
Abstract
Poly(ADP-ribose) polymerase-2 (PARP-2) is a member of the PARP enzyme family, and, similarly to PARP-1, catalyzes the formation of ADP-ribose polymers in response to DNA damage. While PARP-1 overactivation contributes to ischemic cell death, no information is available regarding the role of PARP-2. In this study, we evaluated the impact of PARP-2 deletion on histopathological outcome from two different experimental models of cerebral ischemia. Male PARP-2−/- mice and wild-type (WT) littermates were subjected to either 2 h of middle cerebral artery occlusion (MCAO) followed by 22 h reperfusion, or underwent 10 mins of KCl-induced cardiac arrest (CA) followed by cardiopulmonary resuscitation (CPR) and 3-day survival. After MCAO, infarct volume was reduced in PARP-2−/-mice (38% ± 12% of contralateral hemisphere) compared with WT (64% ± 16%). After CA/CPR, PARP-2 deletion significantly increased neuronal cell loss in the hippocampal CA1 field (65% ± 36% ischemic neurons) when compared with WT mice (31% ± 33%), with no effect in either striatum or cortex. We conclude that PARP-2 is a novel executioner of cell death pathways in focal cerebral ischemia, but might be a necessary survival factor after global ischemia to mitigate hippocampal delayed cell death.
Introduction
Poly(ADP-ribose) polymerase-2 (PARP-2) is a recently discovered member of the growing family of PARP enzymes (Amé et al, 1999, 2004; Berghammer et al, 1999; Shieh et al, 1998). All family members ribosylate target proteins and use NAD+, but PARP-1, the best studied PARP enzyme, and PARP-2 are the only family members known to be activated by DNA strand breaks (Ménissier de Murcia et al, 2003; Amé et al, 1999, 2004; Schreiber et al, 2002). Both enzymes initiate immediate, transient modification of nuclear proteins, leading to cell cycle arrest and DNA repair (D'Amours et al, 1999; Huber et al, 2004; Schreiber et al, 2002; Virag and Szabó, 2002). Thus, both enzymes play an important role in maintenance of genomic integrity, with both overlapping and redundant functions (Ménissier de Murcia et al, 2003). However, PARP-2 has been thought to be the less active isoform, contributing only 5% to 10% of total PARP activity in response to DNA damage (Amé et al, 1999; Schreiber et al, 2002). DNA-binding domains and, to a lesser degree, catalytic sites are different in PARP-1 and −2, suggesting that they have distinct DNA target sites and substrate proteins (Amé et al, 1999, 2004; Oliver et al, 2004). Recent studies also emphasize interactions between the isoforms, by forming heterodimers and sharing common partners to initiate single-strand break repair (SSBR) and base excision repair (BER) (Dantzer et al, 2000; Schreiber et al, 2002), and maintenance of centromere and telomere integrity (Dantzer et al, 2004; Ménissier de Murcia et al, 2003; Saxena et al, 2002).
It is now well understood that overactivation of PARP-1 in response to prooxidant insults and extensive DNA damage is detrimental to neurons (Huber et al, 2004; Szabó and Dawson, 1998; Virag and Szabó, 2002). Inhibition of PARP-1 activation by genetic deletion or pharmacological inhibition confers protection in a large number of in vivo and in vitro models of ischemia/reperfusion (Virag and Szabó, 2002). Poly(ADP-ribose) polymerase-1 deficiency or inhibition is clearly neuroprotective in focal cerebral ischemia in male (but not female) animals (Eliasson et al, 1997; Endres et al, 1997; Goto et al, 2002; Nakajima et al, 2005; Tokime et al, 1998; Komjáti et al, 2004; McCullough et al, 2005), whereas mixed results have been observed in global cerebral ischemia (Moroni et al, 2001; Nagayama et al, 2000; Plaschke et al, 2000; Kofler et al, 2002).
It is not known whether PARP-2 plays a similar role as PARP-1 in ischemic brain injury. We evaluated this hypothesis and determined if PARP-2 deletion is neuroprotective by subjecting PARP-2 knockout mice (PARP-2−/-) and wild-type (WT) littermates to different experimental cerebral ischemia procedures. Mice underwent either middle cerebral artery occlusion (MCAO) as a model for focal ischemia (Eliasson et al, 1997; Goto et al, 2002) or cardiac arrest and cardiopulmonary resuscitation (CA/CPR) as a model for global ischemia (Kofler et al, 2004). We chose to use both experimental paradigms since PARP-1 inhibition has not been equally effective under both focal and global ischemic conditions (Nagayama et al, 2000; Moroni et al, 2001; Kofler et al, 2002).
Materials and methods
This study was conducted in accordance with the National Institutes of Health guidelines for the care and use of animals in research, and protocols were approved by the Animal Care and Use Committee at Johns Hopkins Medical Institutions (focal ischemia model) and Oregon Health and Science University (global ischemia model).
