
Abstract
We previously showed that inhibition of protein kinase C delta (PKCδ) improves brain perfusion 24 hours after asphyxial cardiac arrest (ACA) and confers neuroprotection in the cortex and CA1 region of the hippocampus 7 days after arrest. Therefore, in this study, we investigate the mechanism of action of PKCδ-mediated hypoperfusion after ACA in the rat by using the two-photon laser scanning microscopy (TPLSM) to observe cortical cerebral blood flow (CBF) and laser Doppler flowmetry (LDF) detecting regional CBF in the presence/absence of δV1-1 (specific PKCδ inhibitor), nitric oxide synthase (NOS) substrate (
Keywords
INTRODUCTION
We previously showed that inhibition of protein kinase C delta (PKCδ) via δV1-1 can increase perfusion 24 hours after asphyxial cardiac arrest (ACA). Global cerebral ischemia (via ACA) causes derangement of cerebral blood flow (CBF) responsible for neuronal cell death in the CA1 region of the hippocampus as well as the cortex.
1
Owing to the overall decrease in CBF after ischemia, neuronal cell death
1
can occur in major regions of the brain responsible for learning, memory, and cognitive function.
2
It is thought that during global ischemia, PKCδ (a novel PKC) levels are elevated causing PKCδ to translocate to the nucleus (activation) resulting in cellular damage. In the normal brain, PKCδ levels are nominal whereas global ischemia can cause activation/translocation of PKCδ.
3
Inhibition of PKCδ (via specific inhibitor of PKCδ, δV1-1) can cause a revival of CBF 24 hours after ischemia to counteract hypoperfusion or low CBF found to be suppressed after cardiac arrest from 38% to 65%.1, 4 We previously showed that pretreatment of δV1-1 before ACA can enhance perfusion 24 hours after ischemia, resulting in improved neuronal survival in the hippocampal CA1 and cortex regions in our rat model of ACA.
