
Abstract
Protective effects of estrogen against experimental stroke and neuronal ischemic insult are well-documented, but it is not known whether estrogen prevents ischemic injury to brain endothelium, a key component of the neurovascular unit. Increasing evidence indicates that estrogen exerts protective effects through mitochondrial mechanisms. We previously found 17β-estradiol (E2) to improve mitochondrial efficiency and reduce mitochondrial superoxide in brain blood vessels and endothelial cells. Thus we hypothesized E2 will preserve mitochondrial function and protect brain endothelial cells against ischemic damage. To test this, an
Introduction
Cerebrovascular disorders such as ischemic stroke remain a leading cause of death and disability in industrialized countries. Exposure to estrogen is thought to underlie the lower incidence of stroke in premenopausal women as compared with age-matched men and older women (Bushnell et al, 2006; Liu et al, 2009). In numerous experimental models, estrogen has been shown to decrease brain injury from acute ischemic insult (Alonso de Leciñana and Egido, 2006; Dubal et al, 2006; Gibson et al, 2006), but the underlying mechanisms and possible roles in clinical therapy are still poorly understood (Bushnell et al, 2006; Liu et al, 2009).
Most studies of estrogen protection, and ischemic mechanisms in general, have focused on neurons and astrocytes (Alonso de Leciñana and Egido, 2006; Raval et al, 2006; Nilsen et al, 2006; Dhandapani and Brann, 2007). However, there is growing appreciation of the vascular contribution to stroke risk, brain injury, and recovery, as well as the importance of protection of cerebral endothelial cells as part of effective stroke therapy (del Zoppo and Mabuchi, 2003; Iadecola et al, 2006; Bastide et al, 2007). Yet few studies have addressed the impact of estrogen on cerebral endothelium during ischemic insult (Bushnell et al, 2006; Krause et al, 2006).
Mitochondria play a crucial role in vascular pathology (Davidson and Duchen, 2007) as well as oxidative stress and cell death under ischemic conditions (Sims and Anderson, 2002; Fiskum et al, 2004). In addition to their fundamental role in cellular energy production, mitochondria are a major source of intracellular reactive oxygen species (ROS) and a key participant in cell death pathways such as apoptosis (Adam-Vizi, 2005; Wallace, 2005). Cerebrovascular endothelial cells contain more mitochondria than endothelial cells in other vascular beds as there is a high metabolic requirement to maintain blood–brain barrier (BBB) function (Oldendorf et al, 1977). Thus, cerebrovascular endothelial cells would be predicted to be particularly sensitive to loss of oxygen and glucose; however, little is known about how ischemic insults affect mitochondrial function in cerebral endothelial cells.
Increasing evidence indicates that estrogen exerts protective effects through mitochondrial mechanisms (Duckles et al, 2006; Nilsen et al, 2006; Simpkins and Dykens, 2008). We recently discovered estrogen to suppress mitochondrial ROS production in cerebral blood vessels and isolated endothelial cells, while increasing mitochondrial efficiency (Stirone et al, 2005; Duckles et al, 2006; Razmara et al, 2008). Thus we hypothesized that during ischemic insult to cerebral endothelial cells, estrogen protects mitochondrial function and prevents cellular injury. To test this, we have used an
Materials and methods
Endothelial Cell Culture
An immortalized mouse brain endothelial cell line (bEnd.3) was purchased from American Type Culture Collection (ATCC, VA, USA) and cultured in Dulbecco's Modified Eagle Medium (ATCC) supplemented with 5% fetal bovine serum (ATCC). The endothelial identity of the cultured cells was confirmed by immunocytochemical staining for Von Willebrand Factor, a specific marker of endothelial cells, with 100% positive staining. bEnd.3 cells were seeded in 24-well plates for cell viability and lactate dehydrogenase (LDH) assays; in 12-well plates for ATP assay; in glass-bottom culture dishes (MatTek, Ashland, MA, USA), coated with 0.5 mg/ml poly-L-lysine (Sigma-Aldrich, St Louis, MO, USA), for live imaging; and in T-75 flasks for Western blot studies. Cell cultures were incubated at 37°C in a 5%/95% mixture of CO2 and atmospheric air, and the medium was replaced every 2 days.
Drug Treatment
When bEnd.3 cells reached approximately 90% confluence, the culture medium was replaced with phenol red-free Dulbecco's Modified Eagle Medium (Invitrogen, Carlsbad, CA, USA) supplemented with 1% charcoal-stripped fetal bovine serum to remove possible estrogenic activity in the medium. After overnight incubation in the new medium, cells were treated with 10 nmol/L E2 (Sigma-Aldrich), 10 nmol/L PPT (4,4′,4″-(4-propyl-[1
OGD/Reperfusion
Ischemia-like conditions were induced in the cultures by OGD. To achieve OGD, cultures were transferred to a humidified anaerobic chamber (Reming Bioinstrument, Redfield, NY, USA) under an atmosphere of 5% CO2 balanced with 95% N2. The culture medium was replaced three times with deoxygenated, glucose-free, balanced salt solution (BSS0.0), which contained (in mmol/L) the following: NaCl, 116; CaCl2, 1.8; MgSO4, 0.8; KCl, 5.4; NaH2PO4, 1; NaHCO3, 14.7, and HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], 10 (pH 7.4). The oxygen level inside the anaerobic chamber was monitored with an oxygen sensor. An atmosphere of < 0.5% oxygen (0.3–0.5%) and a temperature of 37°C were maintained throughout the OGD period. Control cells were placed in a normoxic incubator and had their medium changed to BSS5.5, which was identical to BSS0.0 but supplemented with 5.5 mmol/L glucose. At the end of the OGD period, all cultures were placed under normoxic conditions and the media was quickly replaced with glucose-containing, phenol red-free Dulbecco's Modified Eagle Medium or BSS5.5 (live-imaging experiments only). The latter portion of the experiment, in which oxygen and glucose were reintroduced after OGD, is referred to as the reperfusion period. The length of the OGD and reperfusion periods varied depending on the parameter measured.
