
Abstract
Peroxisome proliferator-activated receptors (PPARs) are involved in energy expenditure, regulation of inflammatory processes, and cellular protection in peripheral tissues. Among the different types of PPARs, PPARβ is the only one to be widely expressed in cortical neurons. Using PPARβ knockout (KO) mice, we report here a detailed investigation of the role of PPARβ in cerebral ischemic damage, associated inflammatory and antioxidant processes as well as food intake regulation after middle cerebral artery occlusion (MCAO). The PPARβ KO mice had a two-fold increase in infarct size compared with wild-type (WT) mice. Brain oxidative stress was dramatically enhanced in these KO mice, as documented by an increased content of malondialdehyde, decreased levels of glutathione and manganese superoxide dismutase, and no induction of uncoupling protein 2 (UCP2) mRNA. Unlike WT mice, PPARβ KO mice showed a marked increase of prooxidant interferon-gamma but no induction of nerve growth factor and tumor necrosis factor alpha after MCAO. In WT mice, MCAO resulted in inflammation-specific transient hyperphagia from day 3 to day 5 after ischemia, which was associated with an increase in neuropeptide Y (NPY) mRNA. This hyperphagic phase and NPY mRNA induction were not observed in PPARβ KO mice. Furthermore, our study also suggests for the first time that UCP2 is involved in MCAO food intake response. These data indicate that PPARβ plays an important role in integrating and regulating central inflammation, antioxidant mechanisms, and food intake after MCAO, and suggest that the use of PPARβ agonists may be of interest for the prevention of central ischemic damage.
Keywords
Introduction
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to the nuclear receptor superfamily (Kersten et al, 2000). Several classes of PPARs have been identified, namely PPARα, PPARβ, and PPARγ, and the latter consists of two isoforms, PPARγ1 and PPARγ2 (Kersten et al, 2000). Previous studies have shown that PPARs are expressed differentially in tissues, PPARα being most abundant in the liver and PPARγ in adipose tissue, the pancreas, and phagocytes (Lee et al, 2003). These two PPARs show a less wide tissue distribution than PPARβ, also called PPARδ, which is expressed in the lung, liver, kidney, spleen adipocytes, neurons, and oligodendrocytes (Cullingford et al, 1998). Prior work has documented the central role of PPARs in glucose and lipid metabolism (Kersten et al, 2000). Peroxisome proliferator-activated receptors have been associated with energy expenditure by regulating the expression of mitochondrial uncoupling proteins (UCPs) (Evans et al, 2004; Sell et al, 2004). In particular, PPARα and PPARγ have been shown to modulate UCP1, which participates in the control of thermogenesis in brown adipose tissue (Lee et al, 2003). Besides these main effects, both in vitro and animal studies indicate that PPARs may control the expression of genes implicated in inflammatory responses and consequently alter cytokine production and cell recruitment to the inflammatory site (Hornung et al, 2001). For instance, PPARα knockout (KO) mice displayed a long-lasting inflammatory response to lipopolysaccharide stimulation (Delerive et al, 1999), and PPARα activation results in the repression of inflammatory cytokine production in different cell types (Delerive et al, 2001). In macrophages, PPARγ activation is thought to inhibit cytokine production (Ricote et al, 1998; Greene et al, 2000), whereas PPARβ negatively regulates specific subsets of genes that are activated by T helper 1 cytokines (Welch et al, 2003). In addition, PPARβ induction by inflammatory stimuli may suppress keratinocyte apoptosis during epidermal injury (Tan et al, 2004).
Despite this progress in the understanding of the function of PPARs, their role in central nervous system pathologies remains unclear. Recent evidence suggests that activation of PPARα receptor with selective agonists may confer neuroprotection after focal cerebral ischemia (Deplanque et al, 2003). In contrast, the role of PPARβ, which is abundantly expressed throughout the murine brain (Woods et al, 2003), remains obscure. Among the main biologic phenomena thought to be influenced by PPAR activation, cytokine-mediated inflammatory processes and activation of antioxidant mechanisms are of key importance for cell damage after acute cerebral ischemia. In this context, tumor necrosis factor alpha (TNFα) and interferon-gamma (IFNγ) are known to favor inflammatory processes (Blasko et al, 2001; Li et al, 2001), whereas nerve growth factor (NGF) may protect against inflammatory damage (Guegan et al, 1998) by inducing the activation of antioxidant mechanisms, such as an increase in manganese superoxide dismutase (MnSOD) (Guegan et al, 1999), glutathione (GSH) (Guegan et al, 1998), and/or UCP2 (de Bilbao et al, 2004). Using middle cerebral artery occlusion (MCAO) in mice, the present study explores the effects of a genetic PPARβ deficiency on brain infarct size, activation of the associated inflammatory and antioxidant processes as well as postischemic regulation of food intake.
Materials and methods
Animals, Food Intake, and Treatments
Adult male mice (3 to 6 months) were used in these experiments. The PPARα and PPARβ KO mice (weight 22 to 25 g) were established on a C57BL/6J genetic background. For each mouse strain, appropriate colittermate controls (weight 22 to 24 g) were used. These mice were kindly supplied by Professor W Wahli, University of Lausanne (Tan et al, 2001). The TNFα KO mice (strain Tnftm1Gkl, weight 21 to 23 g) (Pasparakis et al, 1996), IFNγ KO mice (strain B6.129S7-Ifhgtm1Ts/J, weight 20 to 23 g) (Dalton et al, 1993), and their wild-type (WT) counterparts (weight 21 to 24 g), all with a C57BL/6J background, were obtained from the Jackson Laboratory (Jackson-Bar Harbor, Maine, USA). The UCP2 KO mice (C57BL/6J, weight 19 to 21 g) and their WT colittermates (weight 19 to 22 g) were also used as described previously (de Bilbao et al, 2004). All mice strains were backcrossed for at least 10 generations to ascertain the similarity of genetic background with WT mice. Mice were housed in individual cages for food intake and body weight studies, and had ad libitum access to standard laboratory diet and water. Food intake was calculated using the weight of food pellets from the previous day and subtracting the weight of the remaining pellets on the following day (measured at 0900). For all feeding experiments, a minimum of eight ischemic animals (eight WT and eight PPARβ KO) and eight nonischemic animals (eight WT and eight PPARβ KO) were used. Food intake baseline levels were recorded for 3 days before MCAO, and then food intake was quantified daily for 2 weeks. To evaluate the role of central NGF in food intake regulation, 200 ng of NGF 2.5S (Chemicon, Temecula, CA, USA) was injected on the lesion site in PPARβ KO mice (n = 8) 1 day after MCAO, as well as in sham-operated PPARβ KO mice (n = 8). This timing was determined by the maximal NGF effect on food intake observed in WT mice. Nerve growth factor was injected with a microsyringe (5 μl vehicle containing 200 ng of NGF per mouse) after the animals had been anesthetized with isoflurane. The food intake response was studied for 4 consecutive days. Additional experiments were performed in IFNγ KO, TNFα KO, and UCP2 KO mice to explore the role of these proteins on food intake responses after MCAO (eight mice per group, as mentioned above). Because PPARβ KO and UCP2 KO mice showed no hyperphagic phase response after MCAO, we also evaluated food intake in nonoperated PPARβ KO and UCP2 KO mice after 24 h of fasting to examine whether a reduction in food intake induces a compensatory hyperphagia (n = 8 for WT and KO groups).
Induction of Permanent Focal Cerebral Ischemia and Volume of the Infarct
We performed permanent MCAO in PPARα KO (nKO = 6 and nWT = 6) and PPARβ KO (nKO = 18 and nWT = 16) mice. Animals were anesthetized intraperitoneally with xylazine (20 mg/kg)/ketamine (100 mg/kg) in 0.9% NaCl (100 μl/10g body weight), the right temporoparietal region of the head was shaved and a 2-mm incision was made vertically between the orbit and the ear and the skull was exposed. Under an operating microscope, a small burr hole 1 mm2 was made with a high-speed microdrill through the outer surface of the semitranslucent skull over the visibly identified medial cerebral artery at the level of the parietal cerebral artery. Saline was applied to the area throughout the procedure, to prevent heat injury. The inner layer of the skull was removed with fine forceps, the dura and the arachnoid were opened and right permanent MCAO was performed by electrocoagulation (by means of a small-vessel cauterizer), without damaging the brain surface. Permanent inhibition of cerebral blood flow after the lesion was assessed by visual inspection. If the brain surface was visibly damaged or if the middle cerebral artery bled owing to incomplete artery occlusion/coagulation, the animal was not used for the study. The duration of the surgery did not exceed 15 mins in any case (de Bilbao et al, 2000). After 4 days, the animals were perfused through the ascending aorta with a solution of 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.35). Brains were removed and processed for paraffin embedding. Sections (7 μm) of the whole infarct area were cut with a microtome and collected on slides pretreated with 3-aminopropyltriethoxy-silane (Sigma, MO, USA). Sections were counterstained with cresyl violet for the histologic identification of the nuclear boundaries and periinfarct areas, and mounted in Eukitt. For each animal, quantification of the infarcted area was performed on the cresyl-violet-stained sections at five representative levels throughout the rostro-caudal extent of the lesion (A 0.26, −0.22, −0.40, −0.70, and −1.2 −4 mm relative to Bregma) (Franklin and Paxinos, 1997). The rostro-caudal extent of the infarct was the same in both groups of mice. The infarcted area of each section was calculated by subtraction of healthy tissue areas of the contralateral from the ipsilateral side of the section to compensate for the effect of brain edema (Guegan et al, 1998) using a computerassisted image analyzing system (Software «Morphometry», Samba 2005 TITN, Alcatel, Alcatel Grenoble, France). Volumes of infarct (mm3) were calculated for each animal after integration of areas with the distance between each level (de Bilbao et al, 2004).