Experimental Groups
Sensitivity to ischemic brain injury was compared between adult male PARP-2−/- mice and WT littermates. Poly(ADP-ribose) polymerase-2−/- mice were produced by targeted disruption at exon 9 of the PARP-2 gene, as described previously (Ménissier de Murcia et al, 2003). Chimeric offspring (129Sv/C57Bl/6) were subsequently backcrossed once to C57Bl/6 mice. The genotype of all mice was determined by polymerase chain reaction (PCR), as previously published (Ménissier de Murcia et al, 2003). Mice lacking PARP-2 display no visible abnormal phenotype by 18 months of life and are not tumor prone (Ménissier de Murcia et al, 2003).
Focal Cerebral Ischemia Model
Cerebral ischemia was induced by 120 mins of reversible MCAO under halothane anesthesia in oxygen-enriched air, as previously described (Eliasson et al, 1997; Goto et al, 2002; McCullough et al, 2005). Rectal temperature was maintained at approximately 37°C during surgery with a heating pad. A midline ventral neck incision was made, and unilateral MCAO was performed by inserting a 6.0 nylon monofilament into the right internal carotid artery 6 mm from the internal carotid/pterygopalatine artery bifurcation, via an external carotid artery stump. After securing the filament in place and closing the surgical site, the animal was awakened and assessed for neurological damage as follows: 0, no deficit; 1, forelimb weakness and torso turning to the ipsilateral side when held by tail; 2, circling to the affected side; 3, unable to bear weight on the affected side; and 4, no spontaneous locomotor activity or barrel rolling. If no deficit was observed 60 mins after beginning of occlusion period, the animal was removed from further study. Mice with clear neurological deficits were reanesthetized with halothane for suture removal at 2 h of occlusion and allowed to survive for 22 h. Brains were harvested under deep halothane anesthesia, cut into five 2-mm coronal sections, and stained with 2,3,5-tetraphenyltetrazolium chloride (TTC), as described previously (Eliasson et al, 1997; Goto et al, 2002; McCullough et al, 2005). Images were analyzed using image analysis software (Inquiry 3; Loats Associates, Westminster, MD, USA). The area of cortical, striatal, and total infarction was measured in each section and integrated over all slices. Infarct volume was then expressed as the absolute value in mm3 or as a percentage of the corresponding structure of the contralateral hemisphere to correct for edema.
In a separate nonsurvival cohort of animals (WT, n = 2; PARP-2−/-, n = 4), laser Doppler flow over the right parietal cortex (Moor instruments, Wilmington, DE, USA) and mean arterial pressure were monitored to ensure consistency of occlusion and equivalency of physiological variables between groups. Blood samples for arterial blood gas analysis were taken via a femoral artery catheter before MCAO, at 60 mins during ischemia, and 30 mins after reperfusion.
Global Cerebral Ischemia Model
Anesthesia was induced with 3% halothane and maintained with 1% to 1.5% halothane in oxygen-enriched air (FiO2 30%) via a face-mask. Temperature probes were inserted into the left temporalis muscle and into the rectum. Rectal temperature was controlled at near 37°C during surgery with a heating lamp and a heating pad. For drug administration, a PE-10 catheter was inserted into the right internal jugular vein and flushed with heparinized 0.9% saline solution. Cardiac monitoring was performed using an EKG with subcutaneous needle electrodes. Animals were endotracheally intubated using a 22 G intravenous catheter and mechanically ventilated (mouse ventilator Model 687, Harvard Apparatus, Holliston, MA, USA) set to a respiratory rate of 160/min. Tidal volume was adjusted to maintain arterial carbon dioxide tension within the physiological range (35 to 45 mm Hg).
After a 10-mins stabilization period, head temperature was raised by use of a water-filled coil, which was placed around the animal's head and heated to 40°C by running through a water bath (Kofler et al, 2004). Target temporalis temperature was set to 39.5°C to 40°C to produce consistent brain injury. Body warming was discontinued and switched to cooling by moistening the skin with alcohol and by a water-filled pad, which was placed underneath the animal and chilled by running through an ice-water bath. Intraischemic hypothermia was used to improve hemodynamic stability after CPR and to reduce the mortality. Cardiac arrest was induced by injection of 50 µL of cold (4°C) 0.5 mol KCl via the jugular catheter, and confirmed by appearance of asystole on the EKG monitor. The endotracheal tube was then disconnected from the ventilator, and anesthesia was discontinued. Body temperature was allowed to decrease during the arrest period until 30°C, then the cooling devices were removed.