Cell Viability Assays
Cell viability was assessed by the MTT conversion method, which measures reduction of the tetrazolium salt MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenltetrazolium bromide) (Invitrogen) into a blue formazan product, mainly by activity of the mitochondrial enzymes cytochrome oxidase and succinate dehydrogenase in live cells. MTT was dissolved in phosphate-buffered saline and added to the cell cultures (final concentration of 5 mg/ml) after completion of reperfusion. Cultures were then incubated at 37°C for 4h to allow MMT reduction. Cultures then were washed twice with phosphate-buffered saline and the cells were lysed overnight in dimethyl sulfoxide, enabling release of formazan. This product was measured as absorbance at a wavelength of 570 nm using a microplate reader (Molecular Device, Sunnyvale, CA, USA). Data were normalized to measurements from control cultures without OGD, considered as 100% cell survival.
LDH Leakage
Release of LDH into the extracellular space, indicative of cell membrane damage, was detected using a commercial kit from Roche Applied Science (Indianapolis, IN, USA). Immediately after 6-h OGD or OGD/24-h reperfusion, supernatants were collected from culture media and LDH activity was measured according to the manufacturer's instructions. Data were normalized against the total LDH release from full-kill (FK) control cultures that had been treated for 10 mins with 0.25% Triton X-100 (Sigma-Aldrich), a reagent that damages the plasma membrane resulting in total release of LDH.
Western Blot Analysis
Protein levels of ERα and ERβ in bEnd.3 cells were evaluated by Western blotting. Soluble lysate (20 μg) of bEnd.3 cells was mixed with sample buffer and NuPAGE Reducing Agent (Invitrogen) followed by separation on 12% SDS-PAGE gel. Proteins were electrically transferred to polyvinylidene difluoride membranes and probed with anti-ERα antibodies (Affinity Bioreagents, Golden, CO, USA; diluted 1:5000) or anti-ERβ antibodies (Calbiochem/EMD Chemicals Inc., San Diego, CA, USA; diluted 1:1000) overnight at 4°C. Then the membranes were incubated with fluorescent secondary antibodies (Thermo Scientific, Rockford, IL, USA) for 1 h. Protein bands were visualized using the LI-COR Odyssey System (LI-COR Biotechnology, Lincoln, NE, USA).
Live-Imaging Studies
Mitochondrial Superoxide:
Mitochondrial superoxide was measured using the fluorogenic probe MitoSOX Red (Invitrogen). MitoSOX Red selectively enters mitochondria within living cells. When oxidized by superoxide anions in the mitochondria, the dye emits red fluorescence. After 60-min OGD cells were switched to reperfusion conditions and incubated for 20 mins at 37°C with MitoSOX Red (2.5 μmol/L). MitoTracker Green (100 nmol/L) also was used to confirm the localization of MitoSOX Red to mitochondria. Cells were then washed with BSS5.5 to remove excess dye. Fluorescence images were immediately captured with an inverted microscope (TE-200; Nikon, Tokyo, Japan) with a × 40 epifluorescence oil-immersion objective. Images were acquired with a digital camera (DXM1200; Nikon). As positive control, additional cultures were treated for 60 mins with the mitochondrial complex-I inhibitor rotenone (50 μmol/L), known to increase mitochondrial superoxide (Adam-Vizi, 2005). MitoSOX Red fluorescence per cell was quantified by Image J analysis software (version 1.41; NIH).
Mitochondrial Membrane Potential
Mitochondrial membrane potential was measured using tetramethylrhodamine methyl ester (TMRM; Sigma-Aldrich), a cell-permeant, cationic fluorescent dye that is readily sequestered by polarized mitochondria. After 90-min OGD, cells were switched to reperfusion conditions and incubated in the dark with 100 nmol/L TMRM for 25 mins at 37 °C to load the dye into the mitochondria. Then unsequestered dye was thoroughly washed out with BSS5.5. At a time point corresponding to 45-min reperfusion, TMRM fluorescence (excitation wavelength of 540 nm and emission wavelength > 590 nm) in mitochondria was visualized using an inverted microscope (TE-200; Nikon, Tokyo, Japan) with a xenon lamp, filter wheel, and a × 40 epifluorescence oil-immersion objective. Images were acquired with a 12-bit digital CCD camera (Photometrics, Tucson, AZ, USA). To minimize photobleaching of the dye, fluorescence intensity was attenuated with neutral density filters (Omega Optical, Battleboro, VT, USA) and exposure time never exceeded 50 msecs. For each cell, TMRM fluorescence was quantified in the mitochondria-rich perinuclear region, both before and after exposure to the protonophoric uncoupler FCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; Sigma-Aldrich; 5 μmol/L). FCCP was used to dissipate the membrane potential and define the baseline for analysis of mitochondrial potential by TMRM. Data were analyzed (after subtraction of background from a cell-free region) with the Metafluor 7.0 software (Universal Imaging, West Chester, PA, USA). For each cell, TMRM fluorescence intensity was normalized to fluorescence intensity in the same area after exposure to FCCP.
ATP Measurements
After 3 or 6 h of OGD exposure, cells were washed with phosphate-buffered saline and lysed with ATP-releasing buffer containing 100 mmol/L potassium phosphate at pH 7.8, 2 mmol/L EDTA, 1 mmol/L dithiothreitol, and 1% Triton X-100. Cellular ATP levels were measured using a commercial ATP assay kit (Molecular Probes, Eugene, OR, USA), based on luciferase/luciferin enzyme luminescence. Nunc 96-well plates were read using a Novostar Cell-based microplate reader (BMG LABTECH GmbH, Durham, NC, USA). ATP standards were used each time to generate a calibration curve. Based on bicinchoninic acid protein assay, sample protein concentration was determined using a MicroBCA kit (Thermo Scientific).
