
Abstract
The authors hypothesized that glutathione peroxidase-1 (Gpx-1) contributes to the neuroprotection seen in the superoxide dismutase-1 transgenic (Sod-1 tg) mouse. To investigate this hypothesis, they crossed the Gpx-1 -/- mouse with the Sod-1 tg and subjected the cross to a mouse model of ischemia/reperfusion. Two hours of focal cerebral ischemia followed by 24 hours of reperfusion was induced via intraluminal suture. The Sod-1 tg/Gpx-1 -/- cross exhibited no neuroprotection when infarct volume was measured; indeed, infarct volume increased in the Sod-1 tg/Gpx-1 -/- cross compared with the wild-type mouse. Our results suggest that Gpx-1 plays an important regulatory role in the protection of neural cells in response to ischemia/reperfusion injury.
The generation of reactive oxygen species (ROS) in ischemia reperfusion injury has long been implicated in the resultant damage (McCulloch and Dewar, 2001; Dirnagl et al., 1999; Chan, 1996). The detoxification of O2− generated in ischemia reperfusion injury is a two-stage reaction involving, first, superoxide dismutase-1 (Sod-1), which catalyzes the reduction of O2− to H2O2, itself a very damaging reactive molecule via the Fenton reaction (Cuzzocrea et al., 2001). The second stage involves the reduction of H2O2 to H2O by glutathione peroxidase-1 (Gpx-1). Therefore, investigating the relative effects of the ROS and the detoxifying enzymes will improve our understanding of the pathogenesis of neural damage and lead to improved intervention strategies.
Superoxide dismutase-1 transgenic (Sod-1 tg) mice have been shown to be neuroprotective in response to an ischemia reperfusion insult such as stroke (Yang et al., 1994, Kondo et al., 1997). The conundrum faced with the Sod-1 tg mice is that increasing the level and activity of Sod-1 gives rise to H2O2 (Fullerton et al., 1998), itself a toxic ROS (Halliwell et al., 2000), and it is difficult to account for the increase in neuroprotection seen in the Sod-1 tg mice in light of increased H2O2 levels. The mechanism of neuroprotection in the Sod-1 tg mice is unclear, since while the levels of O2− are reduced, the levels of H2O2 would rise. This raises the question that either the H2O2 is not a major mediator of oxidative damage (via the Fenton reaction) seen in the ischemia/reperfusion elicited cell death in the Sod-1 tg mice or that it is being processed at an increased rate.
The present study attempts to determine the relation between Sod-1 and Gpx-1 and elucidate their interactions in ischemia/reperfusion injury. Since Gpx-1 is the major H2O2 detoxifying enzyme in neurons (de Haan et al., 1998), we have focused on its contribution to the balance of ROS detoxification in the ischemia/reperfusion model of injury. In this study, we have generated Sod-1 tg/Gpx-1 -/- crosses to address this issue. The comparison between Sod-1 and Gpx-1 will allow us to directly determine whether Gpx-1 contributes to the attenuation of neural injury seen in the Sod-1 tg mice after ischemia/reperfusion injury.
MATERIALS AND METHODS
All animal procedures were approved by the Animal Ethics Committee of Monash University. Mice of a 129Sv/CF1/BalbC hybrid strain were bred to obtain a colony of homozygous Gpx-1 (de Haan et al., 1998). All procedures were carried out using matched littermate wild-type mice. Mice were studied at 6 to 8 weeks of age (body weight 25 to 32 g). Sod-1 mice were produced on a BalbC/C57Bl6 background, as previously reported (Epstein et al., 1987). The level of hSod-1 to endogenous mouse Sod-1 transcript levels in the brain is 5:1, with a transgene product distribution quite similar to that of the endogenous mouse enzyme (Epstein et al., 1987). The hSod-1 transgenic mice and Gpx-1 -/- have been extensively back-crossed to the C57Bl/6J background, resulting in near genetic confluence.
To develop the novel mutants, female hSOD-1 ++/++ mice were bred to GPX-/- male mice, creating GPX heterozygotes, some of which also overexpressed SOD-1 (GPX+/-, SOD-1++/++ mice). These double heterozygotes were then bred to produce GPX null SOD-1-overexpressing mice (GPX-/-, SOD-1++/++). This new colony was screened by using polymerase chain reaction primers for GPX (5' primer: 5'-AAG GAG GTG CAG GCG GCT GTG AGC G-3',3' primer: 5'-GCG CGG AGA AGG CAT ACA CGG TGG-3') and SOD-1 (5' primer: 5'-CAG CAG TCA CAT TGC CCA A-3',3' primer: 5'-GGT CTC CAA CAT G-3').
