
Abstract
Recent studies suggest that normobaric hyperoxia can be beneficial, if administered during transient stroke. However, increased oxygenation theoretically may increase oxygen free-radical injury, particularly during reperfusion. In the present study, the authors assessed the benefit and risks of hyperoxia during focal cerebral ischemia and reperfusion. Rats were subjected to hyperoxia (Fio2 100%) or normoxia (Fio2 30%) during 2-hour filament occlusion and 1-hour reperfusion of the middle cerebral artery. At 24 hours, the hyperoxia group showed 70% (total) and 92% (cortical) reduction in infarct volumes as compared to the normoxia group. Levels of oxidative stress were evaluated using three indirect methods. First, since oxygen free radicals increase blood—brain barrier (BBB) damage, Evan's blue dye extravasation was quantified to assess BBB damage. Second, the expression of heme oxygenase-1 (HO-1), a heat shock protein inducible by oxidative stress, was assessed using Western blot techniques. Third, an immunoblot technique (“OxyBlot”) was used to assess levels of protein carbonyl formation as a marker of oxidative stress—induced protein denaturation. At 24 hours, Evan's blue dye extravasation per average lesion volume was similar between groups. There were no significant differences in HO-1 induction and protein carbonyl formation between groups, in the ipsilateral or contralateral hemispheres, at 6 hours and at 24 hours. These results indicate that hyperoxia treatment during focal cerebral ischemia—reperfusion is neuroprotective, and does not increase oxidative stress.
Keywords
Tissue hypoxia plays a critical role in the primary and secondary events leading to cell death after cerebral ischemia. Therefore, improving brain tissue oxygenation is considered a logical and important stroke treatment strategy. Hyperbaric oxygen therapy (HBO) has been proven successful in several animal and human stroke studies (Weinstein et al., 1987; Burt et al., 1987; Kawamura et al., 1990; Nighoghossian et al., 1995; Yin et al., 2002). However, the negative results of some HBO studies (Anderson et al., 1991; Roos et al., 1998), and factors such as the limited availability of HBO chambers and poor patient compliance (Anderson et al., 1991), may have decreased the enthusiasm for using HBO as stroke therapy. Furthermore, research during the last 2 decades has provided extensive knowledge about the potential harmful effects of the oxygen molecule (McCord, 1985; Siesjo et al., 1989; Chan, 2001), which has raised serious concerns about the use of oxygen in stroke. Oxygen free radicals are involved in harmful processes like lipid peroxidation (Watson et al., 1984; Mickel et al., 1987), which disrupt cell membranes and increase blood—brain barrier (BBB) damage, particularly during tissue reperfusion. The presence of oxygen is critical for glutamate-induced excitotoxic cell death (Dubinsky et al., 1995), a major mechanism of neuronal injury in cerebral ischemia. Hyperoxia also causes cerebral vasoconstriction (Omae et al., 1998; Watson et al., 2000), which could, theoretically, compromise blood flow to ischemic regions and thereby worsen stroke outcome.
Recently, however, there has been renewed interest in the neuroprotective effects of oxygen. Several studies within the past 2 years have provided compelling evidence that HBO is highly effective in attenuating infarct volume after stroke, and in improving functional outcome after brain trauma (Veltkamp et al., 2000; Sunami et al., 2000; Rockswold et al., 2001; Bramlett et al., 2001; Yin et al., 2002). Miyamoto and Auer (2000) have shown that impressive reductions in stroke lesion volumes can be achieved even with normobaric hyperoxia—the simple administration of 100% oxygen. Using serial magnetic resonance imaging and pathological studies in rats subjected to focal cerebral ischemia, we have shown that normobaric hyperoxia attenuates diffusion—magnetic resonance imaging abnormalities and 48-hour infarct volumes (Singhal et al., 2002). Importantly, our study showed that greater benefit could be achieved by reducing the time to treatment, and that the therapeutic time window was quite narrow, a fact often overlooked by earlier studies. The results of these recent studies, especially the normobaric hyperoxia studies, may have important clinical implications. However, it is conceivable that increased oxygen delivery, particularly during the reperfusion phase after stroke, increases the generation of oxygen free radicals. Therefore, it is important to assess the benefit and risks of hyperoxia therapy during focal cerebral ischemia and reperfusion, in infarcted as well as noninfarcted tissues.
In the present study, we evaluated the benefits and risks of early hyperoxia therapy in a standard animal model of transient focal stroke. First, we assessed the efficacy of normobaric hyperoxia in reducing infarct volumes, when administered during a 2-hour period of ischemia followed by 1 hour of reperfusion. We then compared levels of oxidative stress in the ischemic as well as nonischemic hemisphere, in rats treated with 100% oxygen versus those treated with 30% oxygen, in the same model of ischemia—reperfusion. We used three indirect methods to assess oxidative stress. First, since oxygen free radicals can increase BBB damage from processes like lipid peroxidation, we quantified BBB damage using the Evan's blue dye extravasation method (Uyama et al., 1988). Second, using Western blot techniques, we quantified the expression of heme oxygenase-1 (HO-1), a 32-kd heat shock protein that is inducible by a variety of stress factors including oxidative stress and focal cerebral ischemia (Nimura et al., 1996; Geddes et al., 1996). Third, we used an immunoblot technique (“OxyBlot”) to evaluate the amount of protein carbonyl formation. Oxidative modification of proteins by reactive species such as oxygen free radicals results in the introduction of carbonyl groups into protein side chains, and carbonyl formation is an important detectable marker of the oxidation status of proteins (Aksenov et al., 2001).