Statistical Analysis
Statistically significant differences between groups were determined by analysis of variance followed by Newman–Keuls
Results
Estrogen Protection Against OGD/Reperfusion
On the basis of preliminary experiments, bEnd.3 endothelial cells were subjected to 6-h OGD followed by 24-h reperfusion to produce a modest insult upon which to test the protective effects of E2. This protocol resulted in approximately 40% cell death as evaluated by the MTT conversion assay. As shown in Figure 1, pretreatment of cells with a physiological concentration (10 nmol/L) of E2 for either 24 or 48 h significantly reduced OGD/reperfusion-induced cell death (by 46% and 66%, respectively). However, this protective effect of E2 was not seen after E2 pretreatment for shorter periods of time (0.5 or 12 h). The protective effect of E2 on cell viability was confirmed using the LDH leakage assay. LDH leakage from cells pretreated with 10 nmol/L E2 for 48 h was significantly reduced as compared with that from vehicle-treated cells during both the 6-h period of OGD (63% reduction; Figure 2A) and the 24-h reperfusion period (52% reduction; Figure 2B).
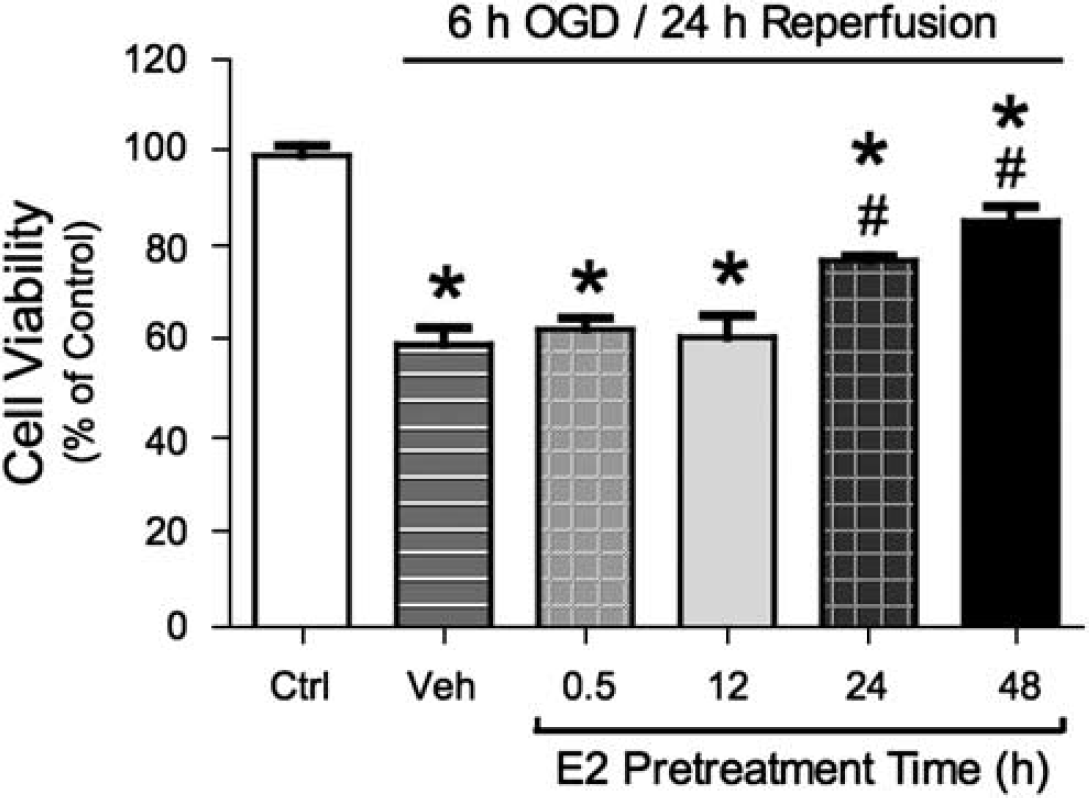
Effects of estrogen on OGD/reperfusion-induced cell death. bEnd.3 cells were pretreated with either vehicle (ethanol 0.02%) or 10 nmol/L E2 for various times (0.5–48 h). Cells were then exposed to 6-h OGD and 24-h reperfusion; all drug treatments were continuously maintained throughout OGD and reperfusion. Cell viability was determined by MTT conversion method. Values shown are mean ± s.e.m. (
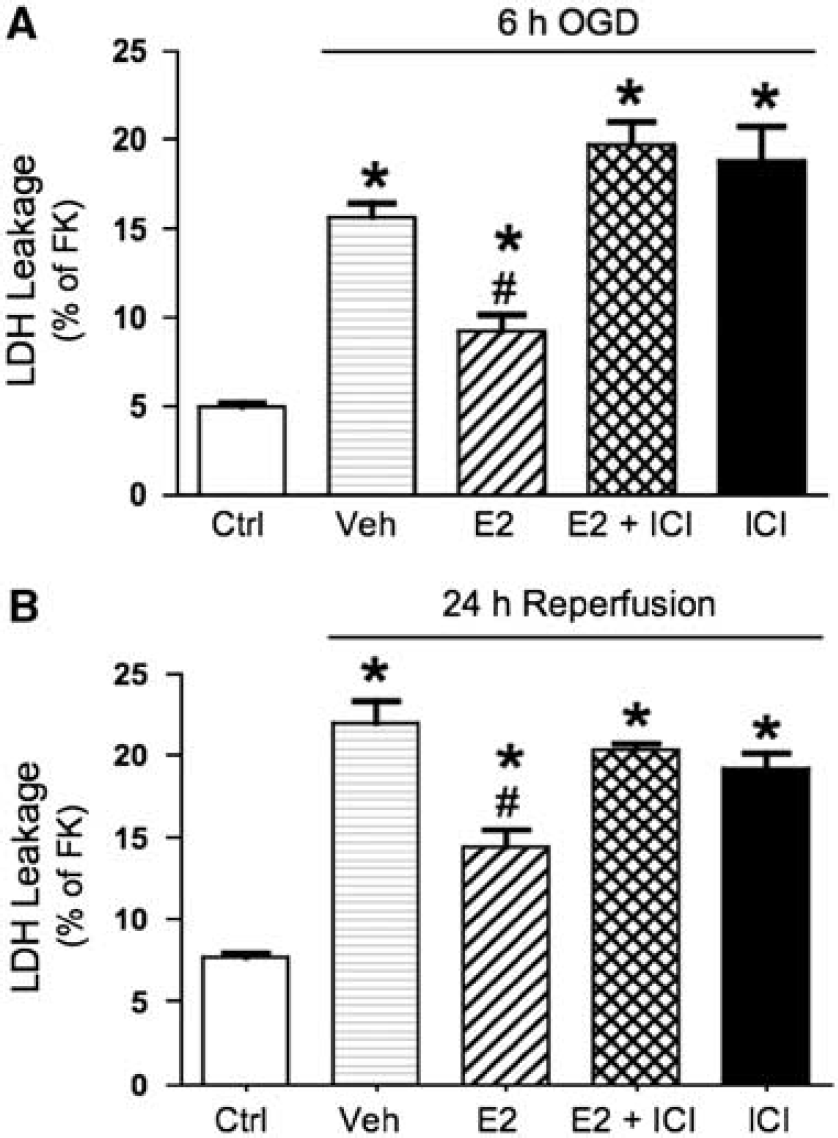
Estrogen inhibits LDH leakage during OGD and reperfusion by an ER-dependent mechanism. bEnd.3 cells were treated with either vehicle (ethanol 0.02%) or 10 nmol/L E2 for 48 h. In some cells, the ER antagonist ICI-182,780 (100 nmol/L) was first applied for 0.5 h and then antagonist treatment was maintained during E2 or vehicle treatment for 48 h. Cells were then exposed to 6-h OGD and 24-h reperfusion; all drug treatments were continuously maintained during OGD and reperfusion. Media were sampled for measurement of LDH activity immediately after 6-h OGD (
Role of ERs