Focal cerebral ischemia was produced by occlusion of the middle cerebral artery (MCA). The MCA occlusion technique was based on that described previously for the mouse (Crack et al., 2001). Evaluation of neurologic deficits and infarct size was also based on that described previously (Crack et al., 2001).
Sod-1 activity was measured using an Oxis Sod-1 activity kit. Briefly, brains were homogenized in 25 mmol/L HEPES, 25 mmol/L sucrose, 1.5 mmol/L MgCl2 and 50 mmol/L NaCl pH 7.2 in a Potter-Elvehjem homogenizer. The resultant homogenate was spun at 10,000 g for 10 minutes. The supernatant was diluted 1:10 and 1:50 for subsequent assay within the linear range. Forty microliters of the diluted sample was added to 900 μL of the assay buffer (2-amino-2 methyl-1,3-propanediol, containing boric acid and DTPA). Mercaptans were quenched by the addition of 30 μL of the mercaptan scavenger (1,4,6-trimethyl-vinylpyridinium trifluoromethane-sulfonate). This was incubated at 37°C for 1 minute. Thirty microliters of the chromogenic reagent was added, and the resultant mix was assayed at 525 nm.
Gpx-1 activity was measured using an Oxis Gpx-1 activity kit. Tissue for the Gpx-1 assay was prepared in the same as way as for the Sod-1 assay. The resultant supernatant was diluted 1:10 and 1:50 for subsequent assay within the linear range. Seventy microliters of sample was added to 350 μL of assay buffer and 350 μL of NADPH reagent. To initiate the reaction, 350 μL of tert-butyl hydroperoxide was added and the reaction was assayed at room temperature at 340 nm.
Data in the text and figures are expressed as mean ± SEM. Group comparisons were analyzed by analysis of variance for unpaired samples. A P value less than 0.05 was considered statistically significant.
RESULTS
To compare the response of (1) Sod-1 tg/Gpx-1 -/- mice, (2) Gpx-/- mice, (3) Sod-1 tg mice, and (4) wild-type mice to ischemia/reperfusion injury, they were subjected to MCA occlusion. Physiologic studies of blood gases and blood pH for all strains were found to be consistent both before and after MCA stroke surgery, suggesting that these parameters had little impact on the outcome of the MCA surgery. There was no difference in posterior communicating artery plasticity between any of the strain groups (data not shown).
All four genotypes (as outlined above) were assessed for infarct size, 24 hours after ischemia/reperfusion. Figure 1 summarizes total hemispheric infarction volume in each mouse genotype, expressed as cubic millimeters and measured by calculating the cumulative areas in the consecutive brain slices. Each genetic modification had a profound effect on stroke volume relative to the wild-type mouse. Compared to the wild-type, the Sod-1 tg mouse had a significantly smaller infarct volume (28.2 ± 2.33), with the Gpx-1 -/- having a substantially increased infarct volume (109.3 ± 6.05) with respect to the wild-type. Interestingly, the combined Sod-1 tg/Gpx-1 -/- had an infarct volume (65.4 ± 10.04) that was similar to the wild-type mouse (50.5 ± 2.5), with this infarct being statistically larger than the Sod-1 tg and smaller than the Gpx-/- (n = 6). Functional deficits were consistent with the severity of the ischemic insult, with the most severe being found in the Gpx-/- group (2.47), with slight functional deficits being ascribed to the Sod-1 tg group (1.1). The wild-type mice and the combined crosses had similar functional deficits, 1.3 and 1.5, respectively.