MATERIALS AND METHODS
Transient ischemia model
All procedures were conducted under an institutionally approved protocol, in accordance with guidelines set by the Guide for the Care and Use of Laboratory Animals (NIH publication 93-23, revised 1985). Adult male Sprague-Dawley rats (250 to 350 g, Charles River Laboratories, MA, U.S.A.) were housed under diurnal lighting conditions and were allowed free access to food and water before and after the surgery. Rats were anesthetized with 1% to 1.5% halothane in a 70:30 mixture of nitrous oxide/oxygen under spontaneous respiration. For measurement of mean arterial blood pressure, heart rate, and blood gases, PE-50 polyethylene tubing was inserted into the right femoral artery. Temperature was continuously monitored with a rectal probe and maintained at 37.0°C with a thermostatically controlled heating pad. Middle cerebral artery ischemia was induced via a standard intraluminal approach (Longa et al., 1989). In brief, a surgical cutdown was performed via the ventral surface of the neck to expose the internal and external carotid arteries. The external carotid artery, occipital artery, and pterygopalatine artery were ligated. Surgical clips were applied to the common and internal carotid arteries, and the external carotid artery was cut. A 4.0 nylon monofilament suture coated with silicon was inserted into the external carotid artery. After releasing the internal carotid artery clip, the suture was advanced up the internal carotid artery until the tip was placed about 17 mm from the internal—external carotid bifurcation. Continuous laser Doppler flowmetry (LDF) (Perimed, North Royalton, OH, U.S.A.) was used to ensure adequacy of middle cerebral artery occlusion (MCAO) and monitor regional cerebral perfusion. For placement of the LDF probe, a burr hole 2 mm to 3 mm in diameter was created in the right parietal bone (2 mm posterior and 6 mm lateral to bregma). After 2 hours, the monofilament suture was withdrawn to allow for cerebral reperfusion. LDF values were recorded for 1 hour after reperfusion, after which rats were returned to their cages. Rats that did not demonstrate a significant reduction to less than 30% baseline LDF values during MCAO, or rapid restoration of the LDF signal during reperfusion, were excluded.
Rats were divided into two groups, Normoxia and Hyperoxia. The normoxia (control) group received 30% Fio2 until the end of the 1-hour reperfusion period. The hyperoxia group received 30% Fio2 during the surgical preparation, and 100% Fio2 from immediately after MCAO until the end of the 1-hour reperfusion period. Arterial blood gases, heart rate, mean blood pressure, and temperature were recorded at frequent intervals.
Twenty-four-hour infarct volumes
Sixteen animals (n = 8 per group) were used in this experiment. At 24 hours after MCAO, rats were killed. Their brains were harvested, cut into 2-mm slices, and stained with 2% 2,3,5-triphenyltetrazolium chloride for 15 minutes. Total (hemispheric), cortical, and subcortical (striatal) infarction volumes were measured using digital imaging (Nikon Coolpix 950, Nikon, Melville, NY, U.S.A.) and standard computer-assisted image analysis techniques (Osiris Medical Imaging Software Version 3.5, University of Geneva, Switzerland), in which lesion areas from each axial slice were integrated to yield total volume.
Blood—brain barrier damage
Sixteen animals (n = 8 per group) were used in this experiment using previously published techniques (Asahi et al., 2001). Ten minutes before reperfusion, 2% wt/vol Evan's blue dye (Sigma) in an intravenous dose of 4 mL/kg, was administered via the femoral vein as a blood—brain permeability tracer and was allowed to circulate. Sixty minutes after reperfusion, the rat chest wall was opened under halothane anesthesia and transcardiac perfusion was performed to remove intravascular dye. The brain was harvested and covering meninges and pineal gland were carefully removed. The left and right hemispheres were separated, weighed, homogenized in 2 mL 50% wt/vol trichloroacetic acid (Sigma), and centrifuged at 10,000 rpm for 20 minutes. The supernatant from each hemisphere was diluted fourfold with ethanol. For fluorescent measurement, an aliquot was diluted threefold with the solvent (50% trichloroacetic acid/ethanol 1:3). A microplate fluorescence reader (FL600, Biotek) was used at an excitation wavelength of 620 nm (bandwidth 10 nm) and an emission wavelength of 680 nm (bandwidth 10 nm) to determine the dye's fluorescence intensity (ng/μL). The total dye content (ng) of Evan's blue dye in each hemisphere was derived from concentrations of external standards in the solvent (100—1,000 ng/mL). The left hemisphere dye content was considered to reflect intravascular dye, and the difference between right and left hemisphere dye content was calculated to yield the extravasated dye in the right hemisphere. Evan's blue dye extravasation per gram of brain tissue (ng/g) was calculated. In addition, since the extent of BBB damage is affected by the size of the ischemic lesion, the right hemispheric extravasated dye content was divided by the corresponding stroke lesion volumes to yield normalized values (ng/mm3).
Western blot analysis
HO-1 protein expression was detected with Western blot analysis. At 6 hours (n = 3 per group) and 24 hours (n = 3 per group) after MCAO, rats were killed with an overdose of sodium pentobarbital (100 mg/kg intraperitoneally) and transcardially perfused with chilled (4°C) phosphate-buffered saline, pH 7.4. The brains were rapidly removed. Whole hemispheres were homogenized in lysis buffer containing 30 mmol/L Tri-HCl (pH 7.4), 150 mmol/L NaCl, 1% polyethoxyethanol, 0.1% SDS, 0.5% sodium deoxycholate, 50 mmol/L dithiothreitol, 2 mmol/L MgCl2, 1 mmol/L Na2VO4, 0.5 mmol/L phenylmethylsulfonyl fluoride, 10 mg/mL leupeptin, and 10 mg/mL aprotinin. Lysates were clarified by centrifugation at 14,000 g for 10 minutes. Protein concentration in the supernatant was determined by the Bradford assay (Bio-Rad). Samples were heated at 95°C for 5 minutes before gel loading, and 50 μg of each sample per lane was loaded onto 4% to 20% Tris-glycine gels with equal volumes of SDS sample buffer (Novex, San Diego, CA, U.S.A.). Following electrophoresis and transferring to polyvinylidene difluoride membranes (Novex), the membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 5% nonfat milk for 2 hours at room temperature. Membranes were then incubated overnight at 4°C with an anti—HO-1 antibody (1:2,000, Stressgene, Victoria, BC, Canada), following incubation with peroxidase-conjugated secondary antibodies (1:1,000) and visualization by enhanced chemiluminescence detection system (Amersham-Pharmacia, Buckinghamshire, U.K.). Expression levels of HO-1 were quantified and expressed as fold increase versus normal controls via measurement of optical density using the NIH Image analysis software.