1
Here, we sought out to define the specific mechanism(s) of how inhibition of PKCδ can alleviate these pathologies. The possible endothelium and endothelial-mediated nitric oxide synthase (eNOS) involvement as a target for PKCδ relating to general circulation was first reported by Monti et al.
5
This led to these current experiments in the brain by utilizing inhibitors and substrates related to nitric oxide synthase (NOS). Our results suggest that pretreatment with δV1-1 before ACA can activate eNOS machinery suggesting that the enhancement of perfusion may be attributed to enhanced CBF. This is further supported by the fact that
MATERIALS AND METHODS
Chemicals
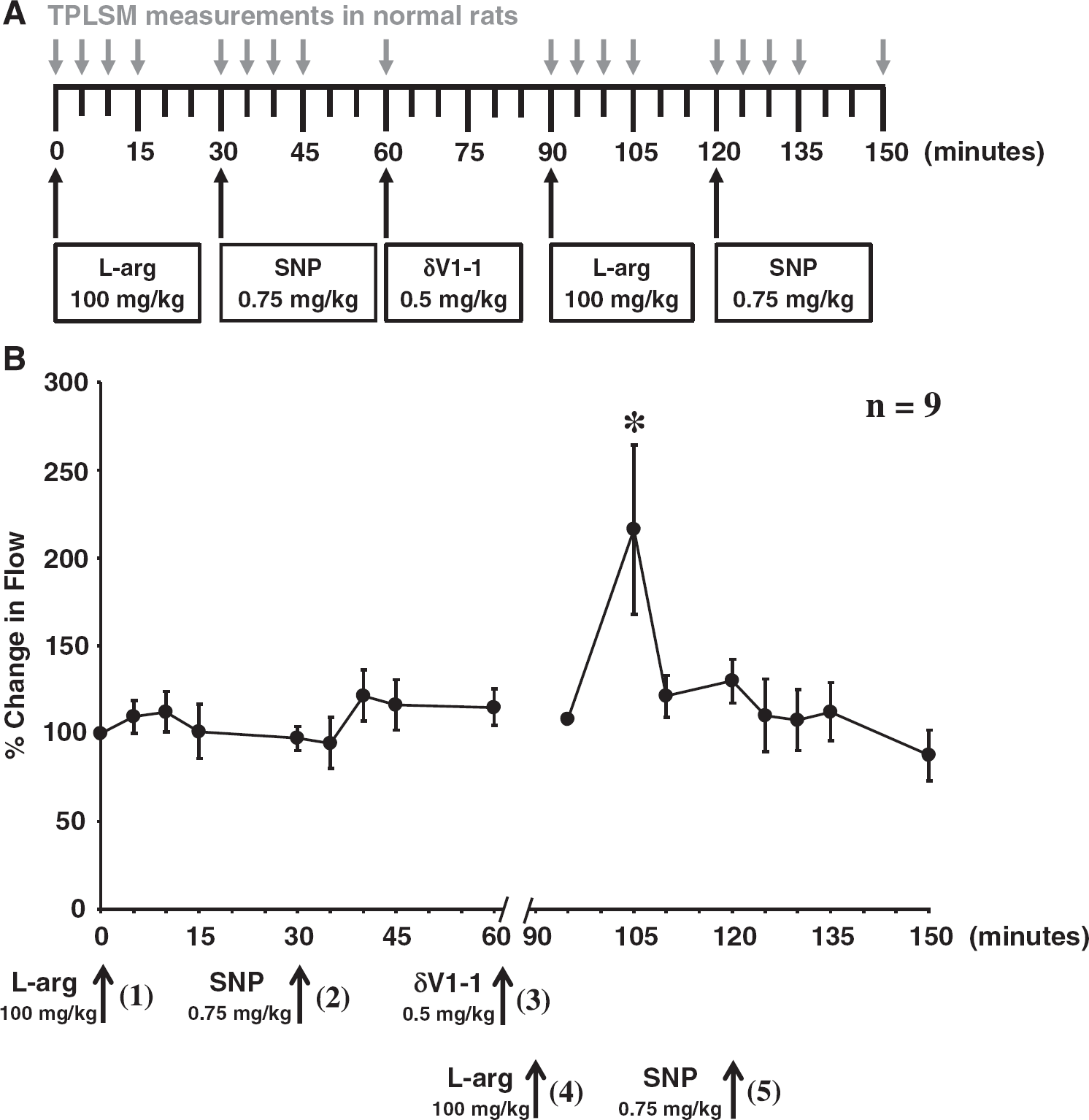
Schematic diagram of the experimental design. (
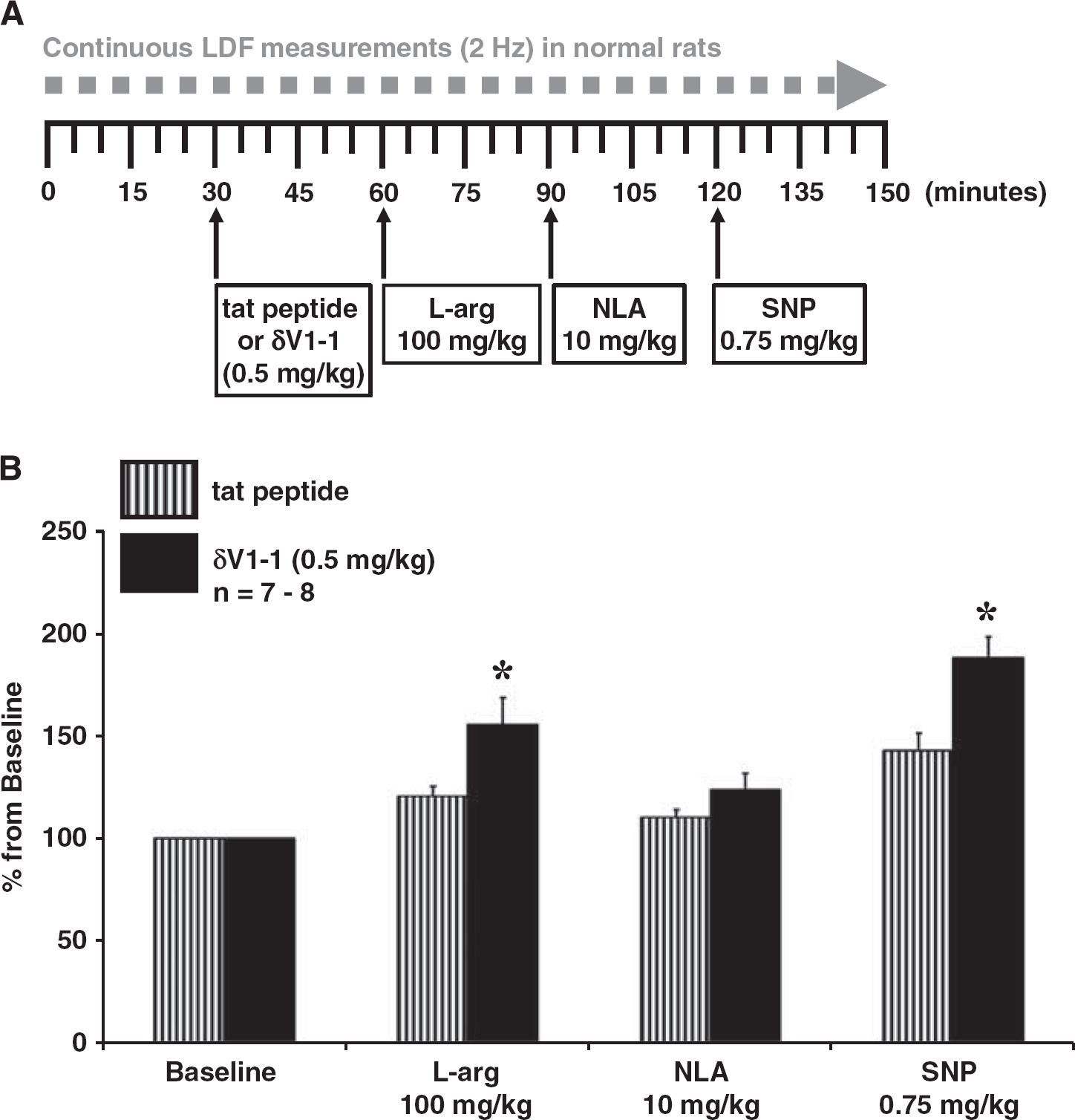
(
Animal Preparation
All procedures were approved by the Institutional Animal Care and Use Committee (University of Miami, Miller School of Medicine) and written up in accordance with ARRIVE. Adult male Sprague–Dawley rats (250 to 350 g) were fasted overnight before surgery. Rats were anesthetized with 4% isoflurane and 30:70 mixture of O2 and N2O, followed by endotracheal intubation. Isoflurane was lowered to 1.5% to 2% for endovascular access. The femoral vein and artery were cannulated using a single-lumen (PE-50) catheter for blood pressure monitoring, blood gas analysis, and intravenous injection of pharmacological agents. The rats were immobilized with vecuronium bromide (2.0 mg/kg, intravenously, administered every 10 minutes) and maintained immobilized throughout the procedure. Head and body temperatures were maintained at 37°C using heating blankets and lamps.