At 10 mins after induction of CA, CPR was begun by injection of 0.5 mL prewarmed epinephrine solution (16 µg epinephrine/mL 0.9% saline), chest compressions (approximately 300/min), and ventilation with 100% oxygen. As soon as restoration of spontaneous circulation (ROSC) was achieved, cardiac massage was stopped. ROSC was assessed by reappearance of electrical activity on the EKG monitor and observation of the chest for visible cardiac contractions to ascertain that electrical activity of the heart was accompanied by appropiate mechanical activity. In case of sustained CA, additional doses of 0.2 mL epinephrine solution were administered in 1-min intervals. If ROSC could not be achieved within 2.5 mins of CPR, resuscitation efforts were abandoned. Simultaneously with the beginning of CPR, the head temperature control system was switched to cooling and finally turned off when a temporal temperature of 37°C was reached. The body was rewarmed using the heating lamp and pad at a rate of 0.3°C to 0.5°C/min. Catheters and temperature probes were removed, and the skin wounds closed. At 30 mins after ROSC, FiO2 was reduced to 50%, and 15 mins later mice were extubated and returned to their homecage for recovery.
At 3 days after CA, brains were harvested under deep halothane anesthesia, immersion-fixed in 10% formalin and embedded in paraffin. Serial coronal sections (10 µm) were cut and stained with hematoxylin and eosin. With the investigator masked to the genotype of the animal, injury to the dentate gyrus, CA1, CA2, and CA3 sectors of the hippocampus at bregma −1.5 mm, rostral and caudal caudoputamen (CP; bregma 0.5 and −1.0 mm, respectively), and cortex (bregma 0.5) were evaluated by light microscopy (× 100 objective). Brain levels and distinct regions were defined using a mouse brain atlas (Paxinos and Franklin, 2001). Neurons with hypereosinophilic cytoplasm and a dark pyknotic nucleus were considered to be nonviable. All viable and nonviable neurons were counted for each microscopic field, and the percentage of nonviable neurons was calculated for CA1, CA2, CA3, and CP. For assessing hippocampal injury, the entire length of each sector was evaluated. For striatal damage, six microscopic fields were investigated on each level, following a distinct pattern with the counting fields evenly distributed over the respective brain area. The entire dentate gyrus and dorsolateral cortical circumference were scanned to estimate injury using the following scoring system: 0 = no injury, 1 = ≤ 10%, 2 = 11% to 20%, 3 = 21% to 50%, 4 = > 50% ischemic neurons.
Statistical Analysis
All data are expressed as mean ± s.d. Physiological variables, infarct volume, and histological injury in CA1, CA2, CA3, and CP were compared between groups by two-way ANOVA with a Student—Newman—Keuls test to correct for multiple comparisons. Injury in brain regions assessed by a scoring system, for example, the dentate gyrus and cortex, is expressed as median and interquartile range (IQR) and analyzed using a Mann—Whitney U-test. Recovery rate from CA and mortality were compared between groups by a χ2-test. Significance was assumed with P > 0.05.
Results
Focal Cerebral Ischemia
Baseline arterial blood pressure and physiological parameters were normal in PARP-2−/- mice and not different from WT mice before, during or after MCAO (Table 1). All variables remained within physiological range throughout the experiment. The intraischemic laser Doppler flow signal was reduced equivalently in all groups during occlusion. Similarly, there were no differences among groups in gross neurological score during occlusion (PARP-2−/-: 2.3 ± 0.5; WT 2.3 ± 0.7). Poly(ADP-ribose) polymerase-2−/- mice were markedly protected from ischemic injury relative to WT littermates, with absolute infarct volumes reduced by > 40% in the cortex (WT, 64.5 ± 21.3; PARP-2−/-, 36.9 ± 14.1 mm3; P > 0.001) and CP (WT, 43.8 ± 10.4; PARP-2−/-, 24.4 ± 5.3 mm3; P > 0.05). Relative stroke volumes corrected for edema are presented in Figure 1.
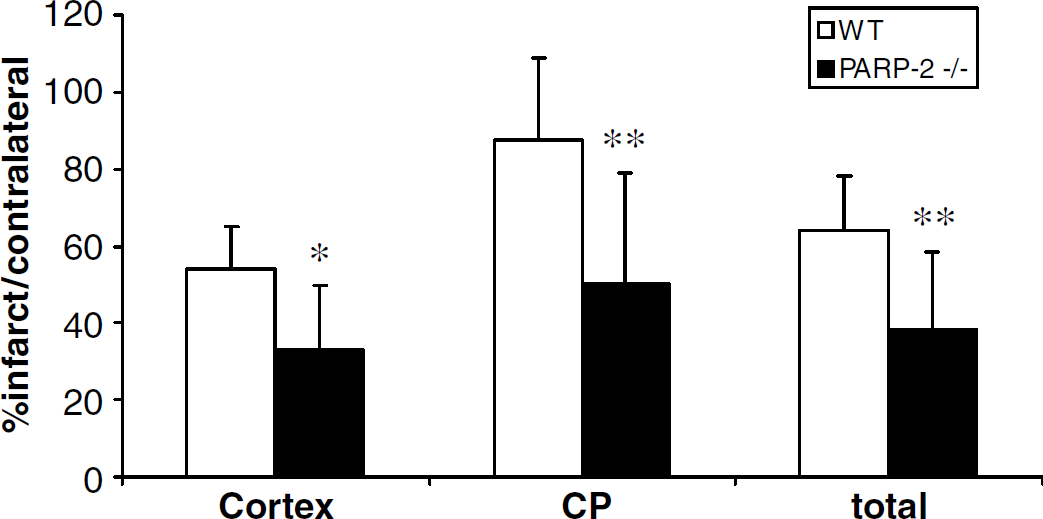
Total and regional infarct volume (percentage of contralateral structure) 22 h after MCAO. Poly(ADP-ribose) polymerase-2−/- mice (n = 11) had less ischemic damage in both cortex and CP compared with WT mice (n = 10). Data are presented as mean ± s.d., *P > 0.01; **P > 0.001.