To evaluate whether local alterations in cerebral vascular anatomy contribute to different susceptibility to injury in PPARβ KO mice, an additional series of five WT and five KO mice were killed on day 4 (D4) after ischemia. Cerebral vasculature was studied in WT and PPARβ KO mice (nonoperated and on D4 after ischemia) after intracardial perfusion with 0.9% NaCl solution followed by perfusion of a mixture of an equal proportion of gelatinous water (5%) and China ink (Sennelier, France) warmed at 40°C (1 ml). Brains were removed and immersed for 24 h in 4% paraformaldehyde at 4°C (Chen et al, 2005). Cerebral vasculature was observed with a Zeiss stero zoom microscope. The absence of cerebral blood flow in the infarct area was assessed by transcranial measurements of CBF that were made using laser Doppler flowmetry Oxford Optronix Ltd (UK) just before and after MCAO. Animals were placed under a sterotactic head frame and then a fine needle probe (MNP110XP, 0.48 mm diameter) was lowered onto the temporal bone surface 0.5 to 1 mm dorsal to the opening giving access to the MCA and wetted with a small amount of physiologic saline. No preoperative or postoperative strain-related differences in CBF were observed and similar reductions in CBF were observed after MCAO in both strains.
Physiologic Parameters: Physiologic parameters including arterial blood pressure (Kent mouse tail blood pressure system RTBP2000, Kent Scientific Corporation, Torrington, USA), plasma glucose (using Roche Glucotrend Active, Rotkreuz, Switzerland), and hematocrit were measured daily in KO and WT mice (n = 4 to 5) before MCAO and on D1 and D4 after injury. Body temperature was measured before, during, and after MCAO by a rectal thermometer probe (Ellab DM 852, Roeovre, Denmark). During surgery, mice were placed on a warm mat and rectal temperature was measured. In the preoperation period, rectal temperature was similar in WT (38.0°C ± 0.1°C) and PPARβ KO (38.0°C ± 0.1°C) mice and remained stable after the operation. Daily body temperature was measured at 1030.
In situ Hybridization for PPARβ mRNA Expression in PPARβ KO and WT Mice after MCAO
On D4 after injury, the expression of PPARβ mRNA was determined by in situ hybridization in WT (n = 4) and PPARβ KO (n = 4) mice and compared with their nonoperated counterparts. Mice were anesthetized with 0.3 ml of a mixture containing 40 mg/ml ketamine and 2 mg/ml xylazine and were immediately perfused intracardially with 20 ml of ice-cold isotonic saline followed by 100 ml of a 4% paraformaldehyde solution. At the end of the perfusion, brains were removed and kept in 4% paraformaldehyde for 3 to 4 days and then transferred to a solution containing paraformaldehyde and sucrose (10%) for 12 h. Sections (25 μm thick) were cut with a sliding microtome (Microtome HM 440E, Microm) and mounted onto slides. In situ hybridization histochemistry was performed using a biotin-labelled oligonucleotide PPARβ probe for mRNA (mouse 61 to 111, 5'-atgggtgacg gagccccgga gctcaatggg ggaccagaac acacgcttcc-3', and its complementary sequence as a negative control) to detect PPARβ (Catalys, Wallisellen, Switzerland). Briefly, 9 pmol/μl of biotin-labelled oligonucleotide was incubated for 2 h at 37°C in a hybridization solution (5 mol/L NaCl, 10 × PE (0.5 M Tris, 1% sodium pyrophosphate, 2% polyvinyl pyrrolidone, 2% Ficoll, SOmMEDTA) buffer, 50% dextran sulfate, 30% formamide) in an OmniSlide Thermal Cycler. After incubation, the slices were washed and biotin labelling was detected using avidin peroxidase complex with diaminobenzidine enhanced by nickel. The specificity of the antisense riboprobe was determined by the absence of a positive signal in sections hybridized with the sense probe.
PCR Determination of mRNAs in PPARβ KO and WT Mice after MCAO
For PPARβ and UCP2 mRNA determinations, ischemic hemispheres were dissected and total RNA was isolated as described previously (Arsenijevic et al, 2000a). Brain tissues were collected (n = 4 per group) at 1, 4, and 7 days after MCAO from WT and PPARβ KO mice, as well as from nonoperated control counterparts. In addition, we assessed mRNA expression for neuropeptide Y (NPY), melanin concentrating hormone (MCH), and orexin, three hormones involved in food intake. For this purpose, the whole hypothalamus was dissected out and the RNA was isolated. The RNA was treated with DNase and then reverse transcribed (Promega, Catalys, Wallisellen, Switzerland). Thereafter, a semiquantitative PCR (Invitrogen, Basel, Switzerland) was performed and the product separated by gel electrophoresis containing ethidium bromide. The bands were then quantified using the Scion Image program (Scion Corporation, MD, USA). Each sample was normalized with its glyceraldehyde 3-phosphate dehydrogenase content. The primers used were as follows: UCP2 (sense 5'-TAC CAG AGC ACT GTC GAA GCC-3', antisense 5'-AGT CCC TTT CCA GAG GCC C-3'; Yu et al, 2000), PPARβ (sense 5'-AGTTCTTGCGCAG TATCCG-3', antisense 5'-AGTGTTGTGAGTGGCTCTAG-3'; Yang et al, 1999), NPY (sense 5'-GGG GCT GTG TGG ACT GAC CCT GG-3', antisense 5'-GAT GTA GTG TCG CAG AGC GGA G-3'; Gallmann et al, 2005), MCH (sense 5'-AAA ATG ATG AGA GCG GCT TCA-3', antisense 5'-CGA GAT TCT GCT TGG AGC CT-3'; Gallmann et al, 2005), orexin (sense 5'-TGG GTA TTT GGA CCA CTG CA-3', antisense 5'-TGG TGT CTG GAG CTC AGG G- 3'; Gallmann et al, 2005), and glyceraldehyde 3-phosphate dehydrogenase, as reference control (sense 5'-TGA AGG TCG GTG TCA ACG GAT TTG GC-3', antisense 5'-CAT GTA GGC CAT GAG GTC CCA CCA C-3'; Subang et al, 1997). All values for the semiquantitative RT-PCR expression of a gene were obtained after normalizing for glyceraldehyde 3-phosphate dehydrogenase. The level of PPARβ and UCP2 was also quantified using real-time PCR (Berthiaume et al, 2004). Lesioned and nonlesioned brain hemispheres (n = 4 for each group of mice) were removed on D4 after MCAO and stored in RNA later (Ambion, TX, USA) at 4°C. Brain mRNA was extracted using the Trizol RNA extraction method. RNA concentration was estimated from absorbance at 260 nm and RNA was transcribed using Expand reverse transcriptase (Roche Diagnostics, Laval, QC, Canada). The mRNA expression level was quantified using quantitative fluorescent real-time PCR (Corbett Research, New South Wales, Australia). Amplification and detection of target mRNA were performed with Platinum Taq polymerase and the intercalating dye SybrGreen I (Berthiaume et al, 2004). The mRNA levels of PPARβ and UCP2 were normalized with beta-actin. The primers used were as follows: PPARβ, sense 5'-AGTTCTTGCGCAGTATCCG-3', antisense 5'-AGTGTTGT GAGTGGCTCTAG-3'; UCP2, sense 5'-TTC TCC TGC GGT CCG GAC ACA ATA-3', antisense 5'- TTGA CTC TCC CCT TGG ATC TGC AG-3'; and beta-actin, sense 5'-CTC TAG ACT TCG AGC AGG AG-3', antisense 5'-AGA GTA CTT GCG CTC AGG AG-3'. The primers were designed using the Vector NTI program. For both experiments, gene expression represented the percentage obtained by comparing operated hemispheres to control hemispheres of nonoperated mice.
