not significantly different between treatment groups across the study period (Table 1). There were no differences in reduction of residual intraischemic LDF signal (expressed as a percent of baseline signal) among treatment groups within the GAD and GABA cohorts (data not shown). Mean ischemic LDF (% baseline signal) in ovariectomized rats with either no hormone or placebo treatment (OVX, 27% ± 8%) or estradiol replacement (E25, 29% ± 12%) was similar, suggesting that the relative ischemic insult was equivalent among all treatment groups.
Physiologic parameters in combined ischemic GABA and GAD cohorts
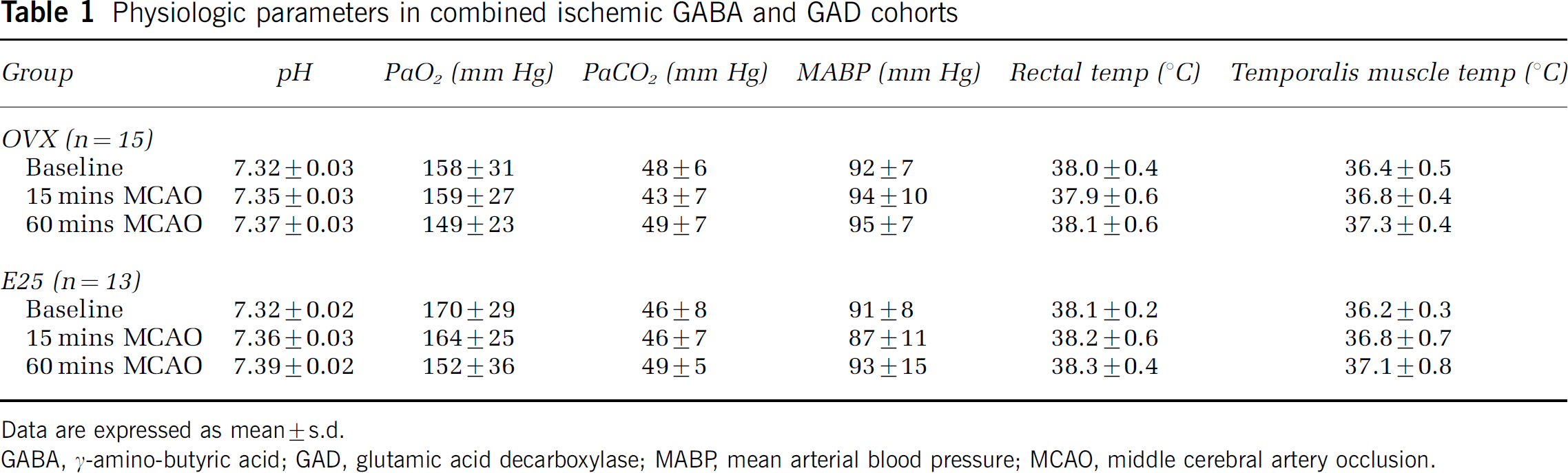
Data are expressed as mean ± s.d.
GABA, γ-amino-butyric acid; GAD, glutamic acid decarboxylase; MABR mean arterial blood pressure; MCAO, middle cerebral artery occlusion.
Figures 2 and 3 show GAD65/67 mRNA levels as determined in nonischemic and ischemic OVX and E25 animals. GAD65/67 mRNA levels were unaffected in naïve rat brain by estradiol treatment in all brain regions sampled (Figure 2). Ischemia alone did not significantly change cortical and striatal GAD65/67 mRNA levels as compared with levels in comparable sampled regions from the contralateral, nonischemic hemisphere (data not shown). However, preischemic estradiol administration in ovariectomized rats reduced GAD67, but not GAD65, mRNA levels in cortical and lateral striatal ischemic brain regions (Figure 3).