Thinned-Skull Window Method
The thinned-skull window method is a modified method from Xu et al. 6 Anesthetized rats were placed on a stereotaxic frame to ensure the stability of the rat. After femoral arteries and vein of the rat were cannulated and carotid ligatures installed, a longitudinal incision extending from the neck region to the frontal area of the rat was made at the midline scalp using microsurgical tools. Utilizing a high-speed microdrill, a thin circular area of the skull (∼2 mm in diameter) was made 1 mm lateral to the bregma. Thinned-skull window was created carefully with a microdrill thinning out the skull with constant irrigation with sterile saline so as to not overheat the skull or the drill bit. The skull is thin enough for visualization of blood vessels via TPLSM when it is approximately half the thickness of the skull or approximately 0.5 mm.1, 6
Two-photon Laser Scanning Microscopy
After thinning the skull, the rat was placed on a TPLSM (Lasersharp2000, Bio-Rad, Hercules, CA, USA). pH, mean arterial blood pressure, pCO2, pO2, glucose, rectal, and head temperatures were constantly monitored throughout the experiment. A 20 × water immersion objective (Olympus XLUMPlanFl) was lowered in proximity to the thinned-skull window. Fluorescent images were captured (every 5 to 15 minutes) at an excitation wavelength of 910 nm with the intravenous introduction of low molecular weight fluorescein–dextran. Cortical cerebral blood vessel images were captured at 20 × and 200 × . Additionally, Z-series (20 ×) and linescan (200 ×) images were obtained throughout the time course of the experiment starting with t=0 (before the introduction of drugs). We have previously experienced no immediate change in physiologic variables or change in CBF or blood vessel diameters upon acute administration of tat peptide or δV1-1. 1 Only microvessels with diameters 5 to 10 μm were considered for TPLSM studies. Upon visual inspection, arterioles can be recognized morphologically based on arterioles branching out from larger vessels, while, venules merge into larger vessels. 7
Linescans for red blood cell (RBC) velocity measurements were analyzed with ImageJ analysis software. 8 Each velocity measurement was calculated by measuring the slope of the dark lines (10 lines) representing 10 RBCs traversing at a point in time of the linescan. The slopes were calculated and averaged. One microvessel was studied per rat with an overall diameter on average of 10 μm. Linescan images were captured and measured as denoted in Figure 1A as ‘TPLSM measurements in normal rats'. Our data were expressed as percent change in flow. This was achieved by utilizing each rat as its own internal control. Baseline measurements before drug treatment were used for normalization of the data. This analysis was necessary owing to the fact that not all microvasculature possess exactly the same vessel size or RBC velocities before ischemia and drug treatments. Statistical analysis was evaluated by one-way ANOVA followed by Tukey's post hoc test.
Laser Doppler Flowmetry
Laser doppler flowmetry measurements were obtained to determine CBF dynamics of cortical blood vessels in rats with no treatment (baseline), tat peptide or δV1-1 (0.5 mg/kg),
Asphyxial Cardiac Arrest
To induce ACA, apnea was induced by disconnecting the ventilator from the endotracheal tube. Six minutes after asphyxia, resuscitation was initiated by administering a bolus injection of epinephrine (0.005 mg/kg, intravenously) and sodium bicarbonate (1 mEq/kg, intravenously) followed by mechanical ventilation. Arterial blood gases were measured before and after ACA. Control animals (sham) were subjected to surgical procedures similar to ACA animals except without induction of ACA. Resuscitation drugs were not used; however, sham animals were treated with isoflurane similar to experimental animals. Based on our prior experiences, administration of epinephrine in sham animals does not have any CBF differences after the blood pressure returns to normal. The rats were immobilized with vecuronium bromide (2.0 mg/kg, intravenously, administered every 10 minutes) and maintained immobilized throughout the procedure. 1
Whole-Blood Nitrite Analysis
Rat whole-blood was extracted before, 15 minutes, and 24 hours after ACA. Nitrite preservation solution was added to the whole blood. Nitrite measurements were determined by tri-iodide-based gas-phase reductive chemiluminescence with an NO analyzer (GE Analytic, Boulder, CO, USA) as described previously. 10 Nitrite concentrations were calculated based on the area under the curve (peak) utilizing a known reference injection of nitrite. Statistical analysis was evaluated by one-way ANOVA followed by Tukey's post hoc test.
