
Abstract
Repinotan is a highly potent 5-HT1A receptor agonist with strong neuroprotective efficacy in animal models of middle cerebral artery occlusion and traumatic brain injury. In this study, we characterized the time window for neuroprotective effects of repinotan in animal models. In the permanent middle cerebral artery occlusion model, repinotan showed neuroprotective efficacy when administered as a triple bolus injection (0.3–100 μg/kg) or an intravenous infusion (0.3–100 μg/kg per hour). A 73% reduction in infarct volume was observed with a 3 μg/kg intravenous bolus, and a 65% reduction was observed with a 3 and 10 μg/kg per hour intravenous infusion. When delayed until 5 hours after occlusion, repinotan (10 μg/kg per hour) reduced infarct volume by 43%. In the transient middle cerebral artery occlusion model, repinotan (10 μg/kg per hour) administered immediately after occlusion reduced infarct volume by 97%, and a delay to 5 hours reduced infarct volume by 81%. In the acute subdural hematoma model, repinotan (3 and 10 μg/kg per hour) reduced infarct volume by 65%. In this model, repinotan (3 μg/kg per hour) administered 5 hours after occlusion reduced infarct volume by 54%. The favorable neuroprotective efficacy, broad dose–response curve, and prolonged therapeutic window observed in all models strongly suggest that repinotan is a promising candidate for treating acute ischemic stroke in humans.
Keywords
Introduction
Ischemic brain damage caused by stroke or traumatic brain injury (TBI) is characterized by an immediate depletion of cellular energy levels. Massive ion fluxes across the plasma membrane and breakdown of the energy-driven membrane potential induce liberation of neurotransmitters into the extracellular space, in particular the excitotoxin glutamate (Rothman and Olney, 1986; Baker et al, 1991; Shimizu et al, 1993; Takagi et al, 1993). Excess of glutamate leads to continuous activation of NMDA, AMPA, and metabotropic glutamate receptors, resulting in massive calcium influx and mobilization of intracellular calcium stores. This initial event triggers at least two fundamental mechanisms leading to cell death during ischemic brain injury; oxidative/nitrosative stress and apoptotic-like cell death. These mechanisms mediate injuries within neurons, glia, and vascular elements, and at the subcellular level they impact the function of mitochondria, nuclei, cell membranes, endoplasmic reticula, and lysosomes (Lipton, 1999; Lo et al, 2003).
Based on the prominent role of glutamate, strategies for development of neuroprotective drugs initially focused on glutamate receptors, which trigger the neurotoxic effects, that is, on NMDA receptors, including their modulatory sites, and AMPA receptors (Boxer and Bigge, 1997). So far, most glutamate receptor antagonists investigated in clinical trials revealed no therapeutic efficacy because of unfavorable risk–benefit ratio or lack of efficacy (Lees, 1997; De Keyser et al, 1999; Muir and Lees, 2003). Glutamate antagonists directly acting at the postsynaptic glutamate receptors show considerable side effects, including hypotension and psychotomimetic effects in humans, and can cause neurotoxic injury in animals (Olney et al, 1989; Kornhuber and Weller, 1997; Yenari et al, 1998). Newer compounds acting via modulatory sites at the NMDA receptor, like the glycine receptor antagonist GV 150526, do not produce neuronal vacuolization or cognitive disturbances. However, these approaches also were not effective. The only agents currently approved for treating acute stroke are thrombolytics, such as tissue plasminogen activator (tPA), which treats cerebral ischemia caused by blockage of an artery in the brain by a blood clot. However, this therapy is limited to a highly selected patient population (Wardlaw et al, 2003).
Consequently, alternative strategies have been devised to attenuate glutamate-mediated toxicity. These approaches include inhibition of second messenger cascades involved in glutamatergic signaling, blockade of ion channels, which may counteract excessive ischemia-induced neuronal depolarization, and mechanisms that induce hyperpolarization via the CB1 (Mauler et al, 2002, 2003a, 2003b) and 5-HT1A receptors (De Vry et al, 1997; Mauler et al, 2001). Recently, we have characterized repinotan hydrochloride (repinotan) as a highly potent and selective 5-HT1A full receptor agonist (De Vry et al, 1998). Repinotan induced a long-lasting but reversible inhibition of neuronal firing in vitro (De Vry et al, 1997) and in vivo (Casanovas et al, 2000), demonstrating the hyperpolarizing properties of repinotan.