Physiological measurements before and during MCAO, and 30 min after reperfusion
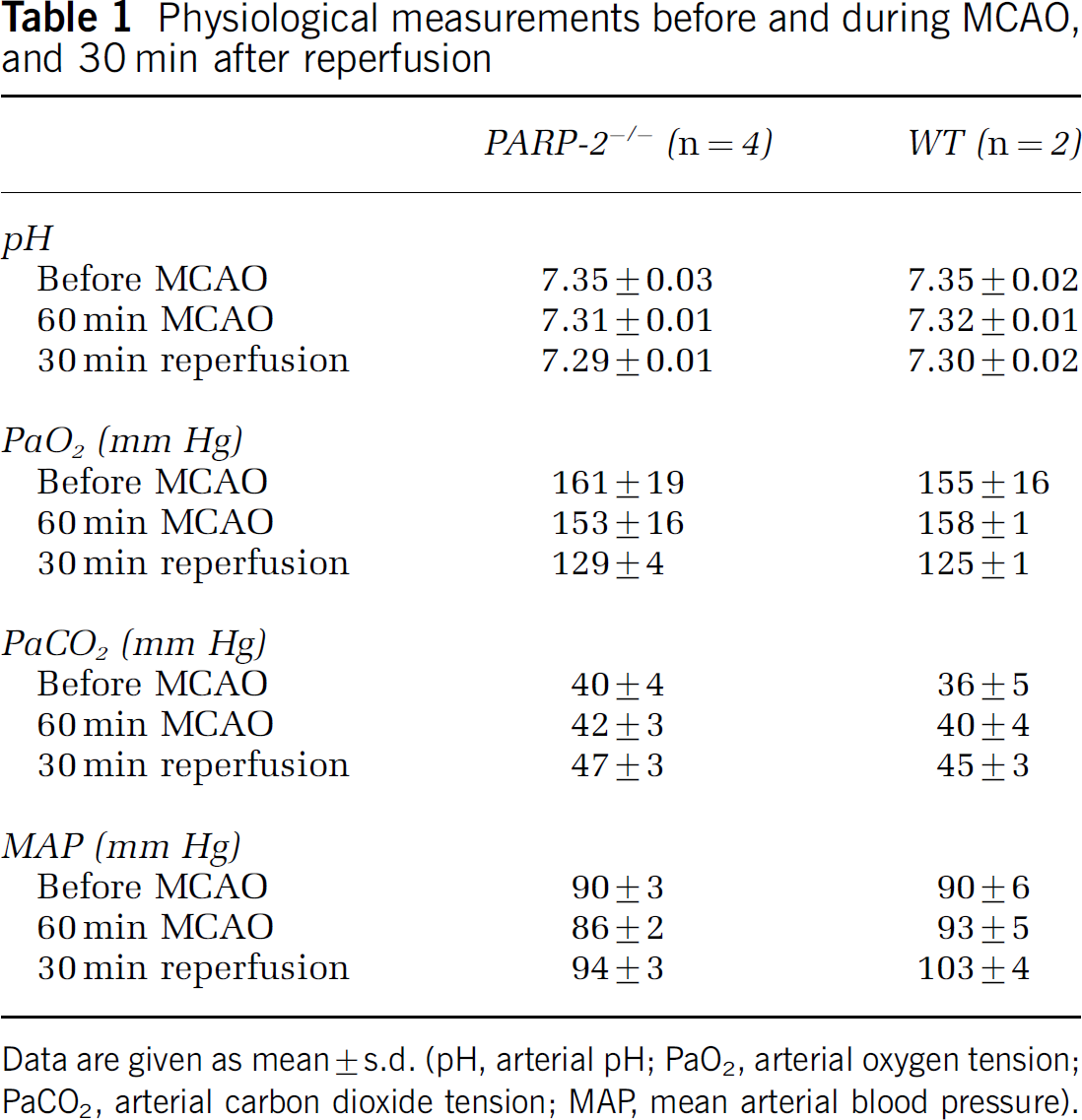
Data are given as mean ± s.d. (pH, arterial pH; PaO2, arterial oxygen tension; PaCO2, arterial carbon dioxide tension; MAP, mean arterial blood pressure).