Western Blot Analysis
Rats were prepared as noted in the ‘animal preparation’ section. Rats were injected with either tat peptide or δV1-1 and 6 minutes of ACA was performed as already mentioned. One hour after tat peptide or δV1-1 injection with or without ACA, the rats were killed and the brain excised. The cortex (the region of CBF measurements via LDF and TPLSM) was extracted and homogenized using a glass homogenizer in RIPA (50 mmol/L Tris pH 8, 150 mmol/L NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 1% SDS). The homogenate was centrifuged at 13,000 × g for 15 minutes at 4°C and the protein fractions were quantified using the Bradford Assay (Bio-Rad Dc Protein Assay, Hercules, CA, USA). Equal amounts of protein (50 μg) were separated on a 10% SDS-PAGE gel and electroblotted to nitrocellulose. Membranes were blocked with 5% bovine serum albumin/tris buffered saline with tween 20 (TBS-T) (for phospho-specific antibodies) or 5% milk/TBS-T (non-phospho-specific antibodies) then incubated in primary antibody at 4°C overnight. Primary antibodies were obtained from Cell Signaling (Boston, MA, USA; phospho-eNOS (Ser1177), nNOS) and Abcam (Cambridge, MA, USA; eNOS, iNOS). Proteins were detected by appropriate horseradish peroxidase-conjugated secondary antibodies (GE Healthcare UK Limited, Buckinghamshire, UK) and enhanced chemiluminescence (ECL) system (Pierce Thermo Scientific, Rockford, IL, USA). Protein loading was determined by re-probing the membrane for β-actin (Sigma-Aldrich). The blots were imaged and analyzed using Bio-Rad Quantity One Analysis Software. Graphical results are expressed as fold change from tat peptide normalized to β-actin. Statistical analysis was evaluated by one-way ANOVA followed by Tukey's post hoc test or Student's t-test for unpaired samples as appropriate with SPSS statistical software (Chicago, IL, USA).
Statistical Analysis
Results were expressed as means±s.e.m. Statistical analysis was evaluated by one-way ANOVA followed by Tukey's post hoc test or Student's t-test for paired or unpaired samples as appropriate with SPSS statistical software. The P0.05 level of probability was accepted as significant.
RESULTS
Protein Kinase C Delta Modulates L -Arginine-induced Enhancement of Cerebral Blood Flow in Cortical Microvessels
We previously showed that inhibition of PKCδ via δV1-1 can cause an increase in CBF 24 hours after ACA blunting hypoperfusion.
1
This leads to our current hypothesis that PKCδ alone can modulate NO machinery in acute or chronic situations. We applied (see Figure 1A, experimental paradigm)
Protein Kinase C Delta Modulates L -arginine and Sodium Nitroprusside-induced Enhancement of Regional Cerebral Blood Flow
The usage of TPLSM to observe cortical microvessels gives a focal perspective on cortical CBF at a point in time; therefore, we also used LDF to obtain a regional perspective on CBF at a high data sampling rate of 2 Hz as opposed to TPLSM (every 5 to 15 minutes after induction of drugs) (Figure 2). The LDF probe was placed in the same position as the TPLSM objective of 1 mm lateral to the bregma. Our LDF results suggest that the introduction of δV1-1 with subsequent
Whole-blood Nitrite Concentration is Enhanced via Inhibition of Protein Kinase C Delta After Cardiac Arrest
In order to determine the possible mechanism of action of PKCδ-mediated enhancement of CBF, we measured nitrite concentration in systemic whole-blood 15 minutes and 24 hours after ACA with tat peptide or δV1-1 pretreated for 30 minutes before the induction of ACA. Animals pretreated with δV1-1 presented with an enhanced nitrite concentration in systemic whole blood (2.50±0.61 μmol/L) 24 hours after ACA as compared with Sham, ACA only, or tat peptide (vehicle)+ACA (Figure 3). Physiologic parameters are provided in Table 1. PO2 for all groups (after ACA) were greater than 150 mm Hg owing to 100% delivery of oxygen and increased ventilator rate (60 to 80 breaths per minute) during and after resuscitation. As nitrite is a metabolite of NO, 20 it is suggestive that NO was the mostly likely cause of the enhanced CBF 24 hours after ACA as previously shown 1 with TPLSM.
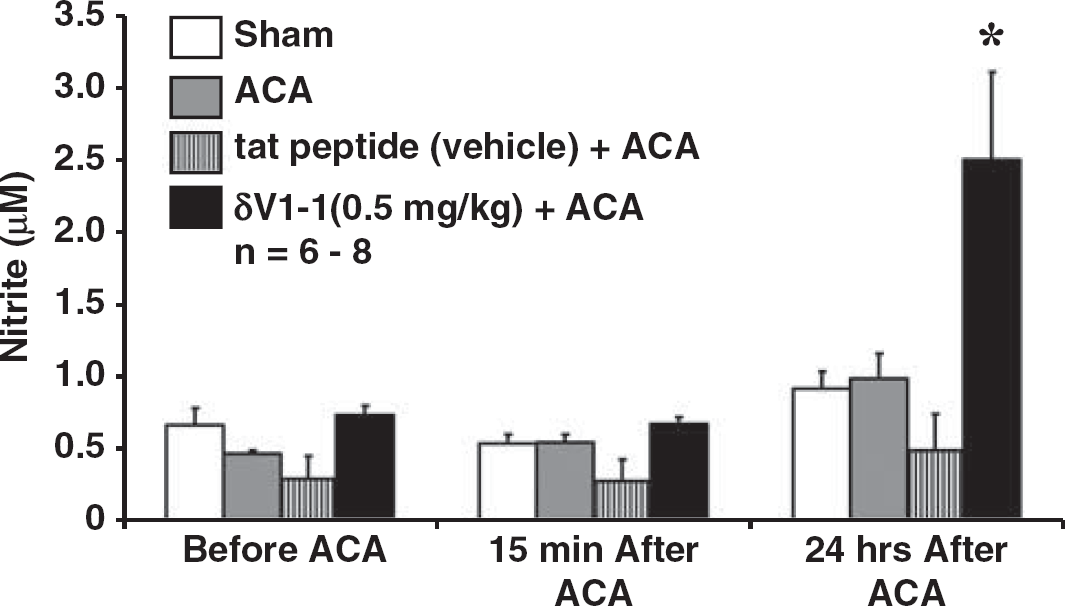
Inhibition of protein kinase C delta (PKCδ) enhanced the concentration of whole-blood nitrite 24 hours after asphyxial cardiac arrest (ACA). Whole-blood nitrite analyses were performed 15 minutes and 24 hours after ACA in the presence of sham (sham surgery=no ACA), ACA only, tat peptide (vehicle, treatment 30 minutes before ACA)+ACA, or δV1-1 (0.5 mg/kg, treatment 30 minutes before ACA)+ACA. Rats subjected to whole-blood nitrite analyses 24 hours after ACA in the presence of δV1-1 presented with an increase in whole-blood nitrite concentration of 2.50±0.61 μmol/L as compared with levels of nitrite 1 μmol/L or lower (see sham, ACA, and tat peptide (vehicle)+ACA) (n=6 to 8, ∗P0.05).