In vivo repinotan displayed neuroprotective efficacy in animal models of stroke (Horvath et al, 1997; Semkova et al, 1998), transient ischemia (De Vry et al, 1997), and TBI (Horvath and Augstein, 1997; Alessandri et al, 1999). Further, repinotan reduces ischemia-induced glutamate release in vivo (Mauler et al, 2001). The aim of the present study was to further characterize the neuroprotective potential of repinotan in models of stroke and TBI.
Materials and methods
Chemicals and Reagents
All chemicals were of the highest commercially available purity and were, if not otherwise indicated, purchased from Merck KGaA (Darmstadt, Germany). Repinotan (R-(−)-2-{4-[(chroman-2-ylmethyl)-amino]-butyl}-1,1-dioxo-benzo[d]isothiazolone hydrochloride) was synthesized by the Department of Chemical Research, Bayer Health Care (Wuppertal, Germany).
Neuroprotection Studies
Animals
Male Long Evans rats (180 to 380 g, Møllegard & Bomholtgaard A/S, Ry, Denmark) were used for the permanent focal middle cerebral artery occlusion (pMCA-O) experiments. Wistar rats (230 to 300 g, Harlan-Winkelmann, Borchen, Germany) were used for the acute subdural hematoma (SDH) experiments. Male Wistar rats (HsdCpb:Wu, 300 to 350 g, Harlan-Winkelmann, Borchen, Germany) were used for the transient middle cerebral occlusion (tMCA-O) experiments. All animals were allowed to adapt to housing conditions for at least 1 week before the study. They were housed in groups of three to five individuals in macrolon cages (type III, Eeco, Castrop-Rauxel, Germany) bedded with sawdust. The animal housing room and the laboratory for surgery were climate controlled and continuously illuminated from 06:00 until 18:00. Room temperature was approximately 21°C, and relative humidity was approximately 50%. Food (Altromin 1324, Altromin Spezialfutterwerk GmbH, Lange, Germany or R/M-H; V1534–00 DDb, Ssniff Spezialdiäten GmbH, Soest, Germany) and water were available ad libitum. Experimental protocols and conditions conformed to German regulations on animal welfare.
Permanent middle cerebral artery occlusion
Under general anesthesia (Forene®, Abbott GmbH, Wiesbaden, Germany or Isofluran-Baxter, Baxter Deutschland GmbH, Unterschleißheim, Germany mixed with≅28% O2 in N2O to 5–1% v/v concentration), the MCA was occluded unilaterally based on the surgical procedure described by Bederson et al (1986). The left temporal–parietal region of the head was shaved, and the skin was disinfected and opened between the orbit and the external ear canal. After a midline incision, the temporal muscle was divided and pulled aside with surgical hooks to expose the lateral aspect of the skull. The facial nerve, major facial arteries and veins, lateral eye muscles, intra- and extraorbital lacrimal glands, and zygomatic bone were left intact. Under an operating microscope, a small burr hole was drilled directly under the zygomatic arc, 1 to 2 mm rostrally to its caudal origin from the aquamosal bone. After careful opening of the dura, the exposed MCA and its branches were permanently occluded between the olfactory tract and the inferior cerebral vein proximal to the lenticulostriate branch by microbipolar electrocoagulation (Bipolator 50, Fischer MET GmbH or Bipol 50, Stockert GmbH, Freiburg, Germany). To avoid recanalization, the occluded vessels were cut and removed. Muscle and skin wounds were closed with surgical suture or with tissue glue (Histoacryl, B. Braun Surgical GmbH, Melsungen, Germany). During surgery and drug infusion, body temperature was monitored using a rectal temperature probe and maintained between 36.5°C and 37.5°C with a heating pad. After recovery from anesthesia, the animals were returned to their home cage. At 7 days after surgery, the rats were decapitated, and their brains were rapidly removed and frozen in 2-methyl butane cooled to −30°C on dry ice. Serial coronal sections (20-μm thick) were cut throughout the entire infarcted area with a standard distance of 500 μm, using a cryostat microtome (Leica CM 3050, Leica Vertrieb GmbH, Bensheim, Germany and Micron HM 500 OM, Microm Laborgeräte GmbH, Walldorf, Germany). Slide-mounted brain sections were stained with cresyl fast violet. Cortical infarct volume was determined with a computer-assisted image analysis system (Optimas, BioScan Inc., Edmonds, WA, USA) by an operator blinded to the group composition. Infarct volumes were expressed in mm3 (mean±1 s.e.m.). For comparison of individual experiments, infarct volumes of treatment groups were expressed as percent of the respective controls, which were set to 100%.