Global Cerebral Ischemia
Injection of KCl led to immediate asystolic CA in all mice. All mice were successfully resuscitated within the predefined time window of 2.5 mins. Body weight, total ischemia time (10 mins arrest + CPR) and epinephrine dose were not different between PARP-2−/- and WT mice (Table 2). Temporalis and rectal temperatures were equally altered in both groups according to protocol. The overall 3-day survival rate was 69%, with no significant differences between groups (Table 2). No differences were seen between survivors and nonsurvivors regarding CA-related parameters, with the exception that nonsurvivors resumed spontaneous breathing later than survivors (10.7 ± 4.0 mins versus 15.9 ± 5.7 mins, P > 0.05), indicating that brain stem ischemia might have contributed to their death.
Body weight, cardiac arrest-related parameters and survival rates
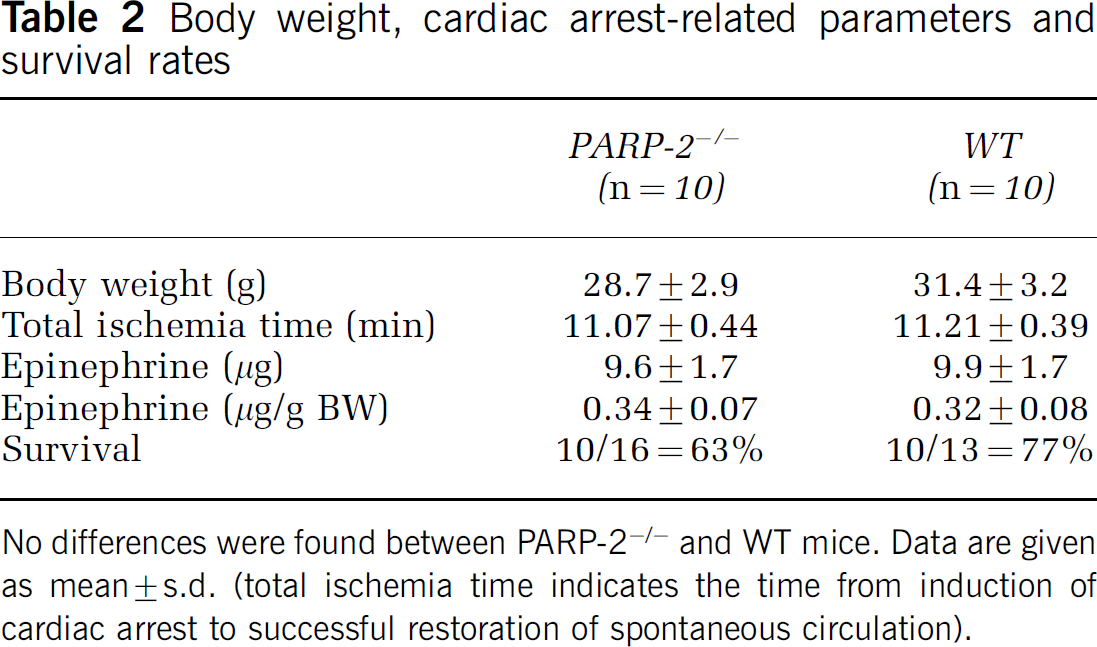
No differences were found between PARP-2−/- and WT mice. Data are given as mean ± s.d. (total ischemia time indicates the time from induction of cardiac arrest to successful restoration of spontaneous circulation).
All mice regardless of genotype sustained histological injury. Analysis of the CA1 subregion of hippocampus revealed that PARP-2−/- mice had more severe injury than WT mice (PARP-2−/-: 65% ± 36%, WT: 31% ± 33%, P > 0.05; Figures 2 and 3). Similar results were found for hippocampal sectors CA2 and CA3 (Figure 2). No difference between genotypes was seen in rostral or caudal CP (Figure 2), dentate gyrus (median score (IQR): WT, 1.0 (1.0 to 1.0); PARP-2−/-, 1.5 (1.0 to 3.0)), or cortex (median score (IQR): WT, 1.5 (1.0 to 3.0); PARP-2−/-, 2.0 (1.0 to 2.0)).