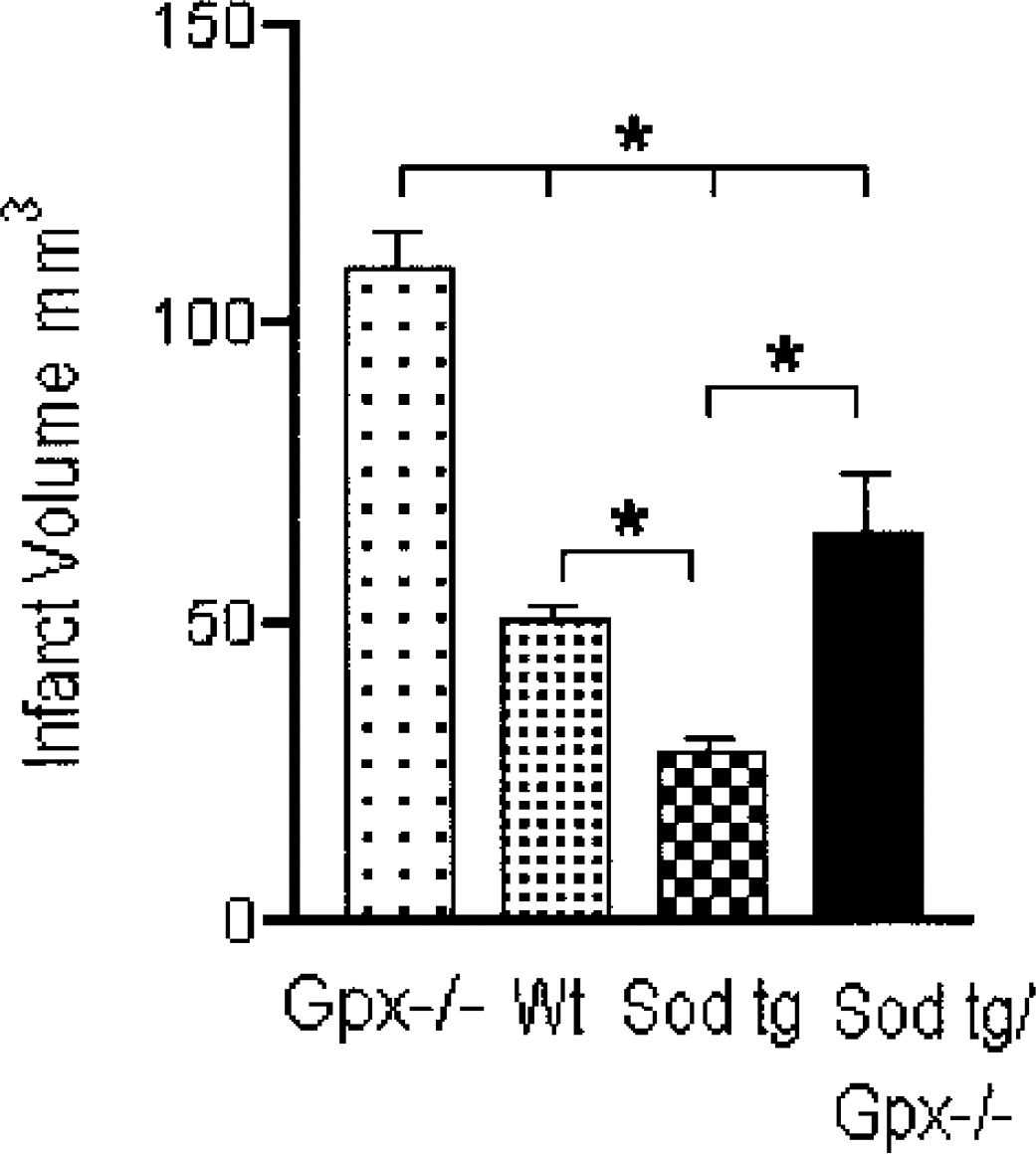
Infarct volume in the Gpx-1 -/- mice, wild-type control mice, Sod-1 tg mice, and the Sod-1 tg/Gpx-1 -/- crossed mice killed at 24 hours after occlusion of the middle cerebral artery. Each bar graph represents six replicates per assessment. Gpx-1, glutathione peroxidase-1; Sod-1, superoxide dismutase-1; Wt, wild-type; tg, transgenic; -/-, knockout.
Homogenates of brain samples from each of the mouse lines were tested for Sod and Gpx activity to confirm the effect of the genetic modifications on the functional proteins. Sod-1 activity was increased 2.66-fold in the Sod-1 tg and 2.22-fold in the Sod-1 tg/Gpx-1 -/- crosses. The Sod-1 activity of the wild-type mouse or the Sod-1 tg mouse was unaffected by the absence of Gpx-1 (Table 1). With respect to Gpx activity, the enzyme assays confirmed the null phenotype (since no activity was detected in the Gpx-1 -/- or Sod-1 tg/Gpx-1 -/mice). The activity of Gpx, however, was significantly increased from 4.5 ± 0.3 to 6.2 ± 0.5 U/mg protein (Table 1). Gpx-1 appears, therefore, to be regulated by and sensitive to increasing levels of Sod or H2O2, whereas Sod-1 is not regulated by or sensitive to Gpx-1 levels.