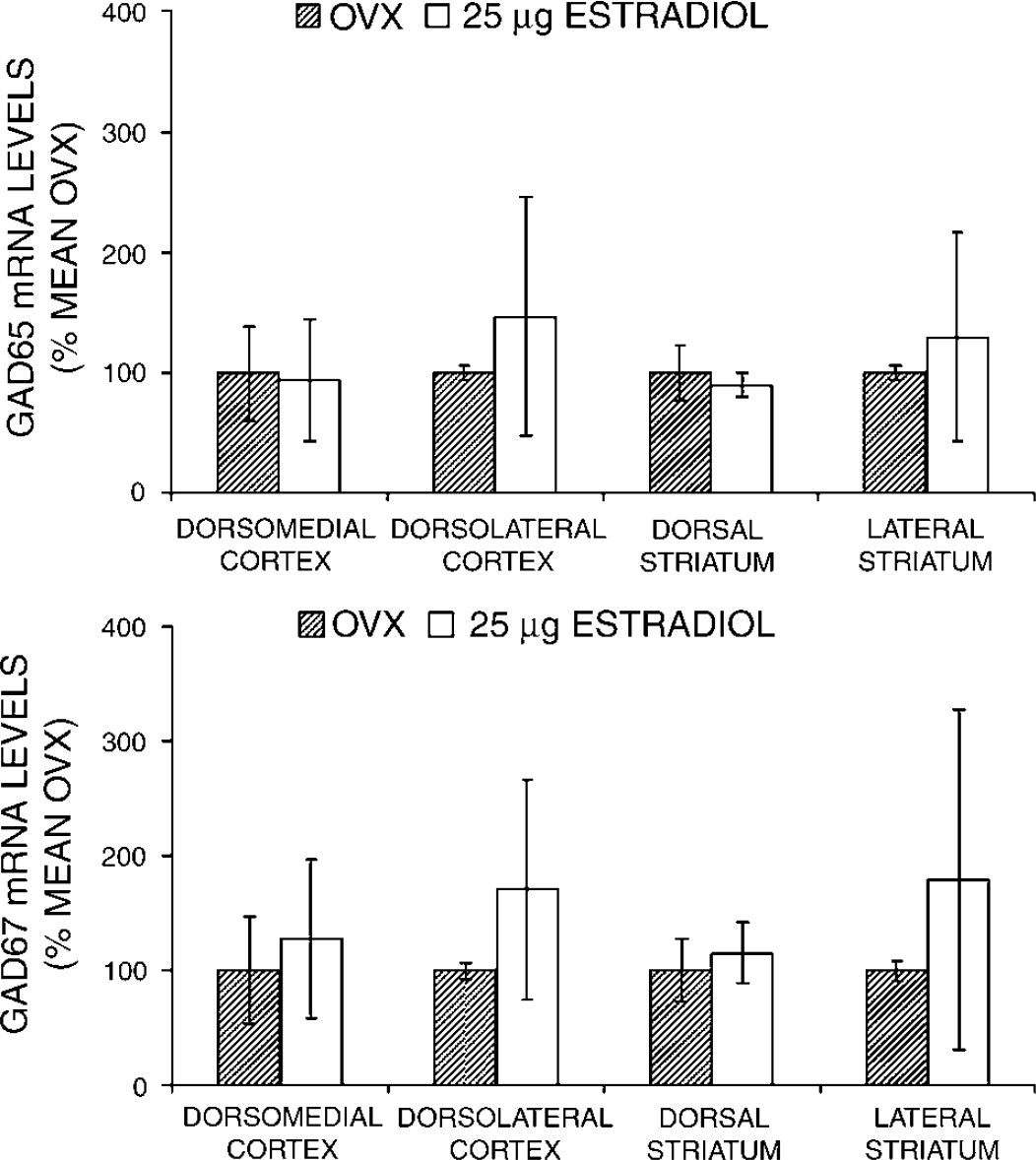
The effect of estradiol on GAD65/67 mRNA levels in selected brain regions was determined by ribonuclease protection assay (RPA) in naïve, ovariectomized female Wistar rats treated with either placebo (OVX) or 25 µg estradiol pellets (E25) (n = 6 per group). All animals were ovariectomized at least 7 to 8 days before hormone treatments. All pellets were placed subcutaneously at 7 to 10 days before euthanasia. GAD65/67 mRNA levels were not increased in naïve rat brain by estradiol treatment. All values are mean ± s.d.