Asphyxial cardiac arrest physiologic parameters from nitrite analyses
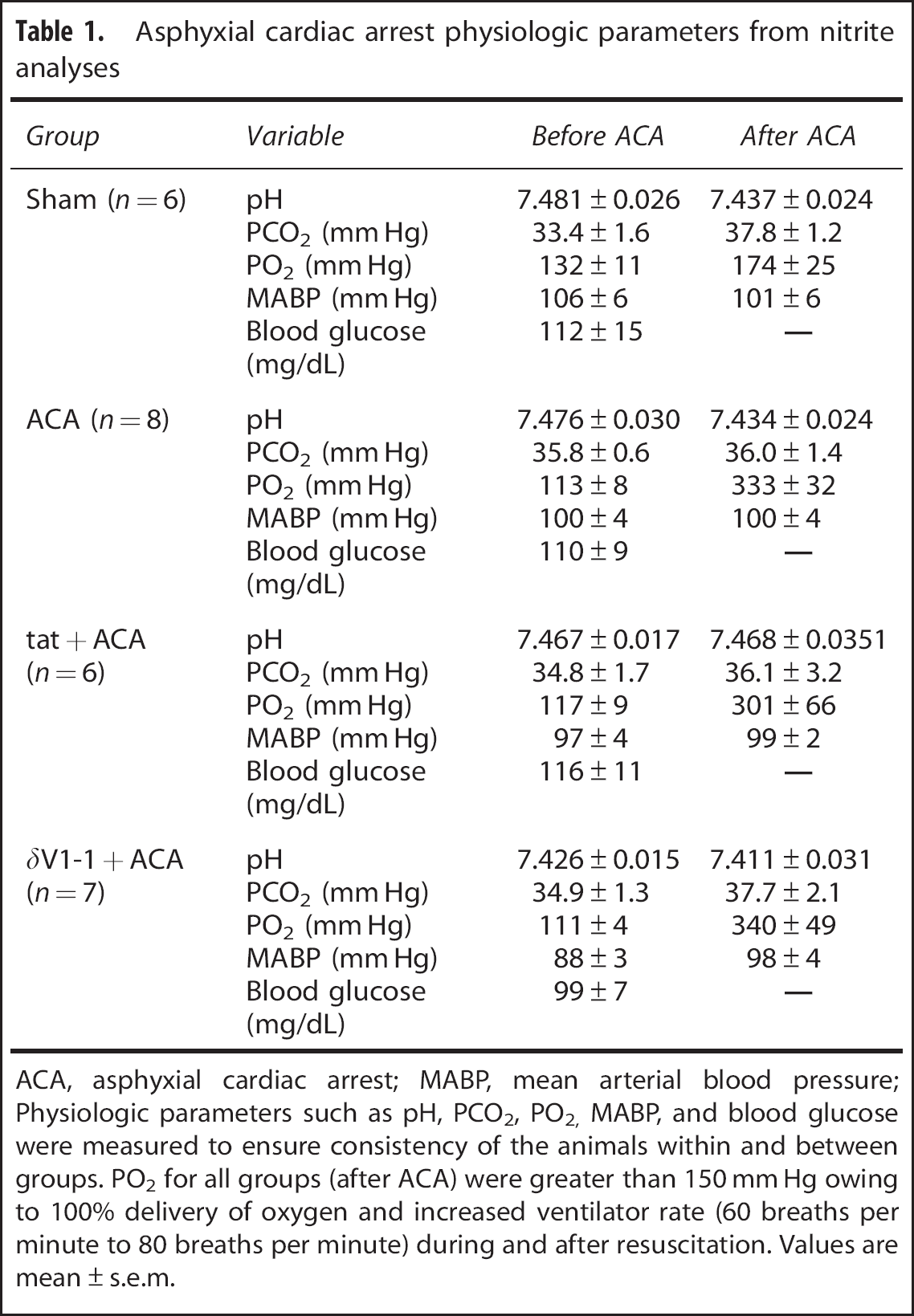
ACA, asphyxial cardiac arrest; MABP, mean arterial blood pressure
Physiologic parameters such as pH, PCO2, PO2, MABP, and blood glucose were measured to ensure consistency of the animals within and between groups. PO2 for all groups (after ACA) were greater than 150 mm Hg owing to 100% delivery of oxygen and increased ventilator rate (60 breaths per minute to 80 breaths per minute) during and after resuscitation. Values are mean±s.e.m.