Effects of estrogen can occur through ER-dependent, as well as ER-independent, mechanisms (Miller and Duckles, 2008). To investigate whether ERs were involved in E2-mediated endothelial protection, an ER antagonist, ICI-182,780, was used. As shown in Figure 2, E2-mediated decreases in OGD/reperfusion-induced LDH leakage were blocked by ICI-182,780, while ICI-182,780 by itself had no effect. Since ICI-182,780 does not distinguish between the two ER isoforms, we then tested selective ER agonists to further explore the role of each receptor. Both ER isoforms, ERα and ERβ, were expressed in bEnd.3 cells as examined by Western blotting (Figure 3A). As shown in Figure 3B, only the ERα-selective agonist PPT mimicked the effect of E2 in decreasing OGD/reperfusion-induced cell death. In contrast, the ERβ-selective agonist DPN had no effect. The concentration used for both PPT and DPN was 10 nmol/L, which has been shown to selectively activate the respective ERs (Harrington et al, 2003; Razmara et al, 2008).
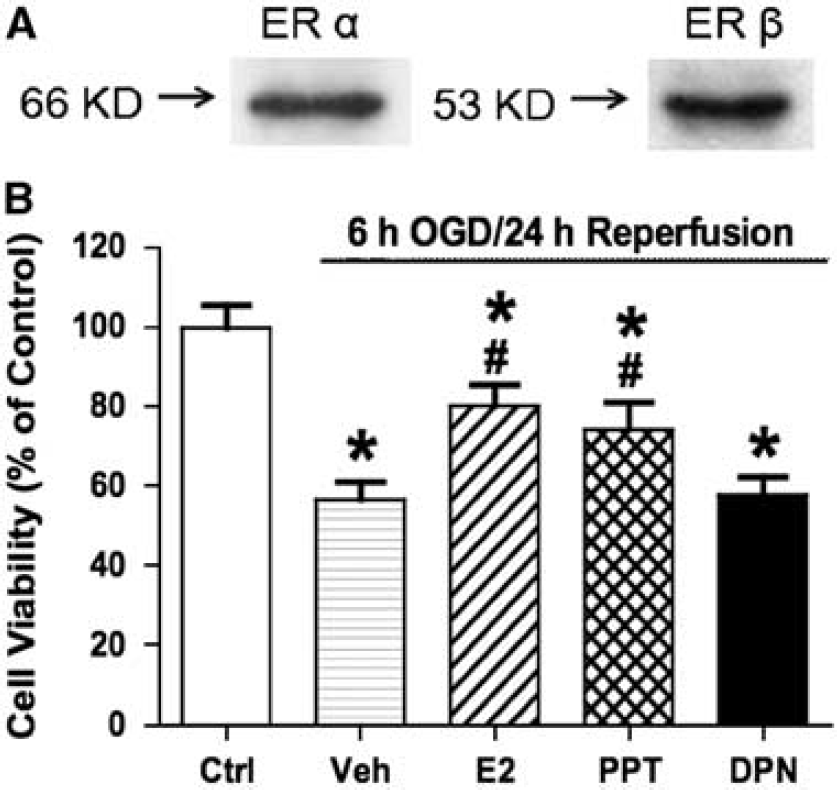
Effects of ER-selective agonists on OGD/reperfusion-induced cell death in bEnd.3 cells. (
Estrogen Preserves Mitochondrial Function
We hypothesized that estrogen would preserve mitochondrial function during the OGD insult. In particular, protection of mitochondria early on in the process would be important for preventing the initiation of cell death pathways and maintaining cell viability. Therefore, effects on mitochondrial superoxide and membrane potential were measured after short OGD exposures (60 to 90 mins) coupled with minimal times of reperfusion (to load the dye indicators).
Effects on Mitochondrial Superoxide:
Mitochondria are major sites for generation of ROS and also are key targets of oxidative insult. Increased mitochondrial superoxide concentration would be a crucial indicator of, and contributor to, mitochondrial dysfunction during OGD. On the basis of our previous finding that estrogen can suppress mitochondrial superoxide production under normal (normoxic) conditions (Razmara et al, 2007, 2008), we hypothesized that estrogen would also suppress increased superoxide generation during OGD. Mitochondrial superoxide was evaluated using MitoSOX Red fluorescence. MitoTracker (Green), which indicates the intracellular location of mitochondria, was used to confirm mitochondrial localization of MitoSOX Red (Figure 4A). As positive control, cells were treated with the mitochondrial complex-I inhibitor, rotenone, to stimulate superoxide formation. This treatment caused substantial increase in MitoSOX Red fluorescence (Figure 4B), thus supporting the use of the technique to reflect levels of mitochondrial superoxide.