Transient middle cerebral artery occlusion
The animals were anesthetized with the inhalation anesthetic isofluran (Forene®, Abbott GmbH, Wiesbaden, Germany or Isofluran-Baxter, Baxter Deutschland GmbH, Unterschleißheim, Germany mixed with≅28% O2 in N2O to 5–1.5% v/v concentration). Transient MCA-O was induced according to a standard surgical procedure (Zhao, 1995) with modifications described elsewhere (Mauler et al, 2003a). After midline opening of the skin and the right lateral neck muscles, the right common carotid artery was exposed. The external carotid artery together with the pterygopalatine artery and the common carotid artery were ligated. The internal carotid artery was closed temporarily with a microvascular clip. A silicone-coated nylon monofilament thread was inserted through a small incision made on the common carotid artery and advanced into the internal carotid artery up to and short over the origin of the MCA and secured in position by encircling sutures.
After 60 mins of ischemia, the occluding filament was withdrawn to allow reperfusion by the ipsilateral intracranial arteries. The skin wound was closed with sutures. During the surgery and continuous intravenous infusion of repinotan or vehicle, body temperature was maintained in the physiological range (37.0°C±0.5°C) with a warming pad. After recovery from anesthesia, the animals were returned to their home cage. Rats that did not show a typical circling behavior soon after the surgery were excluded from further study. After 2 days' survival, cortical and striatal infarct volume determination was performed as described above.
Acute subdural hematoma
Animals were anesthetized with isofluran (Forene®, Abbott GmbH, Wiesbaden, Germany or Isofluran-Baxter, Baxter Deutschland GmbH, Unterschleißheim, Germany mixed with≅28% O2 in N2O to 5–1% v/v concentration). Subdural hematoma was induced according to a standard surgical procedure (Miller et al, 1990) with the following minor modifications (Mauler et al, 2003a). The top of the head was shaved and the skin was disinfected and opened with a longitudinal midline cut. A small part of the periosteum was removed and a burr hole was drilled into the skull with the stereotaxic coordinates: −1 mm caudal, −2.8 mm lateral to the bregma (Paxinos and Watson, 1996). The dura was carefully opened and a specially designed plastic cannula was inserted into the subdural space between the dorsal surface of the brain and the dura. Thereafter, the cannula was fixed in position with tissue glue (Histoacryl, B. Braun Surgical GmbH, Melsungen, Germany). Non-heparinized autologous blood was collected by tail vein puncture and injected via the prefixed cannula directly into the subdural space (total volume of 0.2 mL within 4 mins). The probe was shortened and closed with cyanacrylate glue (Histoacryl). The skin wound was closed with suture clips. During the surgery and continuous intravenous infusion of repinotan or vehicle, body temperature was monitored and maintained within the physiological range (37.0°C±0.5°C) using a warming pad. After recovery from anesthesia, the animals were returned to their home cage. After 7 days, survival the cortical infarct volume determination was performed out as described above.
Drug Application
Repinotan was dissolved in 0.9% NaCl solution. Ready-made solution was administered either as bolus intravenous injection (immediately, 2, and 4 hours after brain injury) or as a 4-hour intravenous infusion; the application volume was 2 and 4 mL/kg per hour, respectively. In all experiments, control animals received the same volume of vehicle as the verum groups.
Data Analysis
Differences of means for neuroprotection and brain edema studies were assessed by analysis of variance (ANOVA) followed, where appropriate, by post hoc least-significance difference (LSD) comparison (SYSTAT Version 10 SPSS Inc.). P≤0.05 was defined as the level of significance. If not otherwise indicated for all in vivo experiments, n was 8 to 12/group.