Endothelial Nitric Oxide Synthase is Enhanced via Inhibition of Protein Kinase C Delta After Cardiac Arrest
As nitrite concentrations were enhanced in the presence of PKCδ inhibitor (via δV1-1), we performed protein analyses via Western blot for eNOS, phosphorylated-eNOS (P-eNOS), inducible NOS (iNOS), and neuronal NOS (nNOS) in the presence of tat peptide or δV1-1 (administered 30 minutes before ACA). Cortex samples (to undergo Western blot analyses, 1 mm lateral to the bregma) were isolated after the animal was killed 24 hours after ACA. Endothelial-mediated nitric oxide synthase but not P-eNOS (Figure 4A), iNOS (Figures 4C and 4D), or nNOS (Figures 4E and 4F) was enhanced in the presence of δV1-1+ACA (1.29±0.12%) as compared with tat peptide+ACA (1.00±0.08%) or δV1-1 only (0.95±0.01%) (without ACA) (Figure 4B). Physiologic parameters are provided in Table 2. PO2 for all groups (after ACA) were greater than 150 mm Hg owing to 100% delivery of oxygen and increased ventilator rate (60 to 80 breaths per minute) during and after resuscitation. These results suggest that inhibition of PKCδ can enhance eNOS levels responsible for vasodilation of cerebral arteries resulting in enhanced CBF 24 hours after ACA.