Brain Cytokine Levels after MCAO in PPARβ KO and WT Mice
The levels of TNFα, IFNγ, and NGF were measured by immunoassay in the brains of operated and nonoperated PPARβ KO and WT mice (nKO = 8 and nWT = 8, each group consisting of four ischemic and four nonoperated animals) on D1 and D4 after MCAO. Brains were aseptically removed and immediately placed on dry ice. The ischemic hemispheres of operated mice and hemispheres of nonoperated mice were put in CHAPS solution and homogenized. The supernatant was collected and frozen at −20°C (Arsenijevic et al, 2000a). Cytokines TNFα and IFNγ, as well as NGF, were measured using immunoassay kits from Amersham and Catalys (Wallisellen, Switzerland). To study possible interactions between these three cytokines, NGF levels were also assessed in IFNγ KO and TNFα KO mice, and in the corresponding WT mice 1 day after ischemia (nKO = 8 and nWT = 8, each group consisting of four ischemic and four nonoperated animals).
Lipid Peroxidation
The levels of malondialdehydes (MDA), indicators of endogenous peroxidation, were measured in PPARβ KO and WT mice on D1 and D4 after MCAO. Mice were anesthetized and perfused intracardially with saline solution. After perfusion, the brain was removed and the ipsilateral and contralateral whole hemispheres of operated and nonoperated mice were washed three times in ice-cold Tris-HCl 20 mmol/L buffer (pH 7.4). They were then gently homogenized for 1 min in a glass tissue grinder (Wheaton, Millville, NJ, USA) in 4 ml of the same buffer. Homogenates were centrifuged at 3,200g for 10 mins at 4°C and the supernatant was processed for MDA levels using a lipid peroxidation assay (de Bilbao et al, 2004). To prevent sample oxidation, 0.5 mmol/L butylated hydroxytoluene (Sigma Chemicals, St Louis, MO, USA) was added to the tissue homogenate. Optical density was measured at 586 nm using a spectrophotometer (Dynatech, Chantilly, VA, USA). Changes in oxidative stress were assessed in brain homogenates in nonoperated (nKO = 8 and nWT = 8) and operated (nKO = 8 and nWT = 8) PPARβ KO and WT mice. For each group of mice, MDA levels of the operated brain hemisphere were compared with those of the hemisphere of nonoperated animals (de Bilbao et al, 2004).
Glutathione Levels
Brain homogenates from WT and PPARβ KO mice, subjected or not to ischemia (1 and 4 days after ischemia) (n = 8 per group), were processed as described previously for reduced GSH (de Bilbao et al, 2004). Reduced GSH levels were measured using a method based on the formation of a chromophoric product resulting from the reaction of 5,5'-dithiobis-(2-nitrobenzoic acid) (Sigma Chemicals, St Louis, MO, USA) and GSH (Sigma, Buchs, Switzerland). For each group of mice, we compared the operated brain hemisphere with the nonoperated hemisphere of control animals. To evaluate whether IFNγ administration influences oxidative stress regulation in ischemic PPARβ KO mice, these animals and their WT counterparts (n = 8 for each group) were pretreated with IFNγ antibody 18 h before MCAO and were killed 4 days later. Interferon-gamma antibody (ATCC HB170 from American Type Culture Collection, MD, USA, provided by C Yesin, CMU, Geneva, Switzerland) was injected at 50 μg per mouse intraperitoneally (Arsenijevic et al, 2000a). Levels of MDA and GSH were also assessed in these mice as described above.
Immunohistochemistry and Double Staining Experiments
Frozen coronal sections (25 μm) from operated (1 and 4 days after ischemia) and nonoperated WT and PPARβ KO mice (n = 4 for each group) were processed for MnSOD immunostaining using sheep anti-human MnSOD (1/100; Calbiochem, San Diego, CA, USA) and rabbit anti-sheep immunoglobulins (Dako, CA, USA) as the secondary antibody. Immunohistochemistry of MnSOD was performed on nonoperated and operated (1 and 4 days after MCAO) WT and PPARβ KO mice. We also tested MnSOD immunostaining 4 days after MCAO in WT and PPARβ KO mice treated with 200 ng NGF 2.5S on the lesion site 1 day after MCAO. Immunohistochemistry of PPARβ was performed using an anti-PPARδ rabbit polyclonal antibody (PA1-832) from Affinity Bioreagents (Golden, CO, USA) and visualized by a secondary goat anti-rabbit fluorescein isothiocyanate (Nordic Immunology, Tiburg, The Netherlands). To label astrocytes, microglia, macrophages, oligodendrocytes, and neuronal cells, mouse polyclonal anti-glial fibrillary acidic protein clone GA5 (1/400; Sigma, MO, USA), rabbit polyclonal anti-Iba1 (1 μ/ml; kindly provided by Professor Y Imai, National Center of Neurology and Psychiatry, Japan), rat anti-mouse F4/80 for macrophage (1/200; Serotec, Oxford, UK), anti-CNPase (2'3' cyclic nucleotide 3' phosphodiesterase) (1/200; Sigma, MO, USA), and the mouse anti-neuronal nuclei monoclonal antibody (NeuN) (1/1,000; Chemicon, Temecula, CA, USA) were used. The glial fibrillary acidic protein, anti-Iba1, anti-macrophages, and NeuN antibodies were detected using Alexa Fluor 546 rabbit anti-mouse (Molecular Probes, Eugene, OR, USA), goat anti-rabbit immunoglobulins Alexa Fluor 568 (Molecular Probes), goat anti-rabbit fluorescein isothiocyanate, goat anti-rat fluorescein isothiocyanate (Nordic Immunology), and goat anti-mouse immunoglobulins linked to AMCA (9-amino-6-chloro-2-methoxyacridine) (Dako). Negative controls included deletion of the primary or secondary antibody. Photomicrographs of immunofluorescent staining were constructed using the Axioskop 2 and the software Axiovision.
Statistical Analysis
Results are expressed as means ± standard error of the mean (s.e.m.). Differences between groups were assessed using a two-way ANOVA followed by the Bonferroni post hoc test. P<0.05 was considered significant.
Results
Physiologic Parameters before and after MCAO
Arterial pressure, plasma glucose, hematocrit levels, and body temperature before and after MCAO on D1 and D4 were not significantly different between WT and PPARβ KO mice at a given time point, as shown in Table 1. There were no significant differences between WT and KO mice between the different days (Table 1). After MCAO, both WT and PPARβ KO mice showed an absence of cerebral blood flow in the infarct area.
Physiologic parameters before and after MCAO in PPARβ KO and WT mice
Hematocrit, plasma glucose, body temperature, and arterial pressure were measured in wild-type (WT) and PPARβ knockout (KO) mice before ischemia, and then on days 1 (D1) and 4 (D4) after MCAO. No significant differences were observed between WT and KO mice on any given day, and there were no significant differences between days in WT and KO. Data are means ± s.e.m. of four to five animals per group. MCAO: middle cerebral artery occlusion; PPARβ: peroxisome proliterator-activated receptor β.