Enzymatic assays
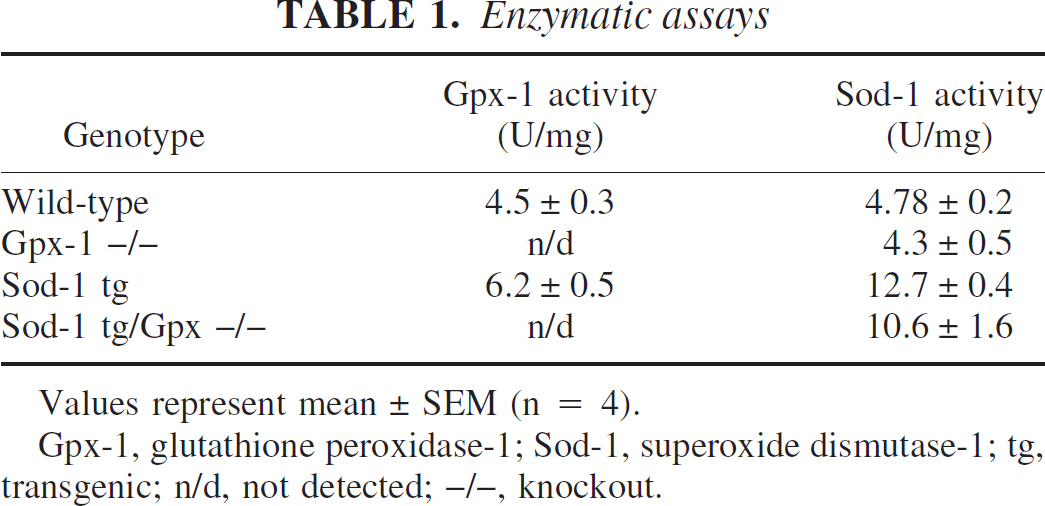
Values represent mean ± SEM (n = 4).
Gpx-1, glutathione peroxidase-1; Sod-1, superoxide dismutase-1; tg, transgenic; n/d, not detected; -/-, knockout.
DISCUSSION
This study strengthens the hypothesis that the antioxidant enzyme Gpx-1 is an important component in the enzymatic defense system against the increase of free radicals that are generated in ischemia/reperfusion injury (Klivenyi et al., 2000, Crack et al., 2001). In crossing the Gpx-1 -/- with the Sod-1 tg mouse and then performing the MCA surgery to induce stroke, we found that a large proportion of the protective effect generated by the overexpression of Sod-1 was negated. These findings imply that the protective effect mediated by the overexpression of the Sod-1 gene is in part dependent on a functional Gpx-1 enzyme clearing the H2O2. This finding enhances the hypothesis that an increase in ROS, in particular H2O2, plays a critical role in the mediation of neural cell death that occurs in response to ischemia/reperfusion injury.
Previous studies have indicated an important role for Gpx-1 in the protection of neural cells from oxidative stress in the form of H2O2 (de Haan et al., 1998) and mice overexpressing Gpx-1 demonstrated a marked neuroprotection after stroke (Weisbrot-Lefkowitz et al., 1998). In addition, we have shown that an imbalance in the antioxidant defense system that results in elevated levels of H2O2 is an important determinant in stroke (Crack et al., 2001). This study highlights the synergistic relation that Sod-1 and Gpx-1 have in dealing with increases of ROS that accompany stroke. By removing Gpx-1 activity in the Sod-1 tg group, the protective effect previously exhibited by the Sod-1 tg was diminished. This cross did not revert to the levels seen in the Gpx-1 -/- alone, however, suggesting that Sod-1 does indeed have a protective role that is independent of the processing of H2O2. This is most likely its role in processing superoxide (Sampei et al., 2000, Kamii et al., 1996) that limits the formation of peroxynitrite.
These findings highlight the control of oxidative damage as a crucial target for therapeutic intervention in stroke pathogenesis. For this intervention to be successful, it must focus not only on dealing with the increases of superoxide but also with the increases in H2O2 and other free radicals. The data presented here suggest that Gpx-1 is one of the key antioxidant enzymes to be involved in the cellular protection invoked in response to ischemia/reperfusion injury and as such may be an important target for potential stroke therapeutics.