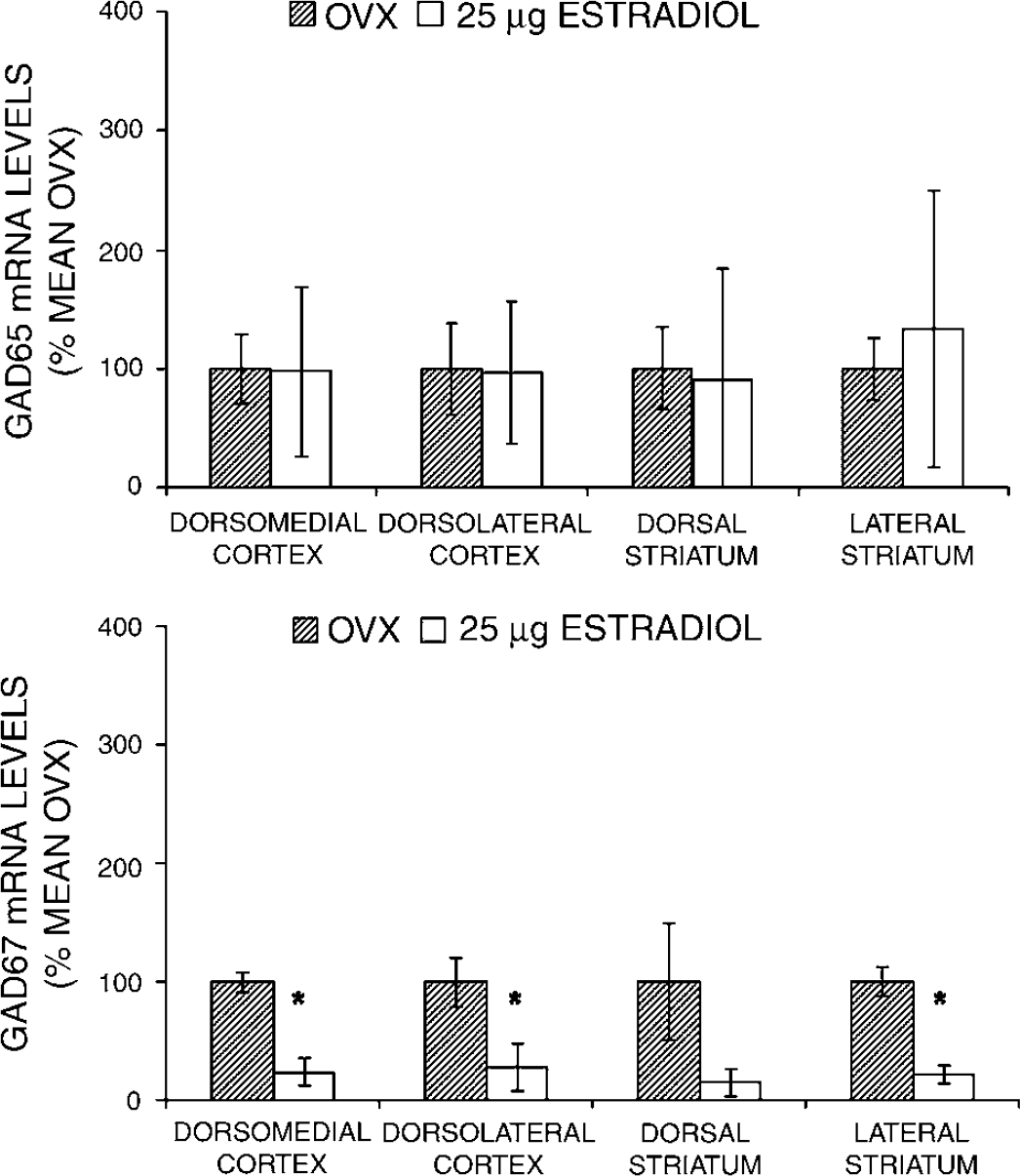
The effect of estradiol and ischemia on GAD65/67 mRNA levels in selected brain regions was determined by ribonuclease protection assay (RPA) in ovariectomized female Wistar rats treated with either placebo (OVX, n = 5) or 25 µg estradiol pellets (E25, n = 2). All animals were ovariectomized at least 7 to 8 days before hormone treatments. All hormone pellets were placed subcutaneously at 7 to 10 days before euthanasia. Preischemic estradiol administration in ovariectomized rats reduced GAD67, but not GAD65, mRNA levels in cortical and lateral striatal ischemic brain regions. All values are mean ± s.d. *P < 0.05 from OVX.