Results
Permanent Middle Cerebral Artery Occlusion
When repinotan was administered as an intravenous bolus, immediately after occlusion, followed by additional injections 2 and 4 hours after occlusion (triple bolus), mean reductions in infarct volume were 53.9%±11.9%, 73%±9%, 54.5%±6.8%, and 41%±10.3% at doses of 1, 3, 10, and 30 μg/kg, respectively (Figure 1A). A decrease in efficacy was observed at lower (0.3 μg/kg; 34.1%±5.6% infarct volume reduction) and higher (100 μg/kg; 36.8%±10.5% infarct volume reduction) doses. A dose-dependent neuroprotective efficacy also could be observed when repinotan was administered as a 4-hour infusion starting immediately after pMCA-O. The highest degree of neuroprotection was observed at doses of 0.3, 1, 3, 10, and 30 μg/kg per hour (47.6%±8.1%, 58.9%±8.4%, 65.4%±9.1%, 65.4%±6.7%, 51.2%±11.0% infarct volume reduction, respectively). A decrease in efficacy also could be observed at lower (0.03 μg/kg; 26.8%±10.4% infarct volume reduction) and higher doses (100, 300 μg/kg; 41.1%±6.5%, 30.5%±9.6% infarct volume reduction, Figure 1B). The therapeutic time window was determined from a series of experiments in which the start of the intravenous infusion was