Protein oxidation detection
To assess the formation of protein carbonyl groups, the OxyBlot protein oxidation detection kit (Integen, NY, U.S.A.) was used according to the manufacturer's detailed protocol. Protein samples were prepared from rat brains harvested at either 6 hours (n = 3 per group) or 24 hours (n = 3 per group) after MCAO. Subsequently, 5 μL protein sample (15 μg) was added with 5 μL of 12% SDS and 10 μL of 1× DNPH solution into a tube. Ten microliters of 1× neutralization solution instead of the DNPH solution served as the negative control. Tubes were incubated at room temperature for 15 minutes. Neutralization solution (7.5 μL) was added to each tube, and one mixed sample per lane was loaded onto 4% to 20% Tris-glycine gels with equal volumes of SDS sample buffer (Novex). Following electrophoresis and transfer to polyvinylidene difluoride membranes (Novex), the membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 1% bovine serum albumin for 1 hour at room temperature. Membranes were incubated overnight at room temperature with the primary antibody stock (1:150), then incubated with secondary antibodies (1:3,000) at room temperature for 1 hour. Blots were developed by an enhanced chemiluminescence detection system (Amersham-Pharmacia). Proteins that underwent oxidative modification (i.e., carbonyl group formation) were identified as a band in the derivatized sample, but not in the negative control. Levels of oxidatively modified proteins were quantified and expressed as fold increase versus normal controls via measurement of optical density using the NIH Image analysis software.
Statistical analysis
All values were expressed as mean ± SD parametric two-tailed Student's t-tests were used to compare results from normoxia and hyperoxia groups. P values less than 0.05 were considered significant.
RESULTS
Physiologic measurements
Table 1 shows physiologic measurements in both groups. Arterial Po2 values increased approximately fourfold in the hyperoxia group soon after MCAO (i.e., after the initiation of 100% oxygen). The other physiologic measurements were not significantly different between groups.
Physiologic measurements
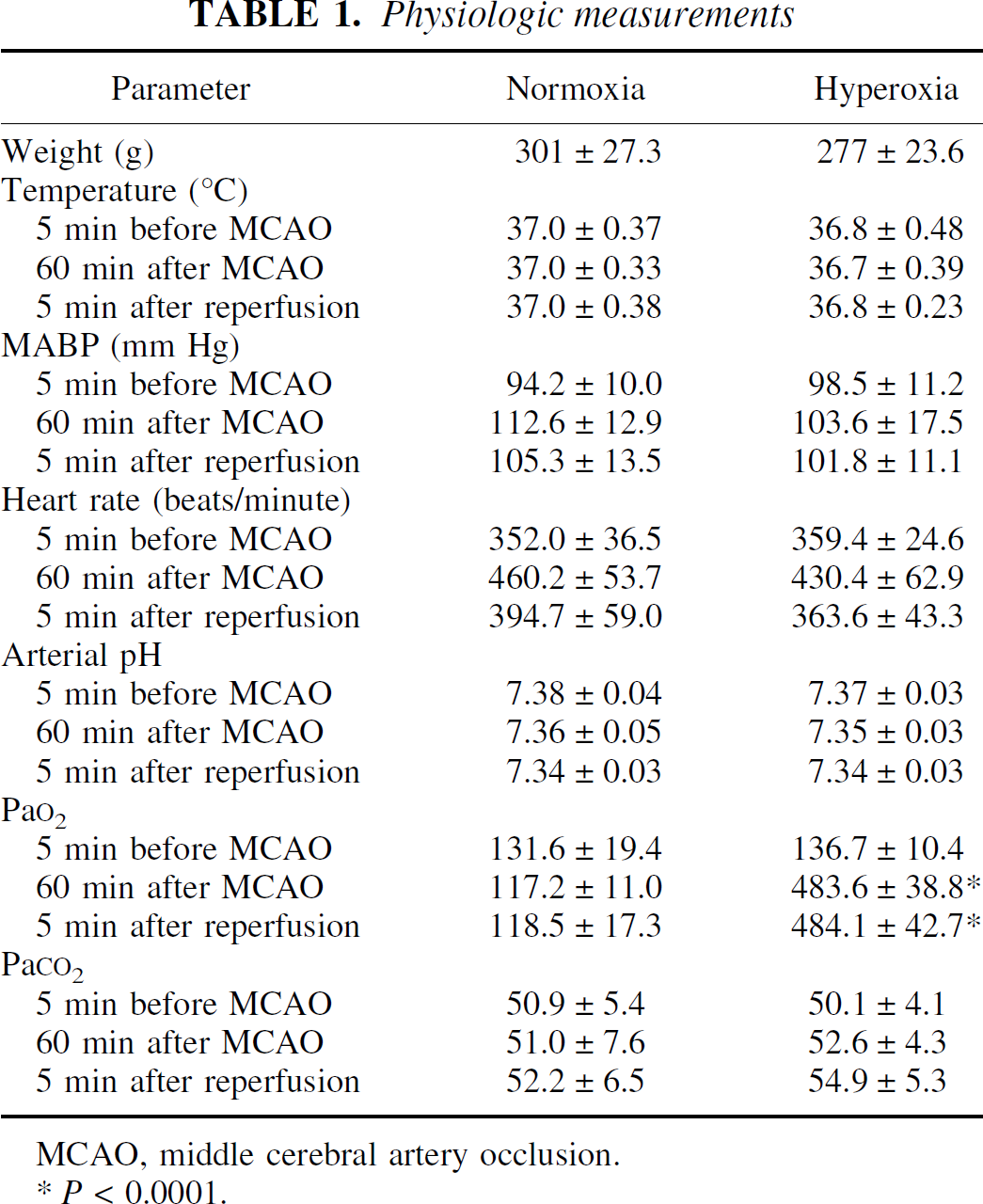
MCAO, middle cerebral artery occlusion.