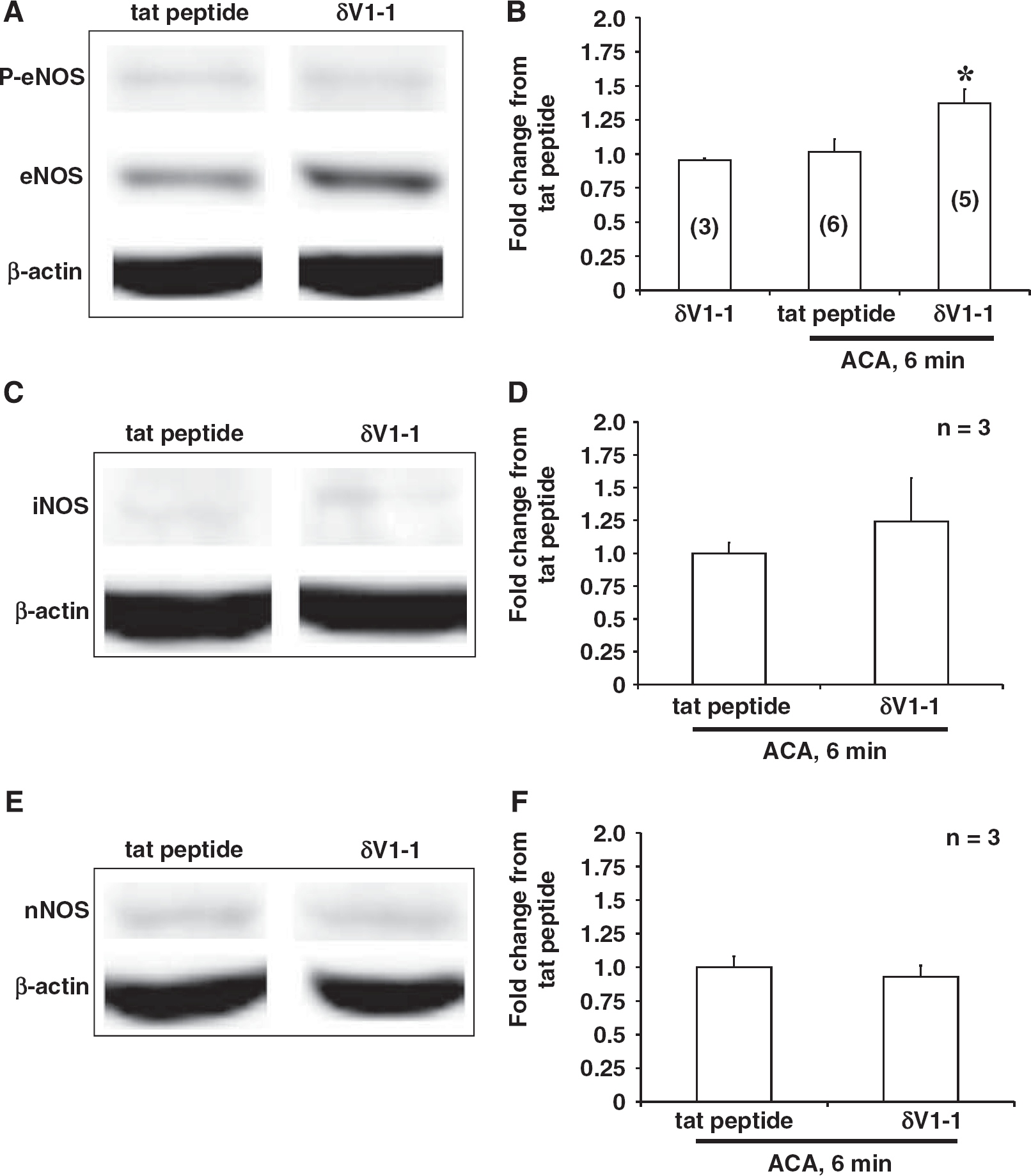
Inhibition of protein kinase C (PKCδ) enhanced endothelial-mediated nitric oxide synthase (eNOS) but not inducible NOS (iNOS) or neuronal NOS (nNOS) cortical protein expression 24 hours after asphyxial cardiac arrest (ACA). (
Physiologic parameters from Western blot analyses
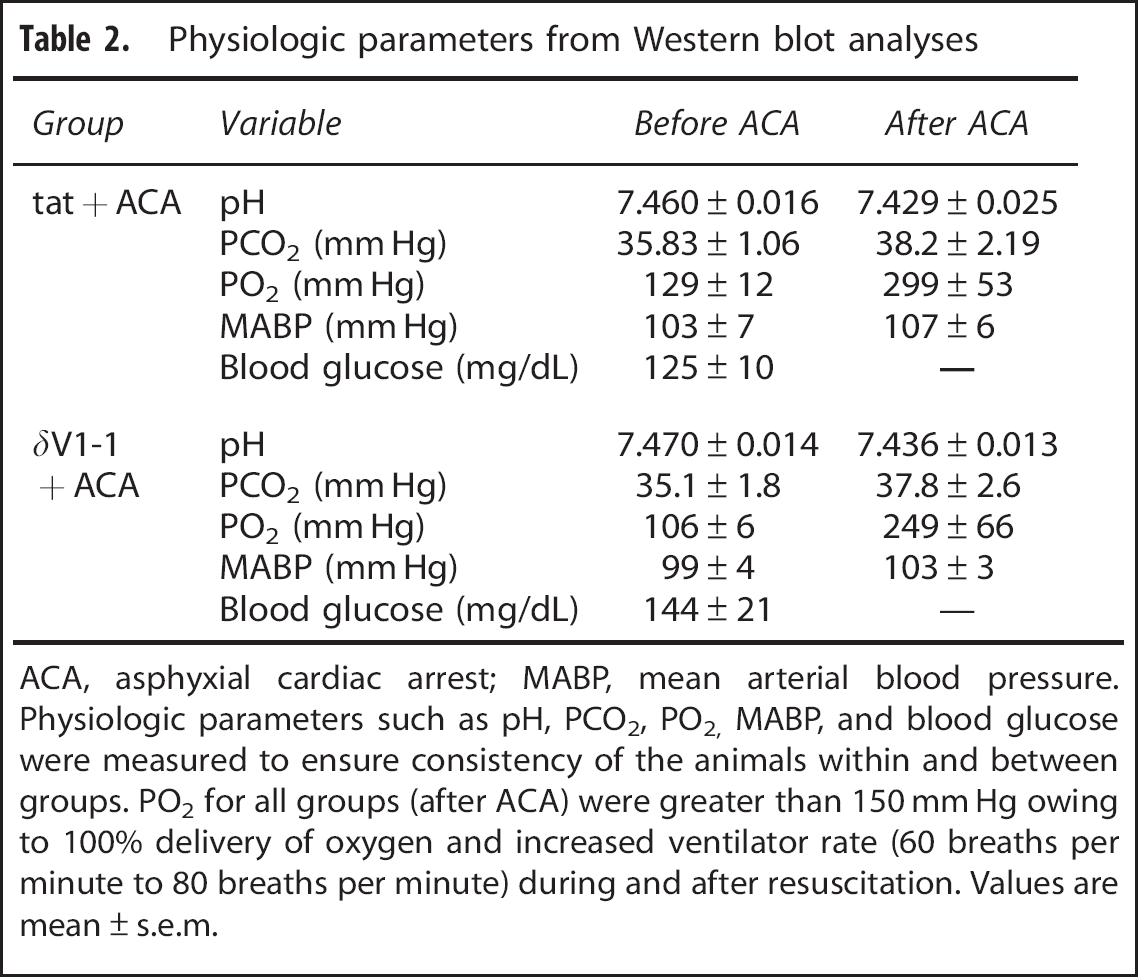
ACA, asphyxial cardiac arrest; MABP, mean arterial blood pressure.
Physiologic parameters such as pH, PCO2, PO2, MABP, and blood glucose were measured to ensure consistency of the animals within and between groups. PO2 for all groups (after ACA) were greater than 150 mm Hg owing to 100% delivery of oxygen and increased ventilator rate (60 breaths per minute to 80 breaths per minute) during and after resuscitation. Values are mean±s.e.m.
DISCUSSION
Previously, we showed that inhibition of PKCδ via δV1-1 24 hours after ACA enhanced perfusion as compared with tat peptide pretreatment.