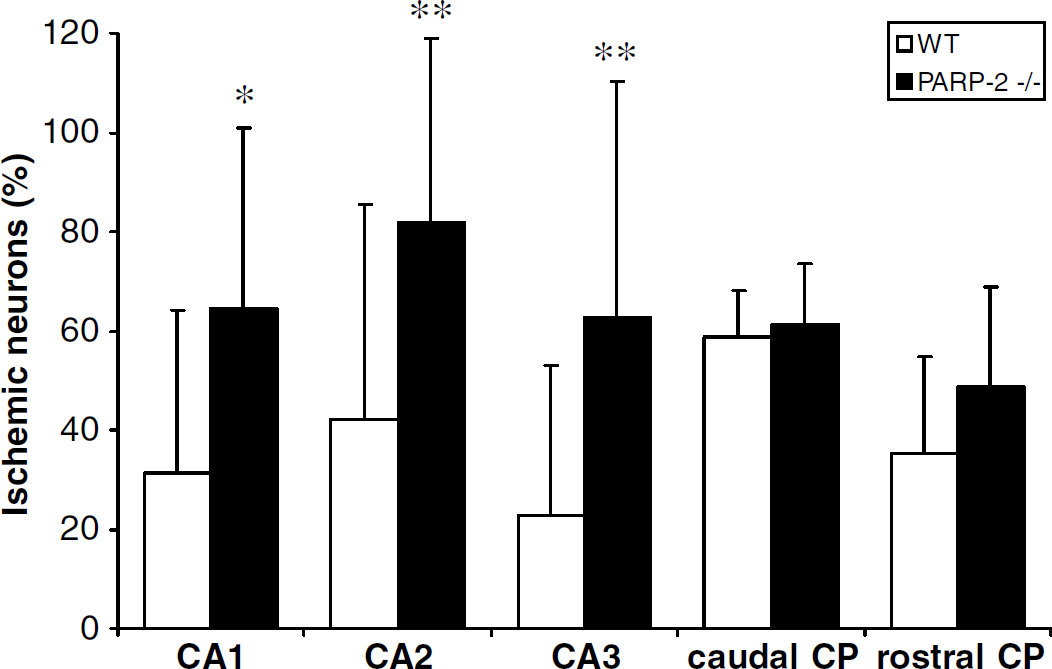
Percentage of damaged neurons after 10 mins of CA and 3 days of reperfusion comparing WT and PARP-2−/- mice (n = 10, respectively). Deletion of PARP-2 markedly increased neuronal cell loss in the hippocampus, but did not affect injury in CP. Data are presented as mean ± s.d., *P > 0.05, **P > 0.01.
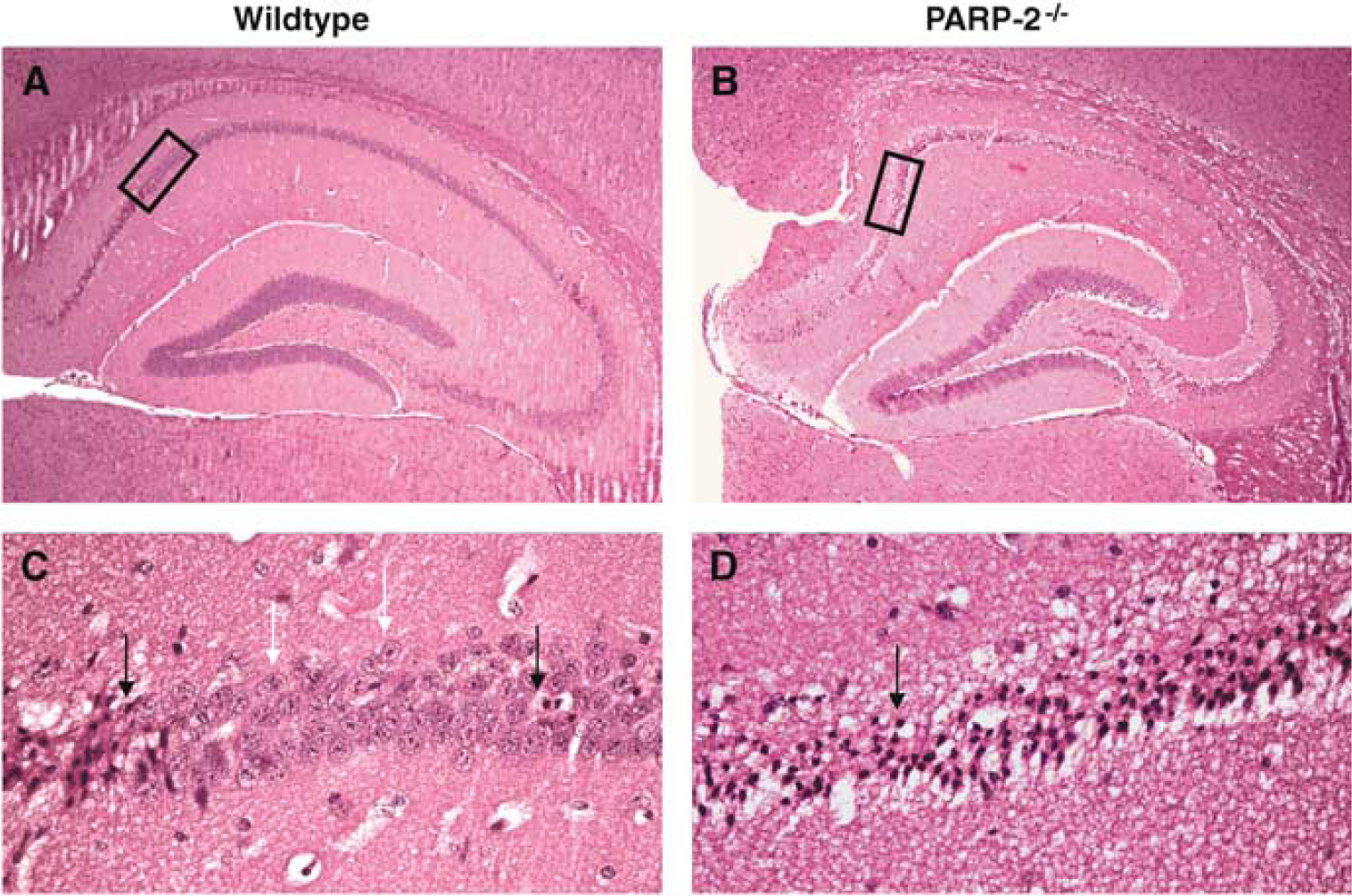
Representative photomicrographs (hematoxylin and eosin) showing ischemic damage in the hippocampus. At 3 days after CA, most of the CA1 neurons in the WT brain (left column) appear intact, whereas in the PARP-2−/- brain (right column) almost the entire hippocampus shows ischemic damage. Black arrows indicate neurons considered ischemic, white arrows normal neurons. Black boxes in pictures (
Discussion
While evidence is accumulating that PARP-2 plays an important role in the maintenance of genomic integrity (Ménissier de Murcia et al, 2003), little is known regarding the role of PARP-2 under conditions of ischemia and reperfusion. Here we show for the first time that PARP-2 has differential effects on cell survival depending on experimental model and mode of cell death. In focal stroke with a largely necrotic pathology, we show that deletion of PARP-2 reduces infarction by approximately 40% of that observed in the WT mouse. In global cerebral ischemia with delayed cell death, neuronal loss is increased, not ameliorated. However, the vulnerability is solely evident in hippocampal neurons. We conclude that PARP-2 is a novel executioner of cell death pathways in focal injury with energy failure, but the enzyme may stabilize DNA repair mechanisms necessary to mitigate hippocampal delayed cell death.
We do not know the mechanism by which PARP-2 deletion provides protection, but initial hypotheses can be drawn from what is known regarding PARP-1. Depletion of NAD+ and subsequently ATP has been proposed as one major mechanism by which PARP-1 overactivation after ischemia/reperfusion is detrimental to the cell (D'Amours et al, 