P < 0.0001.
Regional cerebral perfusion
After ischemic onset, LDF values dropped to approximately 25% of preischemic baselines in all animals. Almost immediately after the end of arterial occlusion, LDF values recovered to 75% to 90% of baseline, indicating successful reperfusion of previously ischemic tissue. There were no significant differences in LDF measurements between groups (Fig. 1).
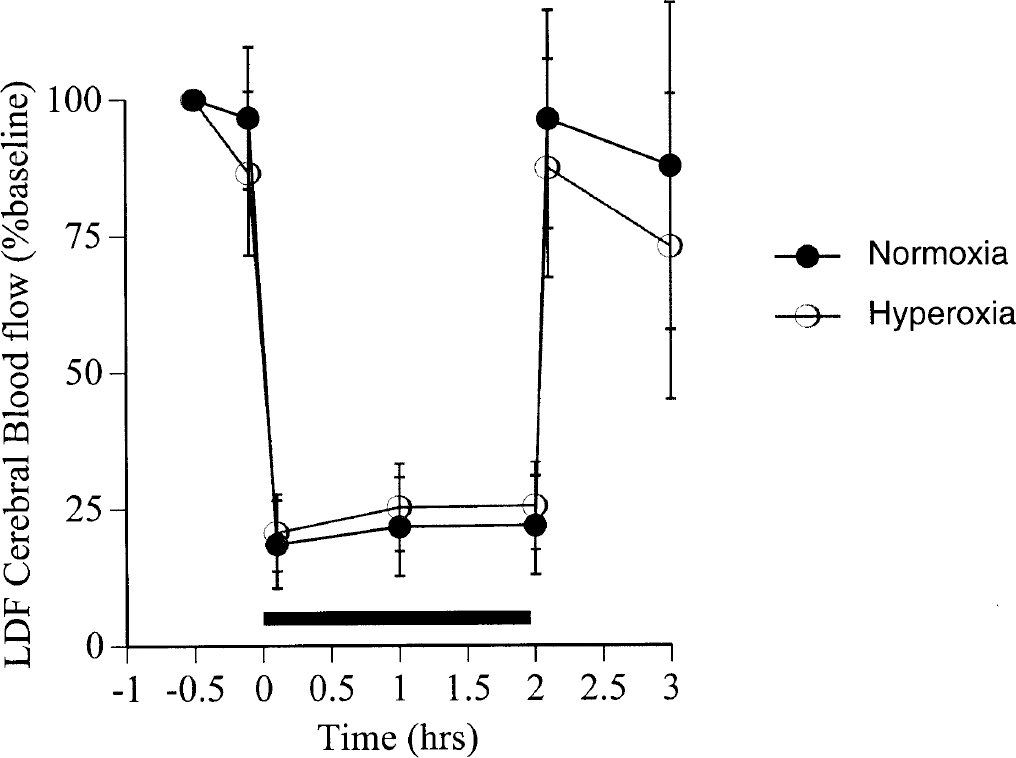
Laser Doppler measurements in the right middle cerebral artery territory in the normoxia group and the hyperoxia group. Regional cerebral perfusion was comparable during the 2-hour period of focal ischemia (black bar) and 1-hour period of reperfusion. Data are expressed as mean ± SD.
Infarction volumes
Figure 2 shows infarction volumes and distributions in both groups. Mean total right-hemispheric infarction volume was reduced by nearly 70% in the hyperoxia group as compared to the normoxia group (64.8 ± 23.5 mm3 versus 209.2 ± 61.4 mm3, P < 0.0001). The neuroprotective effects of hyperoxia were most evident in the cerebral cortex, where there was 92% reduction in infarct volume (10.7 ± 9.7 mm3 in the hyperoxia group versus 129.4 ± 52 mm3 in the normoxia group, P < 0.0001). Subcortical (striatal) infarction volumes were 54 ± 16.1 mm3 in the hyperoxia group and 79.8 ± 31.8 mm3 in the normoxia group; the difference was not statistically significant (P = 0.06).
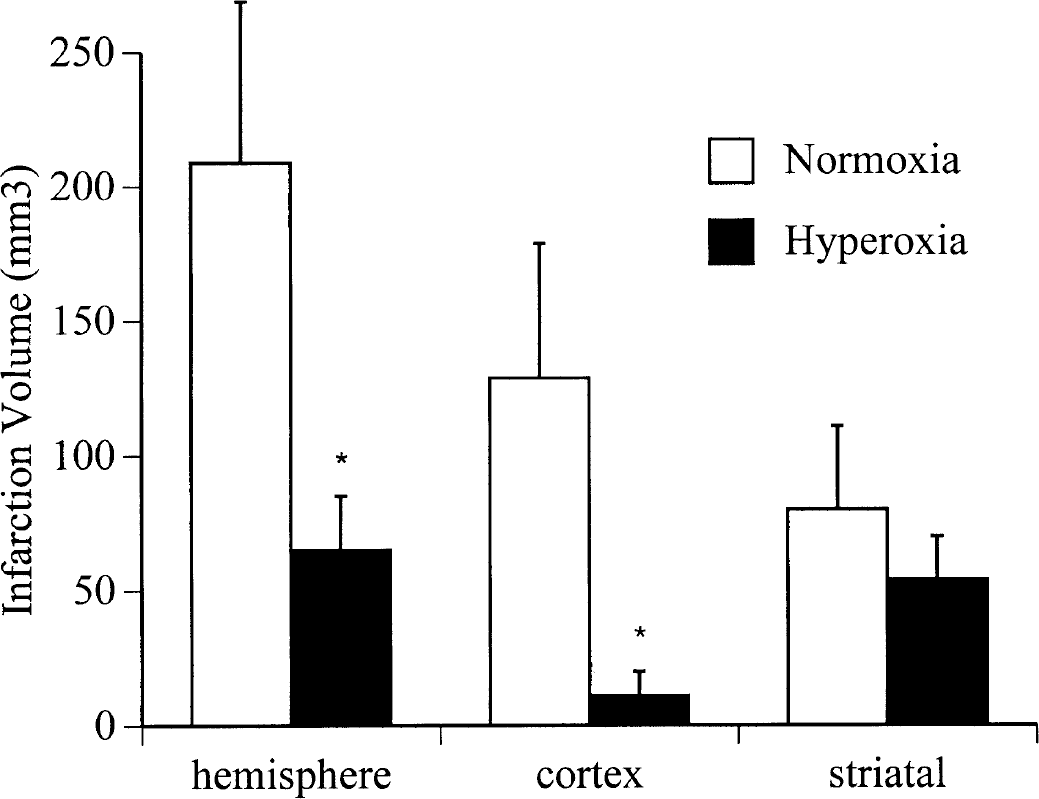
Total (hemispheric), cortical, and subcortical (striatal) infarction volumes at 24 hours after transient right middle cerebral artery occlusion, in the normoxia group and the hyperoxia group. Data are expressed as mean ± SD. Hemispheric infarct volumes were significantly reduced in the hyperoxia group, P < 0.0001. Cortical infarct volumes were significantly reduced in the hyperoxia group, P < 0.0001.