1
This led to subsequent studies aimed at defining how inhibition of PKCδ is enhancing brain perfusion 24 hours after ACA. In this current study, we identified that PKCδ targets directly or indirectly the NO machinery evidenced by enhanced
It is without a doubt that SNP has been denoted as a NO donor causing vasodilation in cerebral as well as systemic arteries. Current dogma has led us to believe that this vasodilation can cause the possibility of increased CBF.21, 22, 23 However, there are many reports that suggest otherwise.24, 25, 26 Sodium nitroprusside-mediated enhancement of CBF depends upon where and what type(s) of vasculature one is measuring from. Traditionally, studies involving ex vivo manipulation of cerebral or systemic vessel preparation has been used to study these types of phenomena utilizing pharmacological manipulations (i.e. in the presence or absence of SNP/NLA etc.) to determine vasodilation or vasoconstriction of cerebral vessels but it does not always directly correlate to enhanced CBF. In this study, we are not claiming vascular tonicity but only CBF perfusion in two techniques employed, the TPLSM of pial circulation and LDF. The objective (from TPLSM) or probe (from LDF) was placed 1 mm lateral to the bregma as published earlier.1, 27 These two techniques are real-time CBF measurements to determine the effects of PKCδ. We used
The therapeutic potential via the modulation of PKCδ varies from vascular restenosis 28 to improvement of endothelial vascular dysfunction via inhibition of PKCδ. 5 It is thought that the inhibition of PKCδ (via δV1-1) can have beneficial effects against ischemia such as attenuation of hyperemia and hypoperfusion in models of global ischemia 1 and vascular protection against endothelial vascular dysfunction via modulation of eNOS. 5 In addition, degradation of PKCδ during reperfusion improves physiologic outcome in the myocardial infarction 29 model altogether suggesting that ischemia causes activation/translocation of PKCδ whereas inhibition or attenuation of PKCδ can improve physiologic outcomes, as it relates to neuronal protection and/or revival of CBF.1, 27
We and others have reported that inhibition of PKCδ via δV1-1 can restore brain blood flow in global or regional ischemia,1, 27, 30 but the mechanism of action remains controversial. In fact, Monti et al 5 suggest in models of cell culture that the effects of δV1-1 in endothelial cells can be complemented with the action of PKCε to modulate and stabilize eNOS preventing endothelial cell dysfunction. In addition, inhibition of PKCδ can enrich endothelial cell survival under hypoxic conditions in human umbilical vein endothelial cells. 31 In coronary postcapillary venular endothelial cells, inhibition of PKCδ with δV1-1 shifts PKCε to the cytoplasm and PKCε can then translocate to the membrane. Protein kinase C ε activation promotes inhibition of eNOS resulting in decreased reactive oxygen species production via Akt phosphorylation. 5 This is contrary to our results. However, using a model of bovine aortic endothelial cells, Rask-Madsen and King, 32 suggested that PKCε can activate Akt and eNOS through the vascular endothelial growth factor pathway. 32 Together, these results from Monti et al 5 and Rask-Madsen and King 32 can provide a possible explanation of our results where inhibition of PKCδ via δV1-1 can activate PKCε translocation causing enhanced eNOS expression. It is also important to note that although not without controversy, many of these studies were performed in cell culture systems using methods to mimic ischemia, such as serum deprivation. As cardiac arrest is a multi-faceted problem consisting of whole-body ischemia that compromises systemic blood parameters and cerebral, renal, and cardiac functions, consequent disruption of CBF, it is possible that our results can provide new additional insight into this problem.
We initially used the TPLSM to detect changes in cortical CBF in the presence of
With the use of
Much of the current understanding regarding the mechanism of activation of NOS has been performed in intact cell cultures suggesting that perhaps there may be differential/additional factors (such as Ca2+ levels responsible for the modulation in various NOS systems) that mediate NOS activation. Others have suggested that the relationship between PKCδ and eNOS activation may be tissue/cell specific. 5 Previously, others have shown that PKCδ phosphorylates serine (1177/1179) resulting in eNOS activation in bovine aortic endothelial cells. 38 Attenuation of PKCδ (via small interfering RNA) decreased NO production in fibroblasts. 39 In both of these studies, the authors used Rottlerin, which at the time was thought to be the only specific inhibitor of PKCδ but others have shown that Rottlerin is not specific for PKCδ. 40 Nonetheless, this leads to our novel findings, which suggest that inhibition of PKCδ (via δV1-1) actually enhances whole-blood nitrite concentration (Figure 3) with eNOS-enhanced levels in the presence of ACA pretreated with δV1-1 (Figure 4). In addition, some investigators have suggested that enhanced P-eNOS in turn, inhibit PKCδ expression in human umbilical vein endothelial cells induced with propofol 41 whereas Ramzy et al 42 suggested that decreased eNOS expression results in increased PKCδ levels in human saphenous vein endothelial cells induced with endothelin-1. 42 Therefore, based on this information from Ramzy et al, 42 inhibition of PKCδ will increase eNOS levels similar to our findings (Figure 4).
In summary, we show that inhibition of PKCδ via δV1-1 can enhance brain perfusion 24 hours after ACA. 1 This enhancement in CBF is most likely mediated via enhanced eNOS levels in the presence of PKC suppression. This is further evidenced by enhanced nitrite concentrations in whole-blood 24 hours after ACA in the presence of δV1-1. More studies are needed to define the physiologic role of eNOS in the context of cardiac arrest beyond activation via phosphorylation as well as defining the therapeutic value of modulation of PKCδ (via δV1-1) against cerebral ischemia.
Footnotes
The authors declare no conflict of interest.