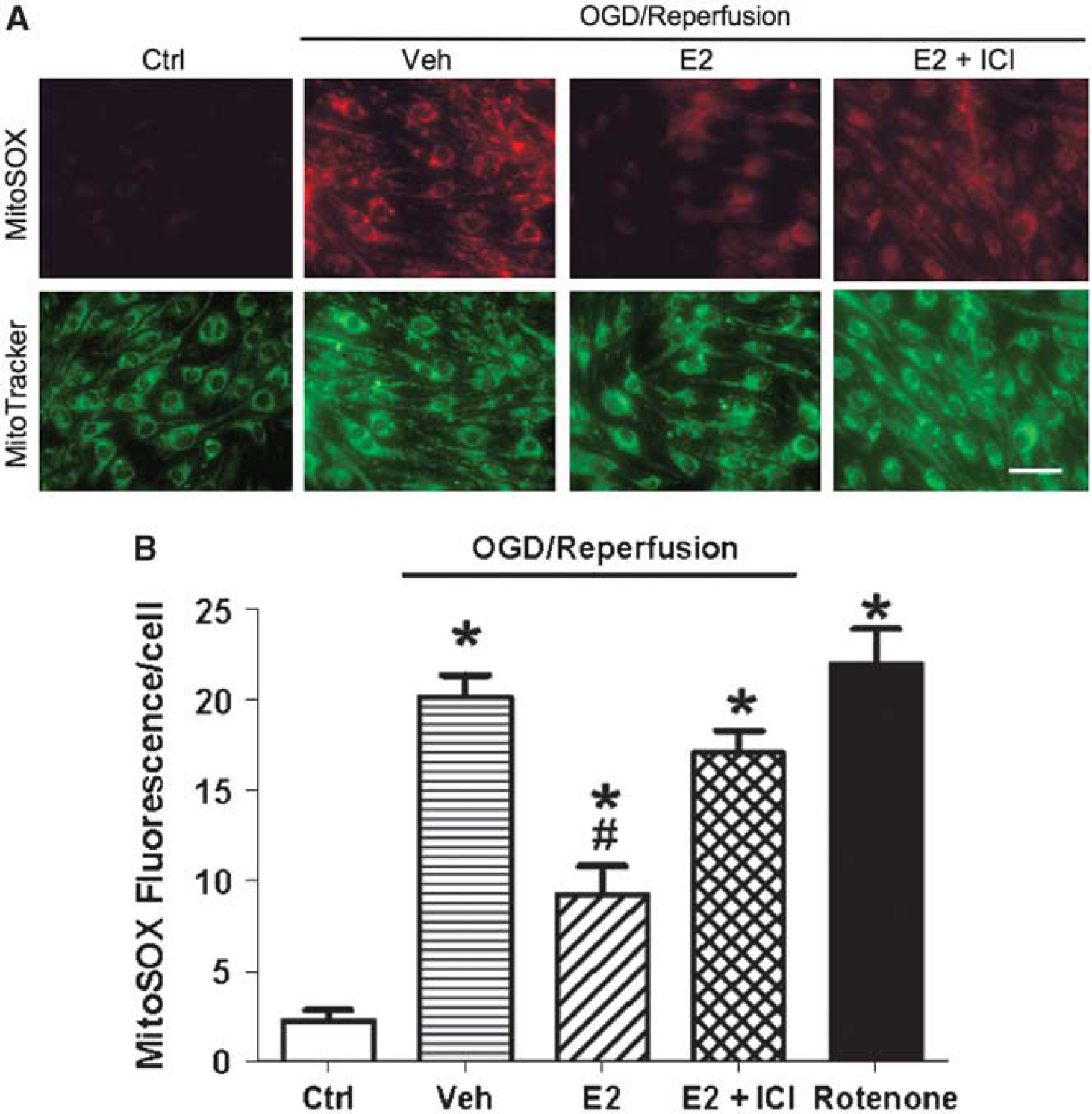
Effects of estrogen on OGD/reperfusion-induced mitochondrial superoxide production in bEnd.3 cells. bEnd.3 cells were treated with either vehicle (ethanol 0.02%) or 10 nmol/L E2 for 24 h. Some bEnd.3 cells were pretreated with the ER antagonist ICI-182,780 (100 nmol/L) for 0.5 h and maintained during E2 or vehicle treatment. Then, cells were exposed to 60-min OGD and 25-min reperfusion, and all drug treatments were continuously maintained during OGD and reperfusion. Mitochondrial superoxide production in live cells was measured at the conclusion of the reperfusion period by fluorescence microscopy using MitoSOX Red dye. As positive control, cells were treated with 50 μmol/L rotenone for 60 mins. (
As shown in the representative fluorescence images (Figure 4A), 60-min OGD/25-min reperfusion caused marked increase in MitoSOX Red fluorescence in vehicle-treated cells as compared with that in control cells not subjected to OGD. However, this effect of OGD/reperfusion was substantially attenuated in cells treated with E2 (24h). In contrast, when cells were treated with ICI-l82,780, in addition to E2, the protective effect of E2 was substantially decreased. Quantification of the MitoSOX Red experiments is shown in Figure 4B. OGD/reperfusion significantly increased MitoSOX fluorescence, and this effect was reduced by 60% when cells were pretreated with 10 nmol/L E2. Addition of ICI-182,780 significantly blocked the protective effects of E2.