Blood—brain barrier permeability
There was patchy but clear leakage of the dye in the right middle cerebral artery territory in all animals. Fluorescence analysis of Evan's blue standards revealed a linear relationship between fluorescence and concentration within the 100 ng/mL to 1,000 ng/mL range (data not shown). The severity of BBB damage in the ischemic hemisphere was reduced in the hyperoxia group as compared to the normoxia group (1,592 ± 822 ng/g vs. 3,955 ± 2,386 ng/g, P < 0.05, Figure 3A). However, after normalizing for infarct volumes, mean Evan's blue dye extravasation was similar between groups (19.7 ± 11.7 ng/mm3 in the hyperoxia group and 14.7 ± 8.9 ng/mm3 in the normoxia group) (Fig. 3B).
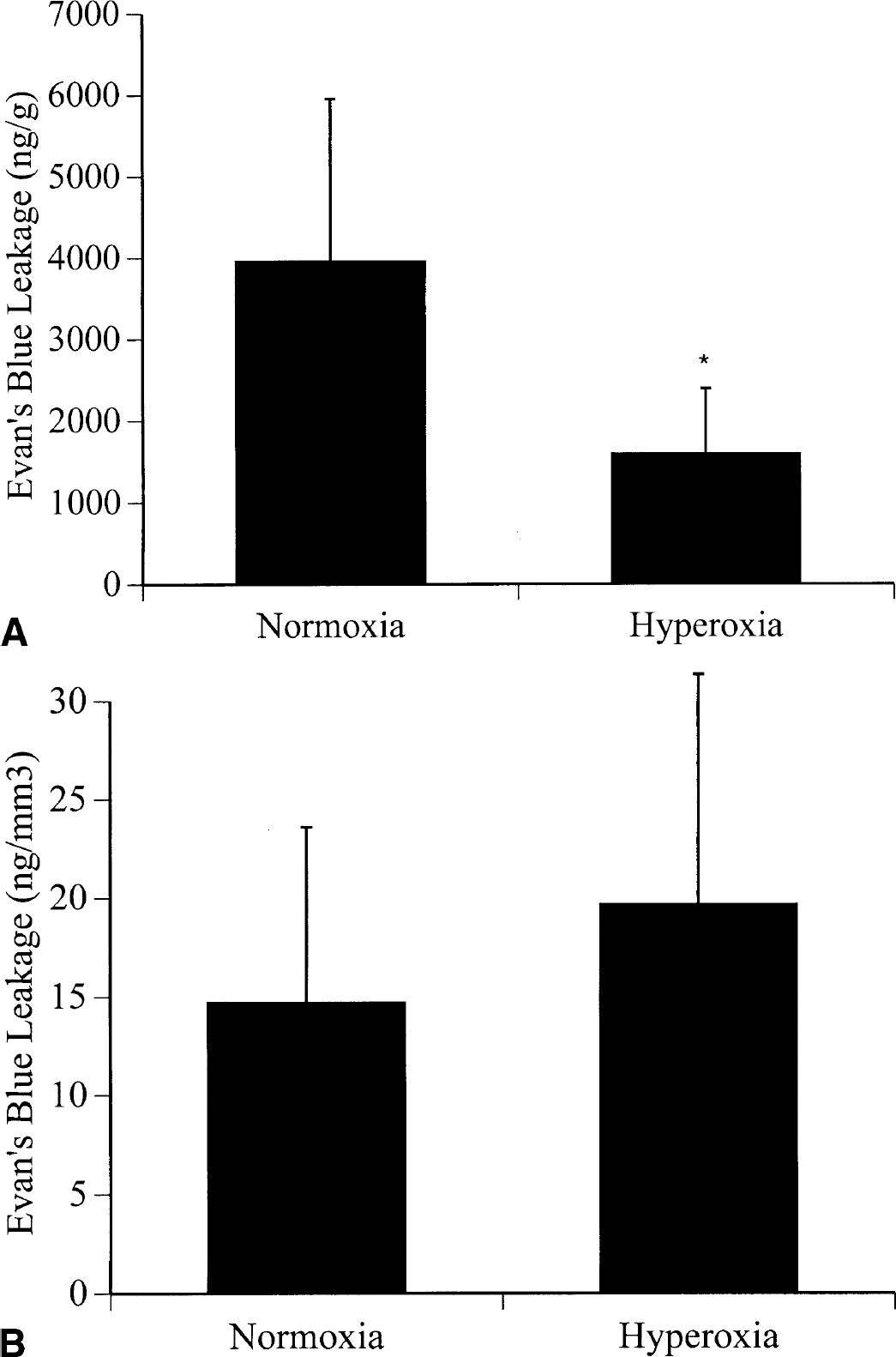
Evan's blue dye extravasation in the ischemic hemisphere at 24 hours after transient right middle cerebral artery occlusion, in the normoxia group and the hyperoxia group.