PPARβ and UCP2 mRNA Expression in WT and PPARβ KO Mice after MCAO
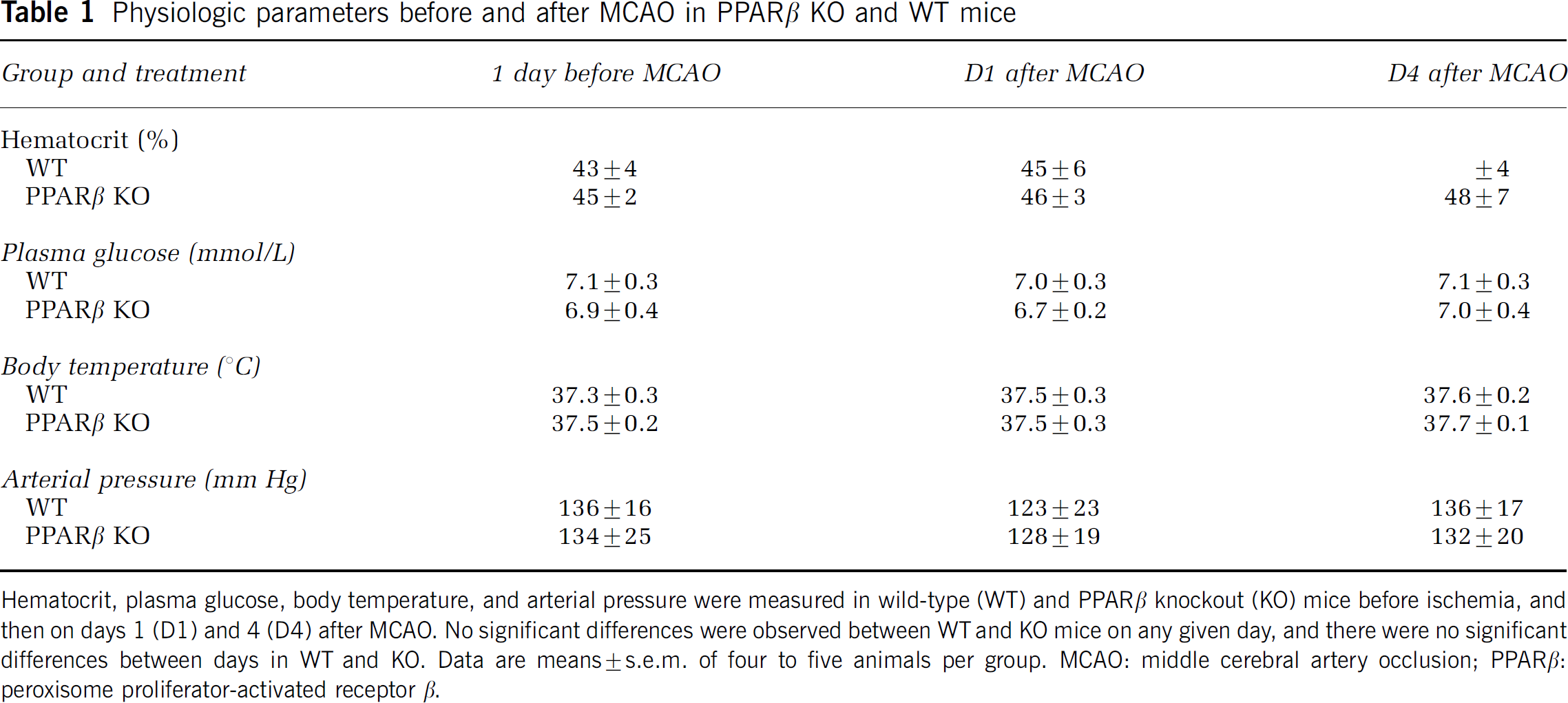
The expressions of PPARβ and UCP2 mRNAs in response to ischemic brain injury were determined by semiquantitative RT-PCR. Peroxisome proliferator-activated receptor β mRNA was significantly upregulated (45% to 58%, P<0.001) in the ischemic hemispheres of WT mice compared with nonoperated animals on D1, D4, and D7 after MCAO (Figure 1A). As expected, PPARβ mRNA was not detected in PPARβ KO mice (Figure 1A). Uncoupling protein 2 mRNA was also significantly increased (30% to 70%, P<0.001) in the ischemic hemispheres of WT mice on D1, D4, and D7 after MCAO (Figure 1B). Interestingly, no significant induction of UCP2 mRNA was observed in the ischemic hemispheres of PPARβ KO mice (10% to 14%, P>0.05) compared with nonoperated animals after MCAO (Figure 1B). Quantitative fluorescent real-time PCR also indicated that PPARβ and UCP2 were induced in WT mice on D4 after MCAO (112% ± 12% and 91% ± 10%, respectively) but not in PPARβ KO mice. In situ hybridization experiments also revealed induction of PPARβ mRNA in the periinfarct area on D4 after MCAO in WT mice (Figure 1C) but not in PPARβ KO mice (not shown). No labelling was observed in the contralateral hemisphere of WT mice. Induced PPARβ mRNA labelling occurred in oligodendrocytes and neurons in the border of the necrotic zone (data not shown) (Woods et al, 2003). Immunofluorescent staining of PPARβ showed that PPARβ protein was induced in the periinfarct area of WT mice but not in PPARβ KO mice 4 days after MCAO (Figures 1D and 1E).
Expression of PPARβ and UCP2 in WT and PPARβ KO mice after MCAO. RT-PCR determination of (
Infarct Size after MCAO is Increased in PPARβ KO Mice
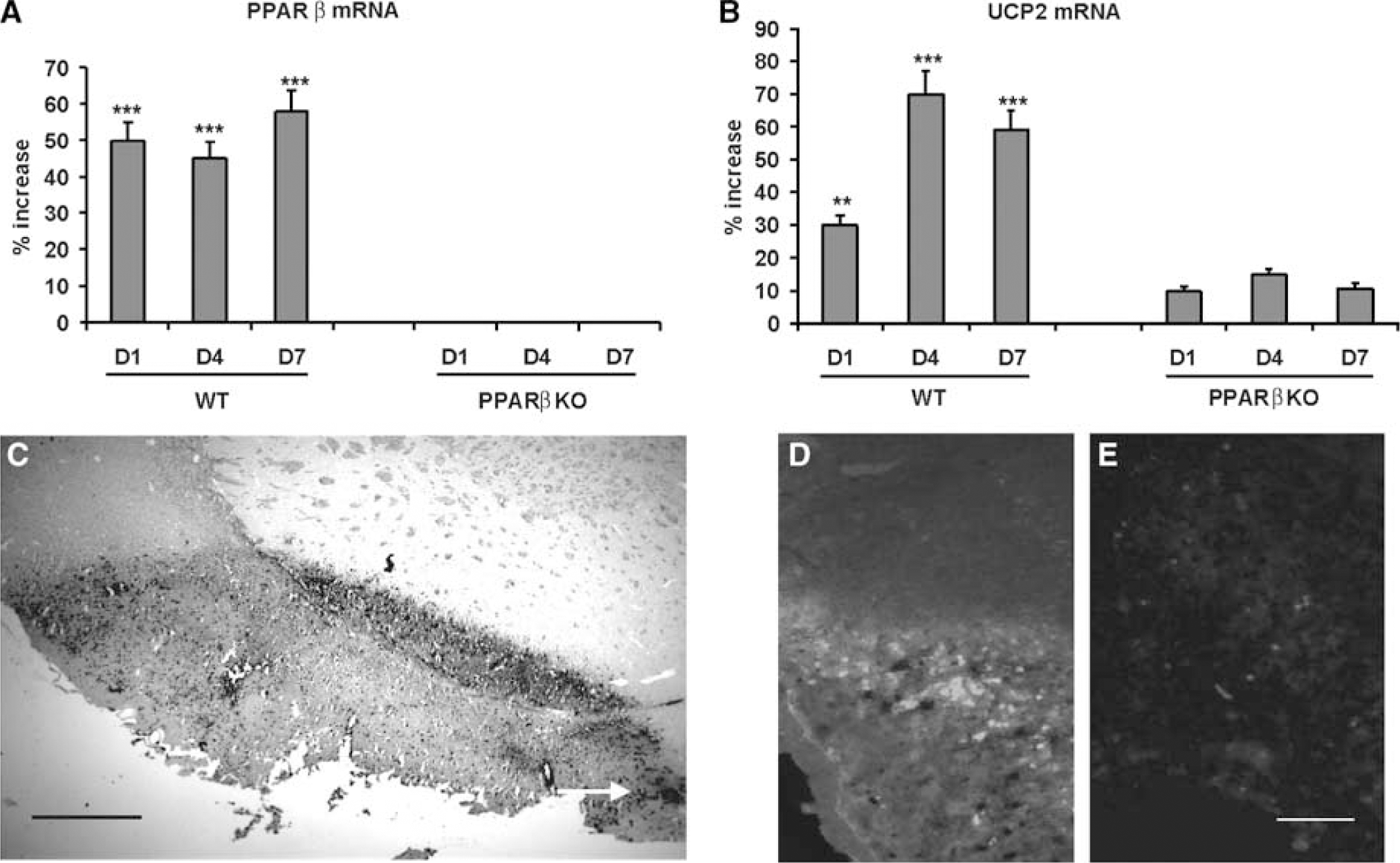
The infarct volume was quantified on D4 after permanent MCAO in WT and PPARβ KO mice, a time at which PPARβ mRNA was markedly increased in the periinfarct area of WT mice (Figure 1A). Infarct volume was increased two-fold in PPARβ KO mice (10.9 ± 0.2 mm3, n = 18) when compared with WT mice (5.3 ± 1.3 mm3, n = 16) (P< 0.001) (Figure 2). No difference was observed in the vascular system (organization of the circle of Willis, position and diameters of the main cerebral arteries) between WT and KO mice. At 4 days after MCAO, no revascularization was observed on the surface of the injured hemisphere in both mice strains (data not shown).