Effects on Mitochondrial Membrane Potential:
Maintenance of mitochondrial membrane potential is crucial for mitochondrial function, including ATP generation. We used the TMRM fluorescent probe to monitor the membrane potential of mitochondria in live cells (Koopman et al, 2008). TMRM is accumulated by mitochondria in proportion to change in membrane potential, and in control bEND.3 cells, it exhibits a typical mitochondrial pattern of perinuclear fluorescence (Figure 5). However, in bEND.3 cells subjected to 90-min OGD/45-min reperfusion (Figures 5A and 5B), mitochondrial TMRM signal was dramatically decreased (to 27% of control levels), consistent with dispersion of the dye to the cytosol after depolarization of the potential. Estrogen treatment (24 h), however, suppressed this effect of OGD/reperfusion by 48% (Figures 5A and 5B), maintaining the average mitochondrial TMRM signal at 62% of control levels. Addition of the ER antagonist, ICI-182,780, blocked the effect of E2, resulting in lower TMRM signals that were similar to those found in vehicle-treated cells after OGD/reperfusion (35% of control levels).
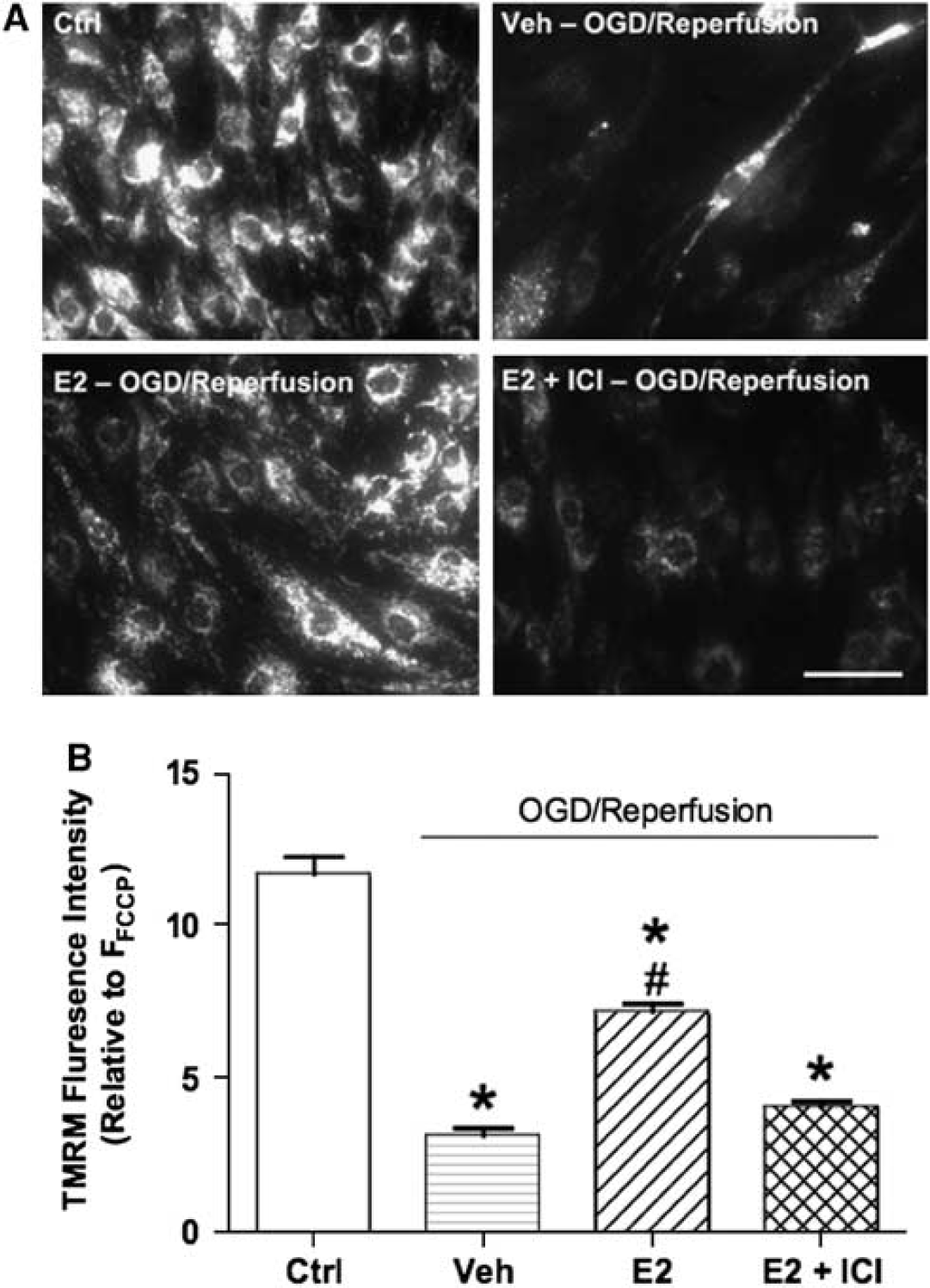
Effects of estrogen on OGD/reperfusion-induced mitochondrial membrane potential loss in bEnd.3 cells. bEnd.3 cells were treated with either vehicle (ethanol 0.02%) or 10 nmol/L E2 for 24 h. Some bEnd.3 cells were pretreated with the ER antagonist ICI-182,780 (100 nmol/L) for 0.5 h first and then antagonist concentration was maintained during E2 or vehicle treatment for 24 h. Then cells were exposed to 90-min OGD and 45-min reperfusion; all drug treatments were continuously maintained during OGD and reperfusion. Mitochondrial membrane potential was measured immediately after 45-min reperfusion. (
Effects on ATP Levels:
As shown in Figures 6A and 3H OGD treatment resulted in 24% decrease of cellular ATP levels as compared with that in control cells not subjected to OGD. Whereas cells pretreated for 24 h with E2 had no significant decline in ATP levels after 3-h OGD, the ATP levels of cells pretreated with E2 plus ICI-182,780 were significantly decreased by 21%. When OGD exposure was extended to 6 h (Figure 4B), ATP levels of vehicle-treated cells and cells pretreated with E2 plus ICI-182,780 were decreased by 40% and 46%, respectively. In contrast, in cells pretreated with E2, ATP levels were only decreased by 25% after 6-h OGD; these ATP levels were significantly higher than that found in cultures treated either with the vehicle or with E2 plus ICI-182,780.