Heme oxygenase-1 expression
In the ipsilateral (ischemic) hemisphere, the optical density of the HO-1 protein band was 18.5 ± 17.8 at 6 hours and 50.7 ± 41.5 at 24 hours in the normoxia group, and 18.7 ± 13.6 at 6 hours and 57.5 ± 47.0 at 24 hours in the hyperoxia group. Despite the nearly threefold higher levels at 24 hours as compared to 6 hours, this increase was not statistically significant in either the normoxia group (P = 0.28) or the hyperoxia group (P = 0.24). In the contralateral (nonischemic) hemisphere, there was no significant increase in HO-1 levels over time (optical density 7.8 ± 4.1 at 6 hours versus 11.3 ± 7.0 at 24 hours in the normoxia group, P = 0.4, and 11.8 ± 6.54 at 6 hours versus 13.0 ± 8.6 in the hyperoxia group, P = 0.8). As shown in Figure 4, there were no significant differences in HO-1 protein expression between the normoxia group and the hyperoxia group at either the 6-hour or 24-hour time points.
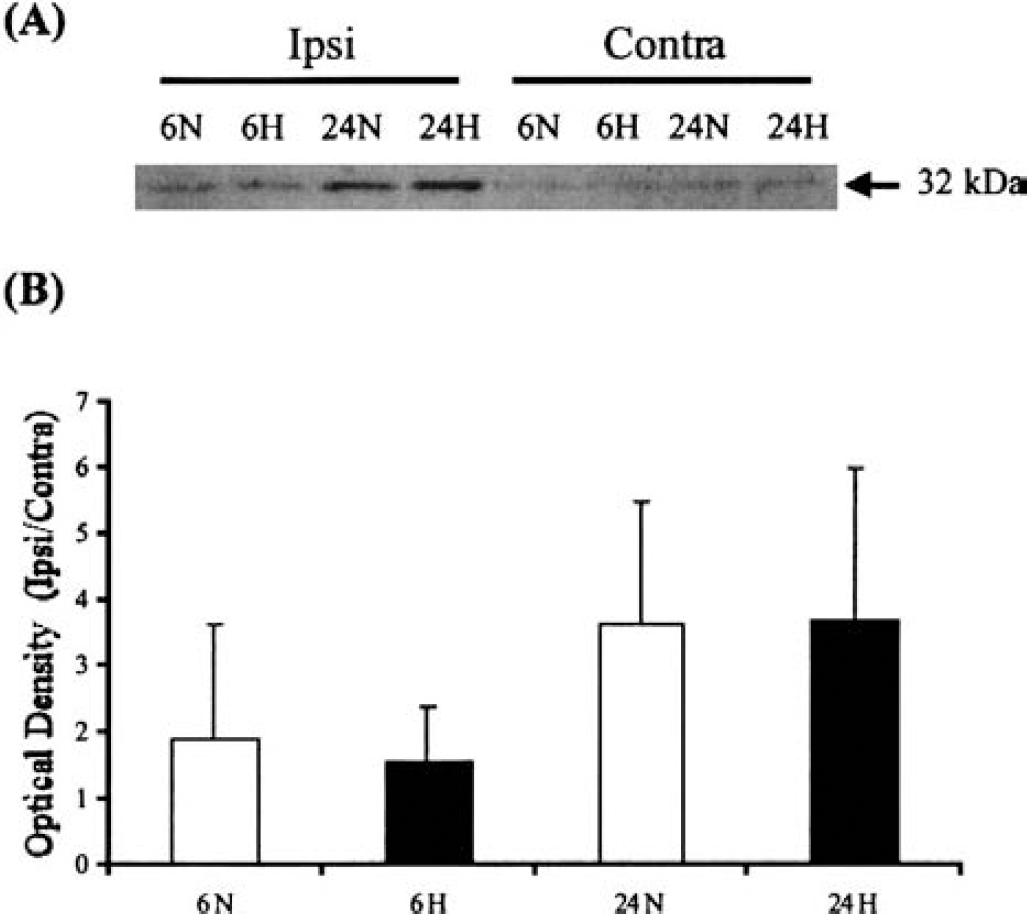
Western blot analysis of heme oxygenase-1 (HO-1) protein expression at 6 hours and at 24 hours after transient right middle cerebral artery occlusion in the normoxia group and the hyperoxia group.
Protein denaturation
In the ipsilateral (ischemic) hemisphere, the optical density of bands signifying protein carbonyls (oxidatively modified proteins) was 22.1 ± 8.8 at 6 hours and 45.8 ± 28.9 at 24 hours in the normoxia group, and 28.8 ± 27.7 at 6 hours and 43.5 ± 35.5 at 24 hours in the hyperoxia group. Despite the nearly twofold higher levels at 24 hours as compared to 6 hours, this increase was not statistically significant in either the normoxia group (P = 0.24) or the hyperoxia group (P = 0.6). In the contralateral (nonischemic) hemisphere, there was no significant increase in protein carbonyls over time (optical density 22.1 ± 10.0 at 6 hours versus 24.7 ± 19.8 at 24 hours in the normoxia group, P = 0.85, and 18.1 ± 9.7 at 6 hours versus 18.4 ± 10.1 in the hyperoxia group, P = 0.97). As shown in Figure 5, there was no significant difference in protein carbonyl formation between the normoxia group and the hyperoxia group, at either the 6-hour or 24-hour time points.