Effect of PPARβ deletion on infarct size. Representative coronal brain sections showing the ischemic infarct 4 days after MCAO in WT and PPARβ KO mice. Brain sections were stained with cresyl violet. The surrounded areas denote the size of the ischemic area. The infarct size was found significantly increased in PPARβ KO mice (P<0.001) (nKO = 18 and nWT = 16) (scale bar: 120 μm).
Importantly, this effect was specific for PPARβ because there was no significant difference in lesion volume between PPARα KO mice and their WT counterparts (5.6 ± 1.6 and 5.8 ± 1.1 mm3, respectively, n = 6 per group) (P>0.05).
Altered Lipid Peroxidation and Glutathione Levels in PPARβ KO Mice after MCAO and their Regulation by IFNγ
In both groups of mice, a significant increase in MDA levels was observed in the ischemic brains when compared with the nonoperated mice (Table 2). Moreover, PPARβ KO mice had significantly higher levels of MDA than WT mice (Table 2). Reduced GSH levels were significantly decreased in the brains of WT and PPARβ KO mice on D1 and D4 after MCAO (Table 2). On D4 after ischemia, PPARβ KO mice had significantly lower levels of GSH than WT mice (Table 2).
Brain MDA and GSH levels
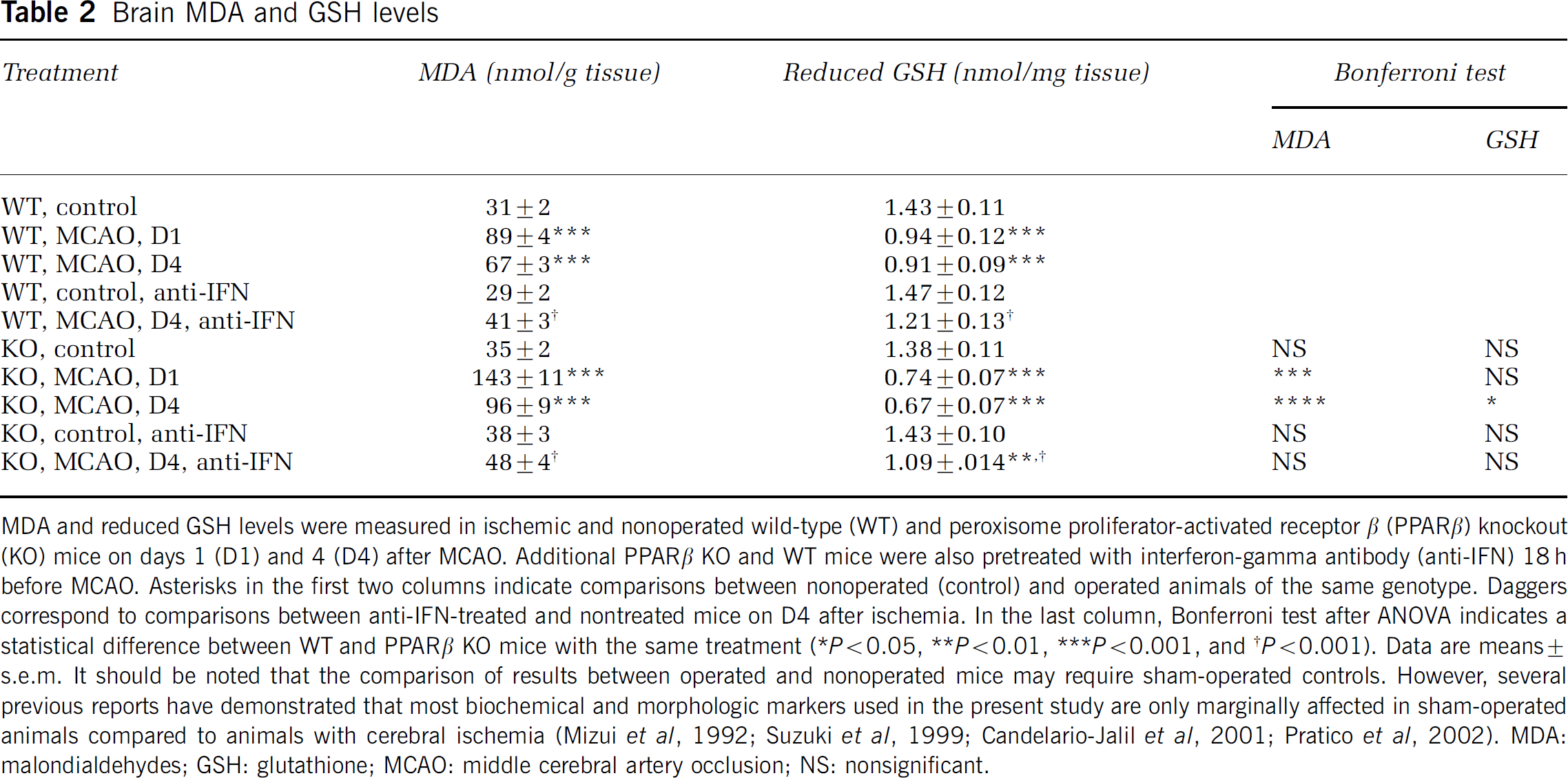
MDA and reduced GSH levels were measured in ischemic and nonoperated wild-type (WT) and peroxisome proliferator-activated receptor β (PPARβ) knockout (KO) mice on days 1 (D1) and 4 (D4) after MCAO. Additional PPAR β KO and WT mice were also pretreated with interferon-gamma antibody (anti-IFN) 18 h before MCAO. Asterisks in the first two columns indicate comparisons between nonoperated (control) and operated animals of the same genotype. Daggers correspond to comparisons between anti-IFN-treated and nontreated mice on D4 after ischemia. In the last column, Bonferroni test after ANOVA indicates a statistical difference between WT and PPARβ KO mice with the same treatment (*P<0.05, **P<0.01, ***P<0.001, and †P<0.001). Data are means ± s.e.m. It should be noted that the comparison of results between operated and nonoperated mice may require sham-operated controls. However, several previous reports have demonstrated that most biochemical and morphologic markers used in the present study are only marginally affected in sham-operated animals compared to animals with cerebral ischemia (Mizui et al, 1992; Suzuki et al, 1999; Candelario-Jalil et al, 2001; Pratico et al, 2002). MDA: malondialdehydes; GSH: glutathione; MCAO: middle cerebral artery occlusion; NS: nonsignificant.
Administration of IFNγ antibody 18 h before MCAO led to a significant reduction of MDA increase and GSH loss on D4 after ischemia in both WT and PPARβ KO mice when compared with nontreated ischemic mice, suggesting that oxidative stress is mediated by IFNγ in both types of mice. When the antibody was administered to mice that did not undergo MCAO, it did not have any effect on MDA or reduced GSH brain levels compared with nontreated mice.
TNFα and NGF Induction is Blunted and that of IFNγ Potentiated in PPARβ KO Mice after MCAO
Tumor necrosis factor alpha was significantly induced in the brain of WT mice on D1 after MCAO (90 ± 8 pg/ml, P< 0.001), and returned to basal levels on D4 (16 ± 3 pg/ml) compared with nonoperated WT mice (19 ± 4 pg/ml) (Figure 3A). In contrast, PPARβ KO mice showed no induction of TNFα on either day after MCAO (15 ± 3 and 18 ± 3 pg/ml for D1 and D4, respectively) when compared with nonoperated animals (17 ± 4 pg/ml, P>0.05 (Figure 3A).