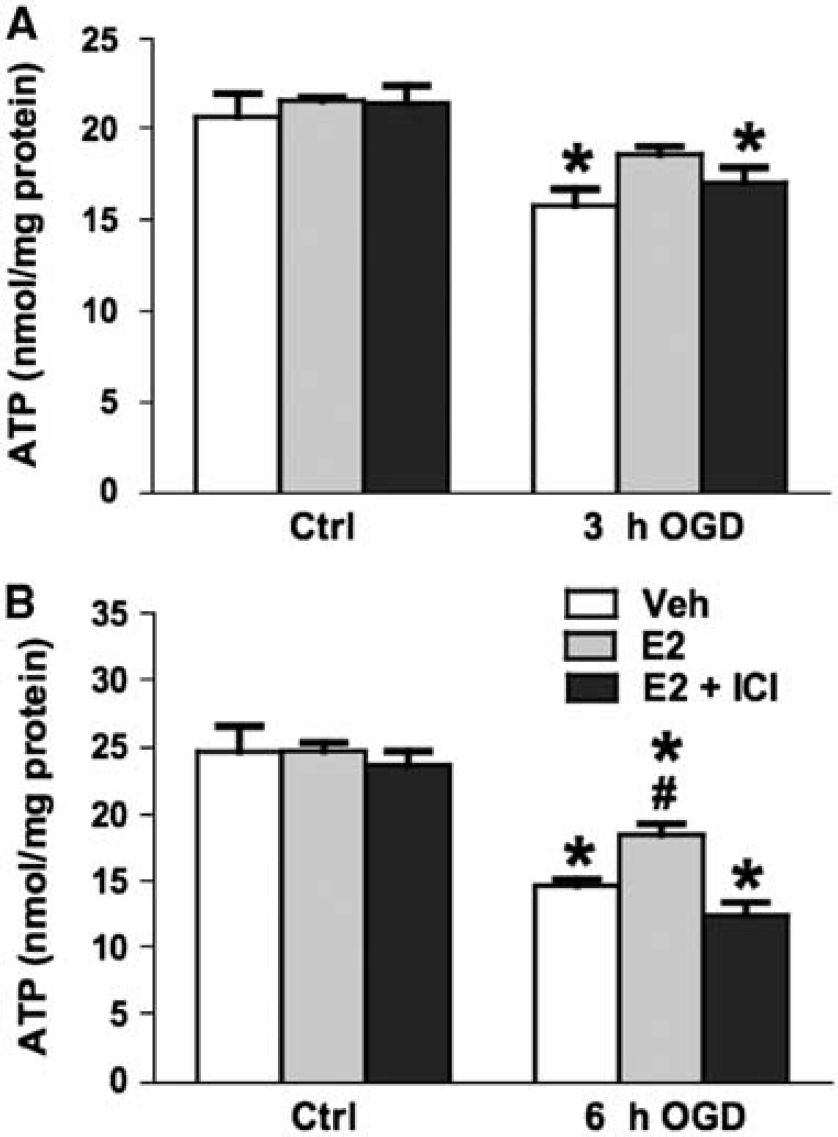
Effects of estrogen on cellular ATP levels after OGD for (
Discussion
The present study demonstrates that estrogen protects endothelial viability and mitochondrial function under ischemic conditions. Endothelial dysfunction is thought to play an important role in the risk and consequences of ischemic stroke. Thus effects of estrogen on cerebral endothelium likely contribute to the well-known protective effects of this hormone against a variety of brain insults, including ischemia. We found that prevention of cerebral endothelial cell death from OGD/reperfusion required pretreatment with E2 and involvement of estrogen ERs, ERα in particular. Moreover, E2 preserved mitochondrial membrane potential, decreased mitochondrial superoxide production, and prevented ATP depletion at early stages of OGD, before cell death. This suggests these effects to be initial events in the mechanism of protection and emphasizes the important role of mitochondria in endothelial effects of estrogen.
A unique aspect of this study is its focus on cerebral endothelium, an important component of the neurovascular unit and a potential therapeutic target for neurological diseases, including stroke (del Zoppo and Mabuchi, 2003; Hawkins and Davis, 2005; Iadecola et al, 2006; Bastide et al, 2007). Cerebral endothelial cells perform essential functions, including maintenance of the BBB and regulation of vascular tone by release of vasoactive factors (Hawkins and Davis, 2005). In addition, the endothelium plays a key role in injury responses such as inflammation, angiogenesis, and release of trophic factors (Davidson and Duchen, 2007). Estrogen has been shown to act directly on cerebral endothelium and affect a number of these functions, including vasodilator release, BBB integrity, and inflammatory responses (Krause et al, 2006; Miller and Duckles, 2008). In animal models, estrogen treatment ameliorates cerebrovascular changes that occur in early stages of brain ischemia, for example, BBB disruption and edema formation (Liu et al, 2005), and endothelial cell–leukocyte adhesion (Santizo et al, 2000). In addition, estrogen helps preserve microvascular blood flow in the ischemic area (Alonso de Leciñana and Egido, 2006). However, the effects of estrogen on cerebral endothelial cell viability have not been studied previously, although it has been shown to protect neurons and astrocytes under ischemia-like conditions (Alonso de Leciñana and Egido, 2006; Raval et al, 2006; Dhandapani and Brann, 2007). OGD is known to induce cell death in cerebral endothelial cells (Andjelkovic et al, 2003; Zhang et al, 2007), and the current study reveals that estrogen can protect against this condition. Reducing endothelial damage during ischemia would directly impact vascular function and indirectly influence the function of other components of the neurovascular unit, thereby decreasing brain injury.