1999; Endres et al, 1997). Since total PARP activity is only reduced by approximately 10% in PARP-2−/- cells, it is questionable if this small decrease can have a sufficiently large effect on the energy state of the cell to improve survival. After focal cerebral ischemia, brain levels of NAD+ were comparable between PARP-1−/- mice and animals treated with a nonspecific PARP inhibitor (Endres et al, 1997), suggesting that NAD+ depletion is mainly caused by activation of PARP-1, but not other isoforms. However, direct information regarding the amount of NAD+ depletion after genotoxic stimuli is lacking for PARP-2−/- cells.
Recently, it has been shown that PARP-1 activation causes AIF translocation and activation of the caspase-independent pathway of apoptosis (Yu et al, 2002; Hong et al, 2004; Komjáti et al, 2004). AIF translocation was not detectable in cortical neurons from PARP-1−/- mice (Yu et al, 2002), suggesting that activation of PARP-2 does not induce AIF translocation. However, no direct evidence is available at the moment to rule out that neuroprotection mediated via PARP-2 deletion involves AIF translocation.
Upregulation of proinflammatory genes via NF-κB coactivation is a third proposed mechanism by which PARP-1 contributes to ischemic cell injury (Hassa and Hottiger, 2002). Administration of a PARP inhibitor attenuated inducible nitric oxide synthase (iNOS) mRNA expression in the brain after transient MCAO (Park et al, 2004). Activation of proinflammatory iNOS contributes to ischemic brain injury, and its inhibition or genetic deletion is associated with reduced infarct size (Park et al, 2004; Moro et al, 2004). Selective reduction of PARP-2 by antisense oligonucleotides was beneficial in a mouse model of chronic colitis by decreasing the production of proinflammatory mediators, for example, tumor necrosis factor (TNF)α, interferon (IFN)γ, and reducing iNOS activity (Popoff et al, 2002). A similar anti-inflammatory effect might have contributed to neuroprotection in the PARP-2−/- mice.