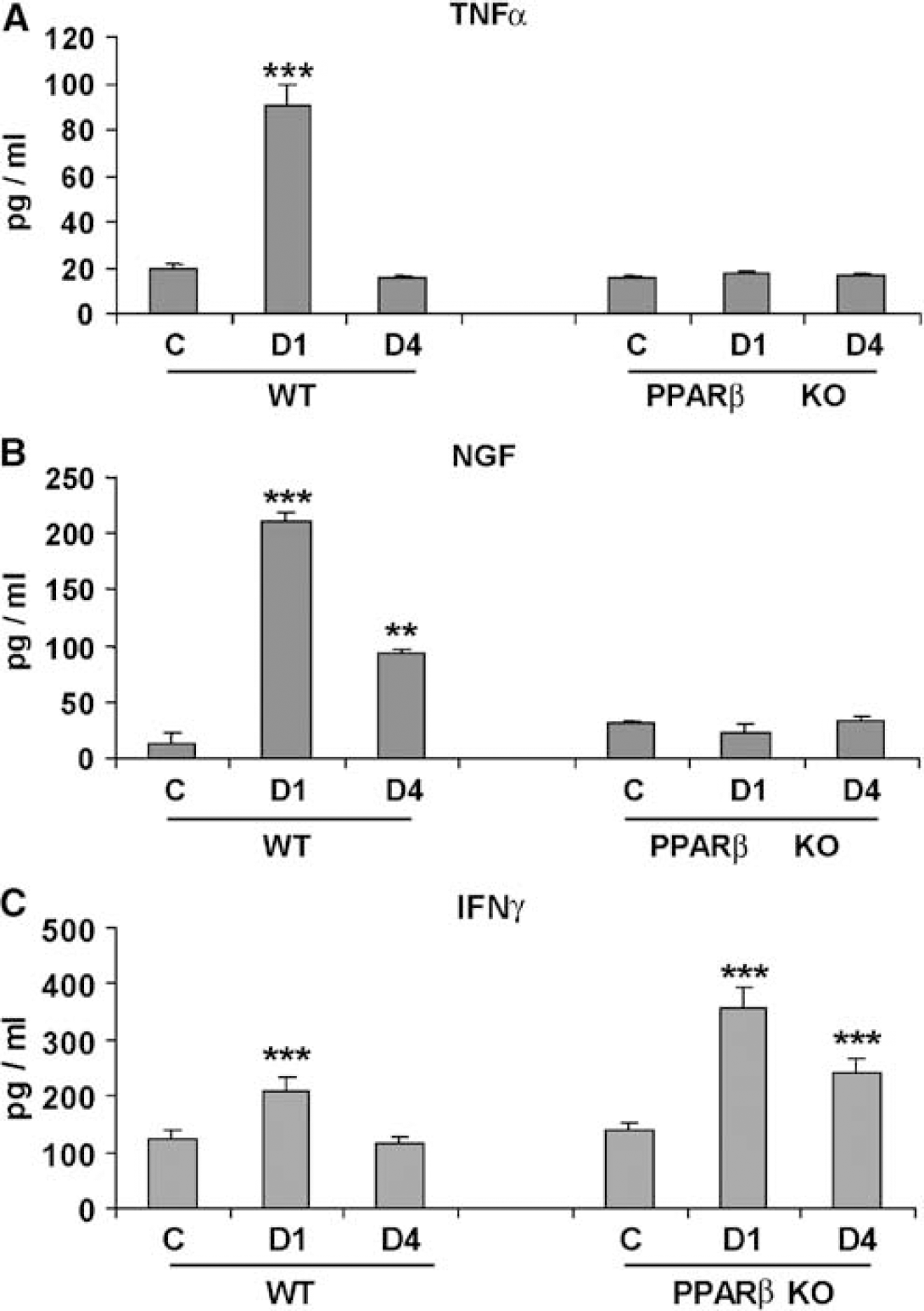
Levels of (
Similarly, NGF levels were markedly increased on D1 (221 ± 7 pg/ml) (P<0.001) and D4 (93 ± 4 pg/ml) (P<0.01) after MCAO in the brain of WT mice when compared with nonoperated animals (21 ± 10 pg/ml) (Figure 3B). Again, the upregulation of NGF levels was completely ablated in PPARβ KO mice (24 ± 7 pg/ml on D1 and 32 ± 5 pg/ml on D4) when compared with nonoperated mice (30 ± 2 pg/ml) (Figure 3B).
Interferon-gamma was significantly increased in the brains of both WT and PPARβ KO mice on D1 after ischemia (211 ± 8 and 357 ± 15 pg/ml, respectively) when compared with nonoperated animals (125 ± 3 and 139 ± 4 pg/ml, respectively) (all comparisons P<0.001) (Figure 3C). At 4 days after ischemia, IFNγ was still significantly increased in PPARβ KO mice compared with nonoperated animals (242 ± 6 and 139 ± 4 pg/ml, respectively) (P<0.001) but returned to basal levels in WT mice (Figure 3C). Interestingly, and although IFNγ basal levels did not differ between nonoperated WT and PPARβ KO mice, IFNγ levels at D1 and D4 were significantly higher in KO (D1: 69%, P<0.01; D4: 74%, P<0.01) than in WT mice (Figure 3C).
MnSOD is Reduced in PPARβ KO mice after MCAO: Reversal by NGF Treatment
Because MnSOD is thought to be the first line of defense against oxygen toxicity, we also examined its presence in WT and PPARβ KO mice before and after MCAO. No immunostaining was detected in WT and PPARβ KO mice before MCAO. Double staining experiments revealed MnSOD-positive neurons and astrocytes in WT mice at D1 and D4 after MCAO. Positive immunostaining was observed in the striatal border and the neocortical border zones of the periinfarct area. In contrast, PPARβ KO mice did not show any staining at the same time period (Figure 4). Importantly, treatment of PPARβ KO mice with NGF 1 day after ischemia resulted in detection of MnSOD labelling at D4 after lesion (Figure 4). In Figure 4, the punctate immunostaining indicates the mitochondrial distribution of the protein in both groups of mice. Treatment with NGF was also associated with decreased infarct size in PPARβ KO mice (from 9.3 ± 0.6 to 6.8 ± 0.4 mm3) (P<0.05, n = 8).
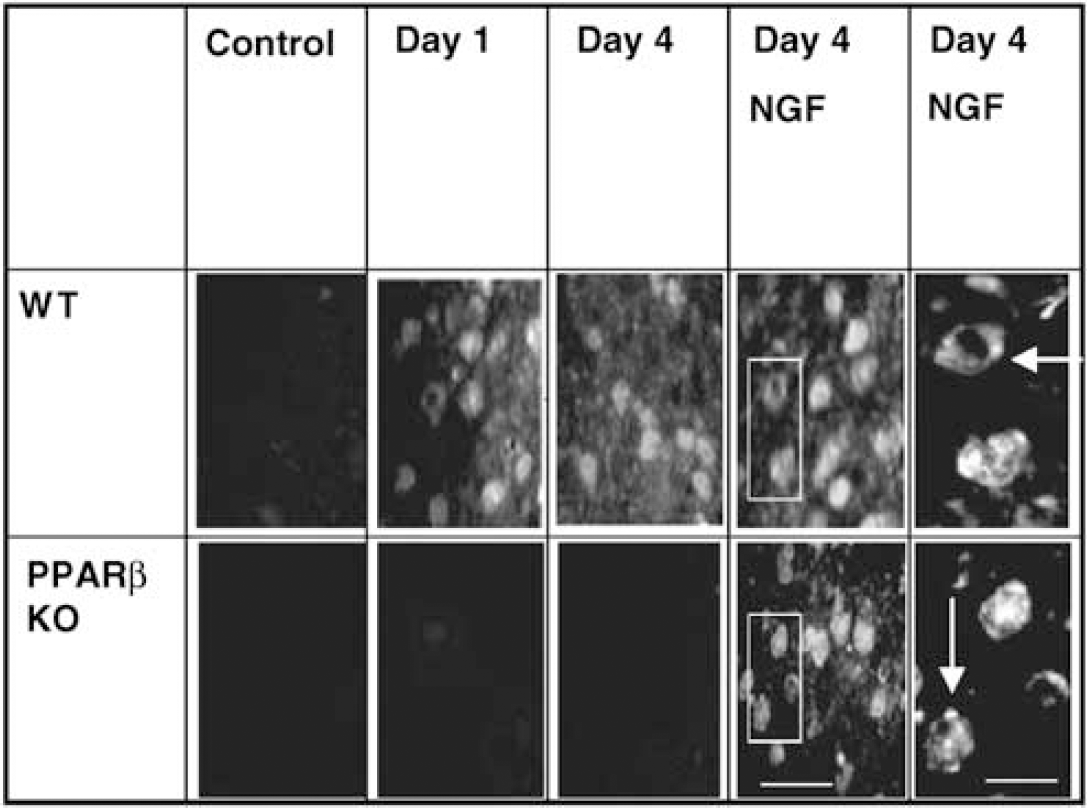
Wild-type but not PPARβ KO mice expressed MnSOD immunostaining in neurons on D1 and D4 after MCAO. Treatment with 200 ng NGF 2.5S on the lesion site 1 day after MCAO resulted in MnSOD induction in PPARβ KO mice (scale bar: 20 μm). The right columns show a higher magnification of the selected areas in which a punctated distribution (arrows) of the protein in mitochondria (scale bar: 7 μm) is observed. No labelling was seen in nonoperated control animals.