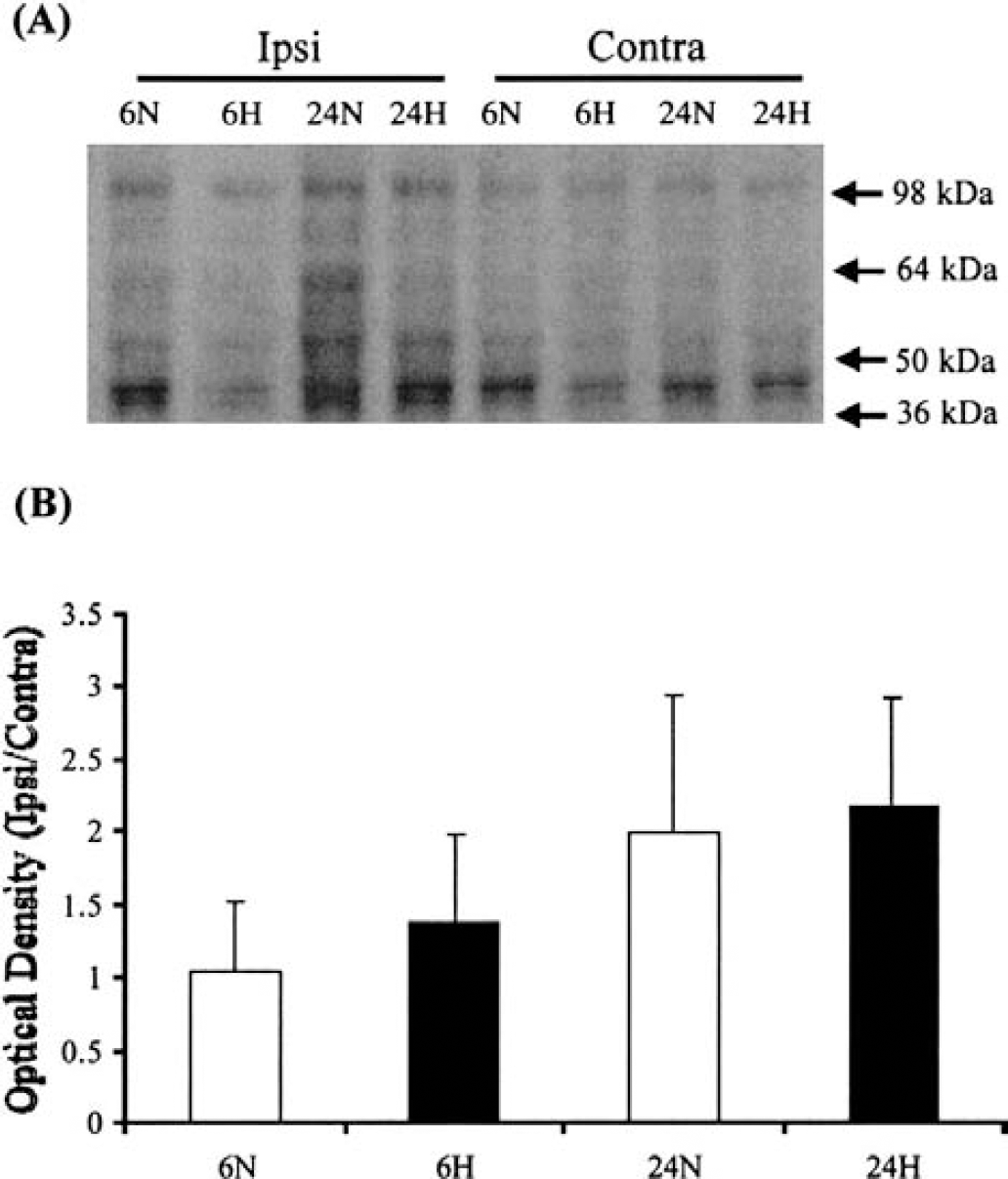
Immunoblot analysis of protein carbonyl groups (markers of oxidatively modified proteins) at 6 hours and at 24 hours after transient right middle cerebral artery occlusion in the normoxia group and the hyperoxia group.
DISCUSSION
The results of the present study clearly show that normobaric hyperoxia therapy, when administered during 2 hours of focal cerebral ischemia and 1 hour of reperfusion, is highly effective in reducing 24-hour infarct volumes. We specifically administered hyperoxia during the initial reperfusion period, since oxygen free-radical generation is known to be a key component of reperfusion injury (McCord, 1985; Siesjo et al., 1989; Chan, 2001). Clinically, reperfusion is a dynamic process, and it is not always possible to delineate the ischemic from the reperfusion period; therefore, the 1-hour reperfusion period in these experiments may be more relevant than the 15-minute reperfusion period in our previous experiments (Singhal et al., 2002). We found no difference in the amount of Evan's blue dye extravasation and levels of HO-1 protein expression or protein carbonyl formation between animals treated with 30% or 100% oxygen. These results suggest that in our experimental stroke model, hyperoxia salvages ischemic brain tissue without increasing the risk of oxygen free-radical injury.