Figure 4 shows steady-state GABA concentrations for the cortical and striatal regions sampled at end-ischemia in OVX and E25 rats. In three out of four regions sampled (lateral and dorsal striatum, dorsolateral cortex) per hemisphere, regional GABA concentrations increased with ischemia, regardless of treatment group. In dorsomedial cortex only, estradiol blocked MCAO-induced increases in GABA concentration.
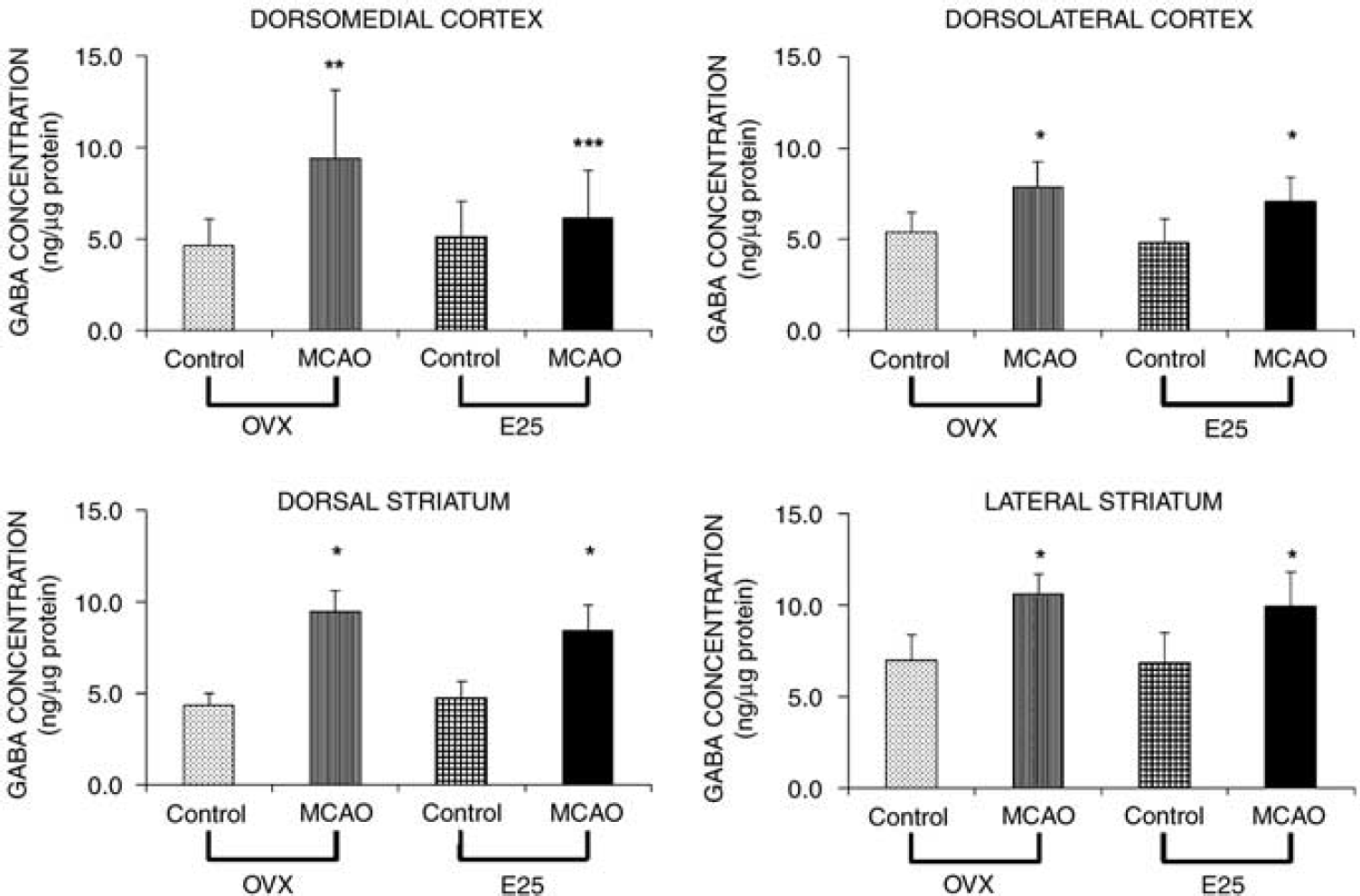
γ-Amino-butyric acid (GABA) concentrations in microdissected brain tissue from ovariectomized female Wistar rats with no hormone (OVX, n = 10) or 25 µg estradiol pellet (E25, n = 11) after 2 h of middle cerebral artery occlusion (MCAO) were determined by HPLC. Control samples were taken from contralateral or nonischemic hemisphere. Middle cerebral artery occlusion samples were taken from ipsilateral or ischemic hemisphere. All animals were ovariectomized at least 7 to 8 days before hormone treatments. All hormone pellets were placed subcutaneously at 7 to 10 days before MCAO. All values are mean ± s.d. *P < 0.001 from corresponding control. **P < 0.01 from corresponding control. ***P < 0.05 from OVX MCAO.
Discussion
Our overall objective was to determine if estradiol is neuroprotective in ischemic ovariectomized female rat brain through effects on GAD65/67 mRNA levels and subsequent changes in GABA concentrations. The current literature suggests that estrogen alters GAD and GABA in the brain, although little work has been performed in the cortex and striatum, especially under ischemic conditions. Glutamic acid decarboxylase expression and GABA concentrations were evaluated at end-ischemia (2 h MCAO) because previous work has shown that GABA and other neurotransmitter levels are increasing extracellularly at this time, whereas it is unclear whether a secondary rise occurs during the reperfusion period (Beneveniste et al, 1984; Globus et al, 1988; Graham et al, 1990; Hagberg et al, 1985; Schwartz-Bloom and Sah, 2001). Most of these studies have focused on extracellular GABA measurements or GAD immunohistochemistry or activity, whereas in this study, GABA and GAD mRNA tissue levels were evaluated. Our study also used an estradiol dose that has been previously shown to be neuroprotective in ischemic ovariectomized ischemic rat brain (Rusa et al, 1999).