Lack of Hyperphagic Response to MCAO in PPARβ KO Mice: Involvement of NGF, IFNγ, and UCP2, but not TNFα
The PPARβ KO and WT mice show acute hypophagia on the day of MCAO intervention and then regain appetite. The compensatory hyperphagic phase, lasting from D3 to D5, in WT mice (n = 8, P<0.01) did not occur in PPARβ KO mice (Figure 5A). Injection of NGF on the lesion site of PPARβ KO mice 1 day after MCAO resulted in significant hyperphagia from D3 to D4 after ischemia (P<0.001) (Figure 5B). In contrast to the patterns of food intake observed after MCAO, both WT and PPARβ KO mice displayed a significant hyperphagic response 1 day after a 24-h period of fasting (n = 8, P<0.01) (Figure 5C).
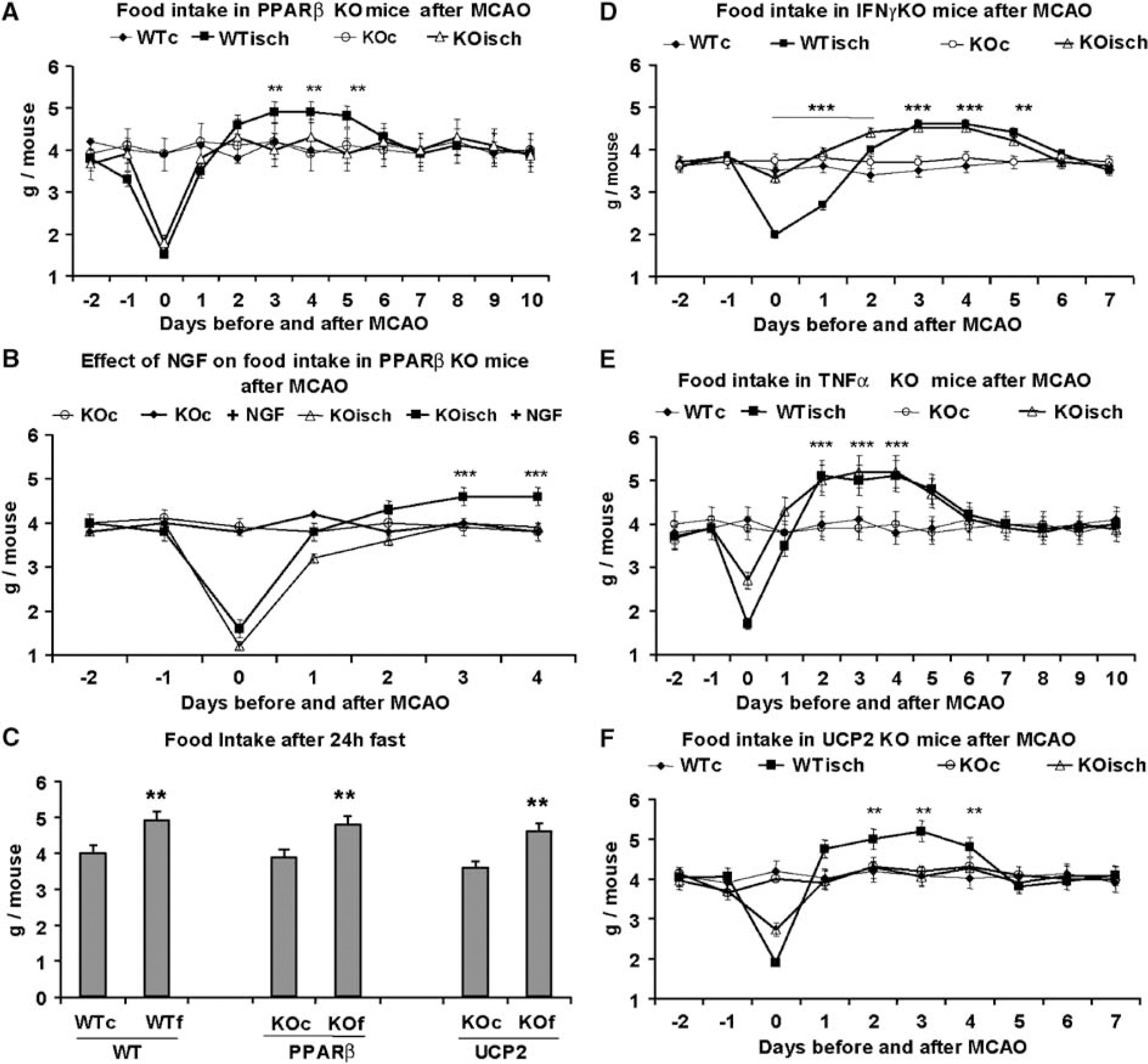
Food intake responses in PPARβ KO, IFNγ KO, TNF KO, and UCP2 KO mice after MCAO. (
Because altered levels of TNFα, IFNγ, and UCP2 were observed in PPARβ KO mice after ischemia, food intake was also measured in TNFα KO, IFNγ KO, and UCP2 KO mice. Interferon-gamma KO mice did not show hypophagia from D0 to D3 after MCAO when compared with WT ischemic mice (P>0.05), but rapidly entered a hyperphagic state compared with WT (Figure 5D). During the period D0 to D2 after MCAO, IFNγ KO mice ate significantly more than WT mice (P<0.001) (Figure 5D). Like their WT counterparts, TNFα KO and IFNγ KO mice showed significant hyperphagia after MCAO compared with their nonoperated controls (from D3 to D5, and D2 to D4, respectively) (P<0.001) (Figures 5D and 5E). In contrast, UCP2 KO mice did not show significant hyperphagia after MCAO when compared with WT (P>0.05) (Figure 5F), but were able to show a significant hyperphagic response 1 day after 24 h fasting (n = 8, P<0.01) (Figure 5C). In all animals, the body weight followed a parallel time course to that of food intake (data not shown).
NPY Expression is Decreased in PPARβ KO Mice after MCAO
Basal levels of neuropeptide mRNAs (NPY, orexin, and MCH) did not differ between PPARβ KO and WT mice (data not shown). The expression of the three neuropeptides showed significant increases 4 days after MCAO in WT mice when compared with nonoperated animals (NPY: 110% ± 5%; orexin: 60% ± 2%; MCH: 69% ± 4%; n = 4 and P<0.001 for the three groups) (Figure 6). However, NPY mRNA induction in PPARβ KO mice was markedly attenuated (22% ± 2%, P<0.001) compared with WT mice (Figure 6). This was not the case for orexin (51% ± 4%, n = 4) and MCH (57% ± 4%, n = 4) mRNAs, which showed a similar post-MCAO increase in both PPARβ KO and WT mice (Figure 6).
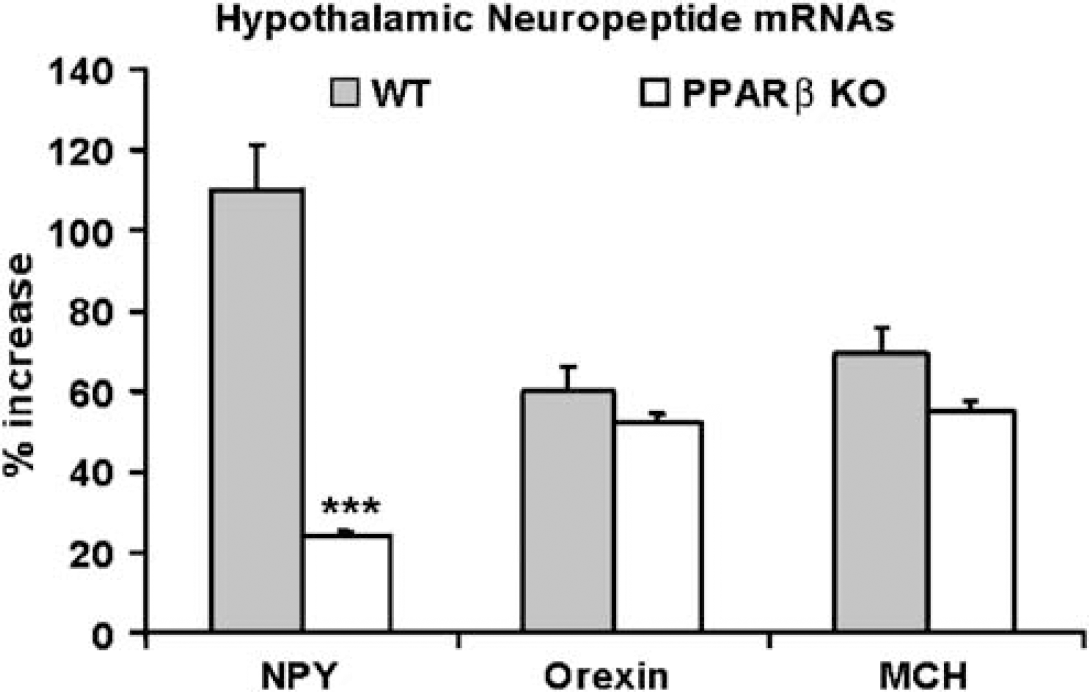
Induction of NPY, orexin, and MCH mRNAs on D4 after MCAO in WT (WT) and PPARβ KO (KO) mice. The induction of NPY was significantly attenuated in PPARβ KO when compared with WT mice (n = 8 for all groups). Each value is expressed as mean ± s.e.m. (***P<0.001).