The contribution of oxygen free radicals toward reperfusion injury after transient cerebral ischemia is well recognized (McCord, 1985; Siesjo et al., 1989; Chan, 2001). In animal studies of global cerebral ischemia, treatment with 100% oxygen has resulted in increased lipid peroxidation and increased mortality (Watson et al., 1984; Mickel et al., 1987). However, it is unclear whether hyperoxia increases the production of oxygen free radicals in models of transient focal cerebral ischemia. In at least two previous studies of transient cerebral ischemia, hyperoxia did not increase the generation of free radicals (Agardh et al., 1991; Sunami et al., 2000). Although we did not directly measure free-radical generation, the methods of evaluating the degree of oxidative stress used in this study have been used previously. Several studies have shown that oxygen free radicals cause lipid peroxidation, thereby damaging cell membranes and increasing BBB damage, and Evan's blue dye has been frequently used to assess the extent of BBB damage (Gartshore et al., 1997; Uyama et al., 1988). The Oxyblot technique detects protein carbonyl groups formed by reactive oxygen species—mediated oxidation of some amino acid side-chains (Aksenov et al., 2001). Brain protein carbonyl groups have been shown to increase at 2 hours and at 24 hours after reperfusion (Liu et al., 1993). HO-1, a microsomal enzyme expressed by glial cells, astrocytes, and neurons, gets upregulated under conditions of oxidative stress and cleaves heme to produce the antioxidant biliverdin (Nimura et al., 1996; Geddes et al., 1996; Turner et al., 1998; Panahian et al., 1999). The time course of HO-1 expression in our study is similar to that of previous studies (Geddes et al., 1996; Turner et al., 1998). Importantly, there was no evidence for increased oxidative stress in the either the ipsilateral or the contralateral (noninfarcted) hemisphere in the hyperoxia-treated animals (who had a nearly fourfold increase in Pao2 as compared to normoxia-treated animals), indicating that hyperoxia is safe and well tolerated, at least for the short durations used in this study.
The timing and duration of oxygen therapy are probably the most important factors responsible for the favorable results of our study. We started 100% oxygen immediately after MCAO, and continued treatment until 1 hour after reperfusion. Our recent study indicates that the therapeutic time window for hyperoxia therapy is approximately 30 minutes in this experimental model, and that shortening the time to treatment can enhance the benefit with hyperoxia (Singhal et al., 2002). Furthermore, hyperoxia can salvage ischemic tissue even if administered solely in the early reperfusion period (Auer and Flynn, 2001). A recent human stroke study failed to show benefit with nasal oxygen (Ronning and Guldvog, 1999); however, oxygen was administered as late as 24 hours after admission, and patients were given 3 L/min oxygen by nasal cannula, which is a considerably smaller dose than that used in this study. Careful analysis of previous hyperoxia studies shows that in most successful studies, hyperoxia was initiated early after ischemia onset, and maintained for a significant duration of ischemia (Weinstein et al., 1987; Kawamura et al., 1990; Miyamoto and Auer, 2000; Veltkamp et al., 2000). Recent data suggest that the therapeutic window for HBO may be approximately 6 hours (Yin et al., 2002), possibly due to the higher tissue Po2 values that can be achieved with HBO. The importance of the therapeutic window concept is emphasized by the success of our study (in which normobaric hyperoxia was initiated within the therapeutic time window) and may also explain the failure of some previous HBO studies where therapy was considerably delayed (Roos et al., 1998; Anderson et al., 1991).
Another important factor contributing to our positive results is the fact that ischemia was transient and not permanent. The benefit of hyperoxia has been most impressive in transient stroke models (Miyamoto and Auer, 2000; Veltkamp et al., 2000; Auer and Flynn, 2001; Yin et al., 2002; Singhal et al., 2002). In addition, our recent experiments with serial diffusion-weighted magnetic resonance imaging suggest that the beneficial effects of hyperoxia cannot be sustained in the face of prolonged hypoperfusion (Singhal et al., 2002). Oxygen probably provides temporary metabolic support to ischemic tissues, and timely reperfusion may be essential for tissue recovery. Transient ischemia is not uncommon, particularly after the advent of thrombolytic stroke therapy, and 100% oxygen therapy may prove particularly useful in patients who present early after stroke onset and are successfully thrombolysed.
Although this study indicates that normobaric hyperoxia may be a safe and effective stroke therapy, the mechanism of neuroprotection remains unclear. Unlike HBO, normobaric hyperoxia does not cause a large increase in brain tissue Po2. However, brain tissue Po2 is known to increase in a linear fashion with normobaric hyperoxia (Duong et al., 2001), and since the critical oxygen tension required for mitochondrial function is extremely low (Hempel et al., 1977), even a small increase in tissue pO2 may be adequate to afford neuroprotection. Furthermore, face-mask oxygen can theoretically be initiated very early after stroke onset, and early treatment may be as important as large increases in tissue Po2 levels. Several recent studies have shown that hyperoxia therapy results in favorable alterations in the levels of various metabolites and proteins, including glutamate, lactate, cyclooxygenase-2, bcl-2, and manganese superoxide dismutase (Menzel et al., 1999; Wada et al., 2001; Rockswold et al., 2001; Yin et al., 2002). Yet another potential neuroprotective mechanism is suggested by a trend for increased regional cerebral perfusion in ischemic tissues after initiation of hyperoxia: perfusion data from our previous magnetic resonance imaging experiments shows that in the ischemic periphery, hyperoxic animals have higher residual cerebral blood volume as compared to normoxic animals (Singhal et al., 2002). Oxygen is known to cause vasoconstriction and decreased cerebral blood flow in the normal brain (Watson et al., 2000); however, paradoxical elevations in blood flow have been documented in the ischemic brain (Nakajima et al., 1983). These data suggest that hyperoxia may salvage ischemic tissue by diverting blood flow from nonischemic to ischemic brain—a “Robin Hood” effect.
In clinical stroke, ischemia—reperfusion is a heterogeneous event at the microvascular level with dynamic processes of fibrin deposition and fibrinolysis, unlike the presently used model of mechanical occlusion and reperfusion. Further studies with clot models of stroke, using thrombolytic agents to simulate clinical ischemia—reperfusion, are needed to assess the benefits and risks of hyperoxia in stroke. In addition, it will be useful to assess the effects of oxygen in different animal models since hyperoxic neuroprotection may be strain dependent (Prass et al., 2000). Because free-radical production varies with the duration of ischemia (Henry et al., 1993), it is necessary to reassess the risk—benefit ratio of oxygen therapy with different durations of ischemia. At present, however, our results have practical implications for the management of stroke patients. Current stroke treatment guidelines recommend supplemental oxygen by nasal cannula if the arterial oxygen saturation is less than 90%. In the future, supplemental face-mask oxygen may prove to be a feasible, cost-effective neuroprotective strategy for ischemic stroke victims around the world.