In these experiments, we evaluated GAD65/67 mRNA expression in brain areas representative of cortical and striatal ischemic regions. We had expected that estradiol alone would change GAD transcription in the cortex and striatum. However, GAD65/67 expression in nonischemic brain did not appear to be estradiol-dependent in ovariectomized rats. Grattan et al (1996) also did not observe any changes in GAD65/67 mRNA levels in the cingulate cortex in either proestrus or metestrus rats. This is in contrast to what is known about GAD expression in other brain regions like the hypothalamus and hippocampus, where estrogen has been reported to suppress or increase GAD mRNA levels and activity (Curran-Rauhut and Petersen, 2002; Duvilanski et al, 1983; Grattan et al, 1996; Herbison et al, 1992; McCarthy et al, 1995; Mirkes and Bethea, 2001; Nicoletti et al, 1982; Searles et al, 2000; Weiland, 1992).
Ischemia alone did not affect GAD65/67 mRNA levels in ovariectomized rat cortex and striatum. Fraham et al (2004) observed no differences in the numbers of GAD mRNA-stained cells in the cortex and hippocampus at 7 and 30 days after focal ischemia. This is in contrast to studies that have showed both ischemiainduced increases and decreases in the cellular steady-state level of striatal and cortical GAD mRNA (Folbergrova et al, 1989; Francis and Pulsinelli, 1982; Fukatsu et al, 2002; Najlerahim et al, 1991; Saji et al, 1994; Salin and Chesselet, 1993). In different animal models of cerebral ischemia, GAD protein was observed to be altered after stroke for days (Kang et al, 2002) or even weeks (Salin and Chesselet, 1993; Yamada et al, 1994). These results suggest heterogeneity in GAD expression and activity, likely because of variation in lesion site and size as well as ischemic duration.