While there is convincing evidence that blocking of PARP activation is protective in focal cerebral ischemia, results from global cerebral ischemia studies are less clear. Poly(ADP-ribose) polymerase inhibitors protect cortical neurons against oxygen glucose deprivation (OGD) injury (Eliasson et al, 1997; Moroni et al, 2001), but not OGD-treated hippocampal slices or CA1 pyramidal cells after 5 mins of bilateral carotid artery occlusion in gerbils (Moroni et al, 2001). In forebrain ischemia, PARP inhibitors decrease cell survival in the CA1 region, possibly by blocking DNA excision repair (Nagayama et al, 2000). We have previously reported that PARP-1−/- mice show neuroprotection in CP, but not in CA1, after CA/CPR (Kofler et al, 2002). In this study, PARP-2 deletion worsens outcome in hippocampus after a global ischemic insult, with no effect on the cortex or CP. It is important to note that no compensation by PARP-1 upregulation is seen in PARP-2−/- mice (Amé et al, 1999; Schreiber et al, 2002); thus, the effects observed in our experiments in PARP-2−/- mice cannot be explained by increased PARP-1 activity.
It is well known that the development of neuronal death is delayed in hippocampus compared with other vulnerable brain regions, for example, CP and, to a lesser degree, cortex (Pulsinelli et al, 1982; Larsson et al, 2001). The loss of DNA repair function has been previously reported to negatively affect neuronal survival in hippocampus (Meli et al, 2003; Nagayama et al, 2000). Since DNA-binding domains are different in PARP-1 and −2, it is also possible that the lack of specific ribosylation of one or more unique PARP-2 substrates underlies the detrimental effect observed in the hippocampus of PARP-2−/- mice after global cerebral ischemia.
Poly(ADP-ribose) polymerase inhibitors have become a promising class of neuroprotective drugs to ameliorate tissue injury after focal cerebral ischemia and reperfusion (Nakajima et al, 2005; Tokime et al, 1998; Komjáti et al, 2004). Since the catalytic domain is very similar in PARP family members, PARP inhibitors are not isozyme-specific but block total PARP activity (Tentori et al, 2002). As PARP-1 contributes approximately 90% to total PARP activity in response to DNA damage (Amé et al, 1999; Schreiber et al, 2002) and a similar reduction in stroke volume is seen in animals with either genetic deletion or pharmacological inhibition of PARP-1 (Eliasson et al, 1997; Endres et al, 1997), PARP inhibitors have been assumed to exert protection mainly by blocking PARP-1 overactivation. Surprisingly, we find similar reduction in stroke damage in the present study with PARP-2−/- mice as that obtained with male PARP-1−/- mice and PARP inhibition in our previous studies with this same mouse model (Eliasson et al, 1997). Although PARP-2 has a much lower activity than PARP-1, PARP-2 deficiency is quite effective in restricting ischemic damage. The phenotypes of PARP-1−/- and PARP-2−/- mice are equally severe regarding their susceptibility to genotoxic insults. Despite the presence of PARP-1, PARP-2−/- cells are defective in BER, showing the requirement of PARP-2 for efficient DNA strand break resealing (Huber et al, 2004; Schreiber et al, 2002). It has been suggested that PARP-1 and −2 have to act as heterodimers in BER; thus, the absence of one of each would have the same consequence on repair efficiency (Schreiber et al, 2002). However, recent results show that nuclear colocalization of PARP-1 and −2 is low outside the nucleolus, even on stimulation with DNA-damaging agents, supporting the hypothesis that PARP-1 and −2 might have distinct targets and that their hetero-dimerization might be restricted to the nucleolus (Meder et al, 2005). A double-knockout mouse is not available since the deletion of both PARP-1 and −2 is embryonically lethal (Ménissier de Murcia et al, 2003), but it would be interesting to determine whether our observation can be confirmed by a lack of additional protection in PARP-1−/- or −2r−/- mice treated with a PARP inhibitor.
A limitation to our study is that we did not use longer survival times and therefore can only comment on the acute effects of PARP-2 deletion. Since PARP-1 and −2 are involved in gene repair activity, loss of this function may contribute to delayed cell death in the penumbra region. Previous experiments have shown robust protection in PARP-1−/- mice for at least 3 days after MCAO (Goto et al, 2002). However, since PARP-1 and −2 show a different profile in the CA model, localize to different sites in the nucleus and have a different biochemistry on activation, we cannot assume that protection is sustained in PARP-2−/- mice similar to PARP-1−/- mice. Long-term effects of PARP-2 deletion need to be determined in further experiments.
In conclusion, our results confirm that poly(ADP-ribosyl)ation is an important pathway in the evolvement of focal ischemic cell death and extend this concept by identifying a new executioner isoform. In contrast, the role of PARP activation in global cerebral ischemia is more complex. The development of PARP-1- and −2-specific drugs might be critical to further evaluate this injury pathway in global brain ischemia.