Discussion
The major finding of this study is that the deletion of PPARβ gene in mice dramatically exacerbated the deleterious consequences of focal cerebral ischemia. The enhanced susceptibility to MCAO was revealed by a 50% increase in lesion volume in the PPARβ KO mice compared with their WT counterparts. The greater sensitivity observed after MCAO was specifically related to PPARβ absence because the infarct size was similar in PPARα WT and KO mice. This latter finding is in agreement with a recent study that clearly showed that PPARα receptor deficiency is not associated with an increased susceptibility to focal cerebral ischemia in mice (Deplanque et al, 2003). Although all three PPAR types are present in the brain, PPARβ receptor is the most abundant in cortical cells (Cullingford et al, 1998), further supporting an involvement of PPARβ activation in neuroprotective mechanisms against focal cerebral ischemia. However, and as is the case for all studies comparing WT with KO mice (Durum and Muegge, 1998; Samson and Taylor, 2005), our study does not eliminate the possibility that PPARβ regulation of MCAO lesion size could be indirect. Yet, our model showed that PPARβ deficiency results in increased lesion size, indicating that lack of PPARβ could not be compensated.
This increased susceptibility to MCAO in PPARβ KO mice was accompanied by massive changes in both antioxidant mechanisms and cytokine activation. The increase of oxidative injury in these mice was indicated by the upregulation of MDA content and reduction of MnSOD and reduced GSH levels, two classical brain antioxidants. Importantly, our results also show that, in contrast to WT mice, the transcript of UCP2, a regulator of reactive oxygen species (Arsenijevic et al, 2000b), is not upregulated in PPARβ KO mice after ischemia. In conjunction with previous studies showing that activation of PPARs may induce UCP2 mRNA expression in various tissues (Aubert et al, 1997; Wang et al, 2003), this observation implies that UCP2 may require PPARβ to be induced in brain tissue after ischemic injury. Altered cytokine levels may also contribute to the deleterious consequences of cerebral ischemia in PPARβ KO mice. In particular, TNFα and NGF, two macrophage cytokines known to be involved in the acute phase of brain ischemic tolerance (Guegan et al, 1998; Scherbel et al, 1999), are not induced after MCAO in the brain of PPARβ KO mice. In contrast, the levels of the proinflammatory lymphocyte cytokine IFNγ were abnormally elevated in the brain of PPARβ KO mice after MCAO, suggesting that PPARβ may inhibit the induction of this inflammatory cytokine. Altogether, these results imply that PPARβ may participate in the regulation of both macrophage and lymphocyte cytokines. The lack of NGF induction after MCAO in PPARβ KO mice cannot be explained by the absence of TNFα because TNFα KO mice were still able to induce NGF (data not shown). It remains to be determined whether the higher amounts of IFNγ found in the PPARβ KO mice could partially explain the ablated NGF because IFNγ has been shown to block NGF secretion in neonatal astrocyte culture but not quiescent astrocytes (Awatsuji et al, 1995). Interestingly, our data also provide further evidence indicating that the oxidative status of PPARβ KO mice after MCAO may be mediated by altered cytokine levels. In fact, IFNγ antibody administered before MCAO to PPARβ KO mice reduced MDA increase and GSH loss in the mice. Consistent with previous observations in this field, acute NGF administration after ischemia led to a substantial decrease of infarct volume in WT mice (Semkova and Krieglstein, 1999). Moreover, the decreased infarct volume and upregulation of MnSOD expression observed in the brain of NGF-treated PPARβ KO mice after ischemia suggest that the reduced availability of NGF may be partly at the origin of the higher vulnerability to cerebral ischemia in PPARβ KO mice (Guegan et al, 1999).
Besides the importance of PPARβ in the regulation of central ischemic damage, another important finding of the present study is that PPARβ is also involved in the regulation of food intake after MCAO. The rapid hyperphagic phase induced in WT mice after MCAO was not present in PPARβ KO mice, suggesting that the cellular signalling involved in MCAO-induced hyperphagia is not functional in PPARβ KO mice. Because NGF treatment can restore hyperphagia in these mice, a role for NGF in mediating the hyperphagic response after ischemia is strongly suggested. This finding is consistent with a previous study showing that NGF induces hyperphagia when administered after hypothalamic damage (Berger et al, 1973). The specific role of PPARβ in MCAO-induced hyperphagia may also involve NPY, as indicated by the blunted NPY induction in PPARβ KO mice after ischemia. Whether NGF is necessary for NPY expression, as suggested by other studies (Verge et al, 1995), remains to be determined. In agreement with previous observations on TNFα and IFNγ anorexic effects (Arsenijevic et al, 2000b; Langhans, 2000), the fact that ischemic TNFα KO and IFNγ KO mice entered in a hyperphagic state suggests that these cytokines are not required for hyperphagia after MCAO. In addition, the hyperphagic phase observed in these two genotypes after MCAO is consistent with their associated increased NGF levels. Thus, the absence of TNFα observed in PPARβ KO mice should not play a major role in the lack of hyperphagic response observed in these mice. In contrast, because IFNγ KO mice ate significantly more than WT mice the first 2 days after MCAO, it is possible that the elevated levels of this anorexic cytokine in PPARβ KO mice could partly explain the lack of hyperphagia after MCAO (Arsenijevic et al, 2000b). Because UCP2 KO mice did not display hyperphagia after cerebral ischemia, the absence of UCP2 induction after MCAO could also contribute to the lack of hyperphagia in PPARβ KO mice. Taken together, these new data indicate that induction of PPARβ and UCP2 expression may be critical in the molecular mechanisms regulating food intake after MCAO. This regulation of food intake is MCAO specific because PPARβ KO and UCP2 KO mice are still able to display a hyperphagic response after a period of fasting. The close relationship between PPARβ and UCP2 is further highlighted in that they are expressed in the same hypothalamic nuclei known to regulate food intake (Richard et al, 1998; Woods et al, 2003).
Although our data indicate that PPARβ is involved in infarct size and regulates hyperphagic response after MCAO, the present data do not support a causal relationship between these two phenomena. Middle cerebral artery occlusion induces hypophagia followed by hyperphagia and then a return to basal food intake levels in balb/c, C57bl/J6, sv129, b6, and CFW mice strains (Arsenijevic et al, 2003). Several lines of evidence indicate that the size of the infarct is not necessarily related to hyperphagia. First, an increased infarct volume in the PPARβ KO mice was associated with lack of hyperphagia. Second, IFNγ KO mice have a smaller infarct size than their WT counterparts (data not shown), yet they showed a more rapid onset of hyperphagia. In contrast, our previous work revealed a smaller infarct size in UCP2 KO mice than their WT counterparts in the absence of hyperphagia (de Bilbao et al, 2004).
Permanent distal MCAO model affects the cerebral cortex, whereas food intake response is traditionally thought to be related to the hypothalamus. However, numerous earlier contributions showed that food intake is a complex process involving several cortical areas (Hinton et al, 2004; Kaye et al, 2005; Rolls, 2005). Similarly, lesions in the cerebral cortex may decisively influence food intake (Modo et al, 2000). Furthermore, a recent contribution showed that altered eating behaviors are mostly related to the neural circuits between the cerebral cortex and appetite-regulating centers located in the hypothalamus (Rolls, 2005). Alternatively, appetite regulation may also be influenced by acute inflammatory phenomena related to the activation of cytokines/eicosanoids (Langhans, 2000). Although our data do not provide a definite conclusion about the etiology of feeding disorders in PPARβ KO mice, they strongly support a predominant role of cytokine and humoral immunity mechanisms in this context.
To our knowledge, this is the first study addressing the role of PPARβ in three main biologic events associated with cerebral ischemia, namely morphologic changes, activation of antioxidant and inflammatory mechanisms, as well as food intake. The above-discussed findings clearly suggest a central role of PPARβ in neuroprotection against focal cerebral ischemic damage, as well as in the molecular mechanisms involved in the associated antioxidant, inflammatory, and food intake responses. It may orchestrate energy balance and inflammation during MCAO. Based on these observations, a pharmacological modulation of PPARβ, such as that induced with the use of specific PPARβ agonists (Li et al, 2004), could represent a relevant therapeutic possibility in the field of cellular protection after stroke.