We chose to measure mRNA expression of GAD65/67, because expression of these isoforms in naïve brain is affected by exogenous estrogen (McCarthy et al, 1995) and vary across the estrous cycle (Grattan et al, 1996). Both isoforms are found in most GABA neurons but their intraneuronal distribution differs (Esclapez et al, 1994). GAD65 is more prevalent in axon terminals whereas GAD67 is more readily detectable in cell bodies (Esclapez et al, 1994). In naive brain, GAD65 synthesis also tends to be lower than that of GAD67, (Esclapez et al, 1994). Functional differences between the two GAD isoforms are therefore likely because of different levels of GAD65/67 as well as different intraneuronal localizations.
Estradiol or ischemia alone did not alter GAD65/67 expression in the cortex or striatum, but estradiol in ischemic brain did selectively reduce cortical and striatal GAD67 mRNA levels. This would suggest that estradiol pretreatment may mediate ischemiainduced downregulation of GAD67 mRNA. We did not observe constitutive regulation of GAD67 in the cortex and striatum by estradiol, as expression in naïve ovariectomized rats and the contralateral, nonischemic hemisphere remained constant regardless of estradiol treatment. No hormone responsive elements in the GAD promoter have been described in the literature. However, the selective effects of estradiol in ischemic brain on GAD67 mRNA expression could reflect regional differences in estrogen receptor subtype or receptor densities, as well as the cell types present since estrogen is known to regulate normal gene expression in some brain areas but not in others (Dubal et al, 1999; Stone et al, 1998). Because only GAD67 mRNA levels were depressed, our data do not likely reflect a general depression of all RNA transcription during ischemia. It may be that GAD67 mRNA expression, especially in cortex, is more sensitive to estradiol under ischemic conditions than GAD65 mRNA levels.
Our observation of increased GABA concentrations in selected regions of the striatum and cortex during ischemia is in agreement with other laboratories studying the effects of ischemia on presynaptic and postsynaptic targets of GABA neurotransmission (for a review, see Schwartz-Bloom and Sah, 2001). Only in dorsomedial cortex did estradiol block ischemiainduced increases in GABA concentrations, a region that also showed depressed levels of GAD67 mRNA. As there were no changes in GAD65/67 mRNA levels during ischemia alone, it is possible that the increases in GABA concentrations observed at end-ischemia were because of reduced GABA turnover via GABA transaminase inhibition in specific brain regions rather than increased GABA synthesis (Erecinska et al, 1984; Kobayashi et al, 1999; Mrsulja et al, 1978). One limitation of this study is that we examined only one time point at end-ischemia. Perhaps GABA levels evaluated hours into the reperfusion period would have been decreased in response to the lowered end-ischemic GAD67 mRNA levels that would have resulted in lower levels of GAD67 protein at some later time point. While the distribution of GAD65/67 protein expression and the level of GAD65/67 activity was not determined in this study, changes in GAD mRNA levels have been positively correlated with corresponding changes in GAD enzymatic activity (McCarthy, 1995; Pinal and Tobin, 1998; Segovia et al, 1990).
In summary, estradiol or ischemia alone does not alter GAD65/67 mRNA levels in the cortex and striatum. While estradiol replacement with doses previously shown to diminish stroke damage selectively reduced GAD67 mRNA in several ischemic brain regions, decreased transcription is not necessarily associated with reduced GABA concentrations at the end of ischemia, except in dorsomedial cortex. Estradiol had no effect on GAD65 mRNA expression in sampled ischemic brain regions. Therefore, estrogen's neuroprotective effects in ischemic brain are not likely mediated through its effects on GAD mRNA expression, and changes in GAD mRNA levels did not correspond with hypothesized consequent changes in GABA concentrations. The changes seen in GAD67 mRNA expression are in fact opposite to what one would predict based on the reported protective effects of GABAergic agents (Hagberg and Sandberg, 1998; Schwartz-Bloom and Sah, 2001; Sternau et al, 1989), suggesting that perhaps estrogen could be attenuating GABA metabolism rather than enhancing synthesis, or could be augmenting other aspects of GABAergic transmission such as GAD cofactors, GABA transporters or GABA receptors.