One caveat of the current study is the use of an immortalized mouse brain endothelial cell line, bEnd.3, which may not show all of the differentiated functions associated with cerebral endothelial cells
Previous studies of neuroprotection by estrogen have suggested numerous and diverse mechanisms, genomic and non-genomic, receptor and non-receptor mediated (Alonso de Leciñana and Egido, 2006; Simpkins and Dykens, 2008). In the present study, E2 protection of endothelial mitochondria and cell viability during OGD was receptor-mediated. E2 was effective at a concentration of 10 nmol/L, which is in the range of circulating levels found in animals during the estrous cycle and consistent with the known affinity of E2 for its receptors. Furthermore, all the protective effects of E2 on endothelial cells during OGD/reperfusion were blocked by the ER antagonist, ICI-182,780. In contrast, a study of neuronal cell death found much higher estrogen concentrations to protect against ischemia/reperfusion, albeit through different mechanisms that may not depend on classic ERs (Prokai et al, 2003). Many studies, however, do point to ER mechanisms in neuroprotection; however, both ERα and ERβ subtypes have been implicated (Dubal et al, 2006; Alonso de Leciñana and Egido, 2006; Simpkins and Dykens, 2008; Noppens et al, 2009). ICI-182,780 does not distinguish between these ER subtypes, which are similar in structure and ability to bind E2, but appear to have different cellular distributions and distinct roles (Miller and Duckles, 2008).
Both ERα and ERβ are expressed in cerebral endothelium (Stirone et al, 2003; Razmara et al, 2008; Odenlund et al, 2008), and our data confirm the presence of both subtypes in bEnd.3 cells. However, we found that only an ERα-selective agonist, PPT, mimics the effect of E2 to protect endothelial cells against OGD-induced death. DPN did not have any effect; this compound has moderate selectivity for ERβ over ERα (Harrington et al, 2003). Thus ERα appears to have a crucial role in cerebral endothelial cell protection. This finding is consistent with our previous study showing that PPT, but not DPN, mimics the effects of E2 in reducing mitochondrial ROS production in human brain endothelial cells during normoxia (Razmara et al, 2008).
ERs exert effects on endothelial cells through a variety of mechanisms, such as genomic regulation and cell signaling pathways, and at different locations within the cell, for example, nucleus, plasma membrane caveolae, and mitochondria (Krause et al, 2006; Miller and Duckles, 2008); which of these pathways underlies suppression of endothelial cell death remains to be elucidated. As discussed below, effects on mitochondrial function and oxidative stress are clearly important. It is suggestive that pretreatment for more than 12 h was needed before significant protective effects of E2 against cell death were observed. These findings indicate that time is needed for E2 to act, most likely in a genomic manner, and tend to rule out mechanisms involving rapid alterations in cell signaling through membrane ERs (Miller and Duckles, 2008). Interestingly, estrogen has been shown to mimic the effects of ischemic preconditioning in protecting hippocampal neurons (Raval et al, 2006). Ischemic preconditioning of cerebral endothelial cells is also protective (Andjelkovic et al, 2003; Zhang et al, 2007), suggesting the possibility that estrogen evokes similar mechanisms to make the cells more resistant to detrimental effects of hypoxia/aglycemia. Our findings are in agreement with reports showing that long-term pretreatment with estrogen is required to protect neuronal cells from injury-induced cell death (Wilson et al, 2000; Nilsen et al, 2006). However, some studies have found estrogen to diminish stroke injury in animals when administered at the time of insult or during reperfusion (Liu et al, 2009), so clearly multiple mechanisms may be involved within the brain.
A key finding of the current study is the marked effect of E2 treatment on mitochondrial function in cerebral endothelial cells subjected to OGD/reperfusion. Mitochondrial dysfunction is implicated in the etiology and development of vascular pathology and age-related diseases such as ischemic stroke (Sims and Anderson, 2002; Wallace, 2005; Davidson and Duchen, 2007). Recent studies indicate that estrogen also affects mitochondrial function in non-endothelial cells of the neurovascular unit, specifically neurons and astrocytes (Nilsen et al, 2006; Araújo et al, 2008; Irwin et al, 2008; Simpkins and Dykens, 2008). Mitochondrial ROS production, loss of mitochondrial membrane potential, and decreased ATP are early triggers for mitochondrial dysfunction and eventual cell death in ischemic injury (Davidson and Duchen, 2007). In cultured bEnd.3 endothelial cells, we found that E2 attenuated these early mitochondrial events during OGD, thus suggesting a critical mechanism for maintaining endothelial viability. However, at this point it is not known whether E2 acts directly or indirectly on mitochondrial function to produce the results observed.
Mitochondrial regulation is emerging as a key role for estrogen (Duckles et al, 2006; Simpkins and Dykens, 2008). The precise mechanisms are not yet understood, but evidence suggests involvement of both genomic and non-genomic actions, as well as possible ER actions, within the mitochondrion itself. In particular, E2's effects on mitochondrial ROS, a major source of cellular ROS, may be a general mechanism underlying the protective effects of this hormone (Duckles et al, 2006; Simpkins and Dykens, 2008). In rat cerebral blood vessels, E2 decreases mitochondrial ROS production as well as increasing the expression of electron transport proteins, strongly supporting an increased capacity for vascular energy production (Stirone et al, 2005). Similar effects of E2 are found in cerebrovascular endothelial cells in culture (Razmara et al, 2008). Interestingly, both ERα and ERβ have been implicated in suppressing mitochondrial ROS in different cell types (Razmara et al, 2008; Flynn et al, 2008; Yang et al, 2009). In primary human brain endothelial cells, an agonist for ERα, but not ERβ, inhibited mitochondrial superoxide generation under basal, normoxic conditions (Razmara et al, 2008). This finding is consistent with the effects of PPT in the current study, although a possible role for ERβ cannot be excluded.
In conclusion, the present study demonstrates that estrogen protects cerebral endothelial cells subjected to OGD through ER-dependent mechanisms. These findings clearly show the direct protective effects of estrogen against ischemic injury
Footnotes
Acknowledgements
This study was supported by the United States National Heart, Lung and Blood Institute (Grant R01 HL-50775), and Chinese Government Scholarships for Postgraduates (20073020). and 973 Program of the Ministry of Science and Technology in China (No. 2004CB518902). We thank Vincent Procaccio and Saege Atea Hancock for technical support and Douglas Wallace and the Center for Molecular and Mitochondrial Medicine and Genetics for use of facilities.
The authors state no duality of interest.