delayed after pMCA-O. Delaying the start of the infusion by 1 hour did not result in reduced neuroprotective efficacy of repinotan (Figure 2), compared with immediate treatment, and a 3-hour treatment delay resulted in a 48% reduction in infarct volume at 3 μg/kg per hour (Figure 2). When repinotan (3 μg/kg per hour) administration was delayed to 5 hours after pMCA-O, mean infarct volume was reduced by 25% (NS). However, when the repinotan dose was increased to 10 μg/kg per hour, mean infarct volume reduction was 43% (P<0.05) (Figure 2).
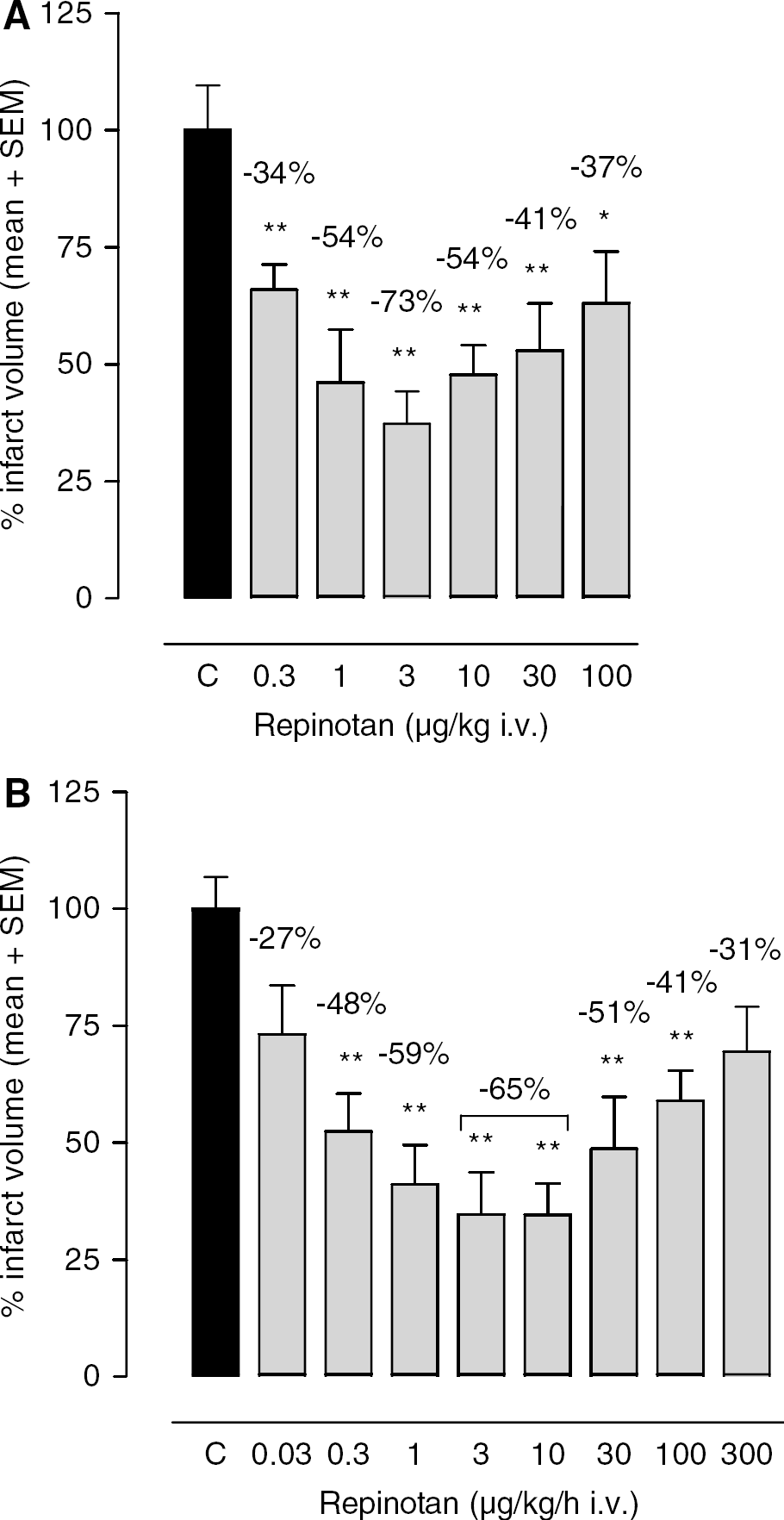
Neuroprotective efficacy of repinotan in the rat permanent MCA-O model. Repinotan was administered (
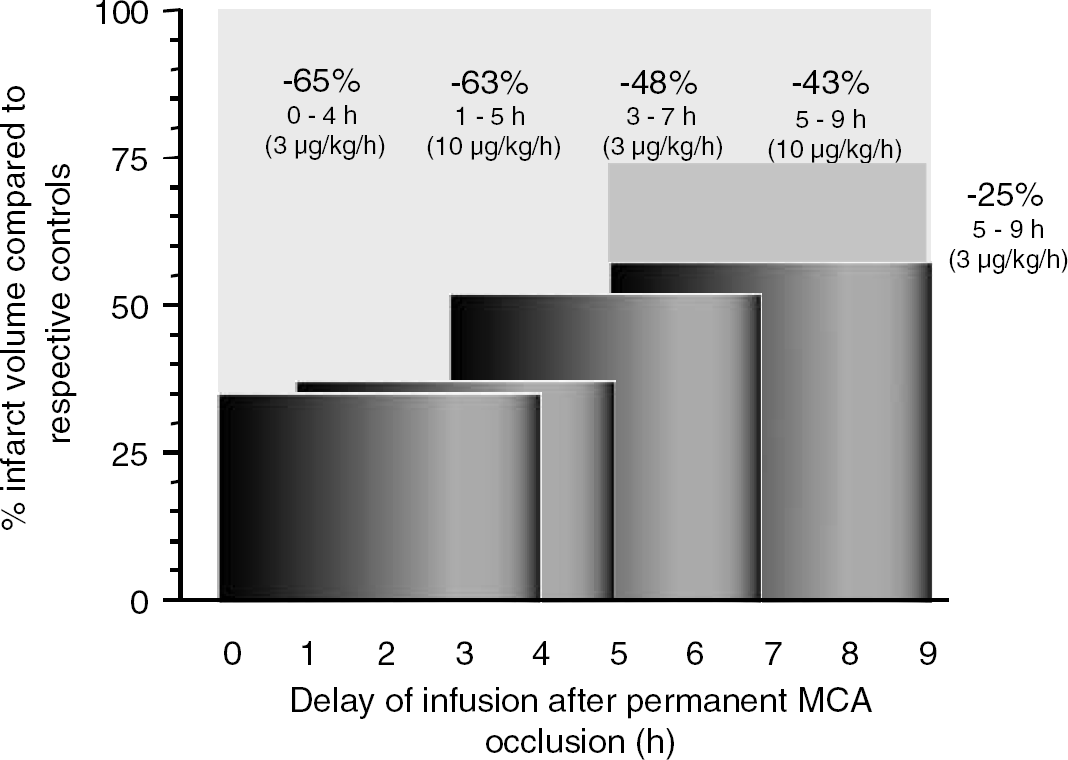
Therapeutic time window for intervention with repinotan after intravenous infusion in the rat model of pMCA-O. Repinotan was administered as a continuous 4-hour infusion either immediately, 1, 3, or 5 hours after occlusion of the MCA. Doses used were 3 μg/kg per hour for the 3-hour delayed treatment and 10 μg/kg per hour for the 1 and 5-hour delayed treatment.
Transient Middle Cerebral Artery Occlusion
Cortical brain damage resulting from reperfusion injury induced by 1 hour occlusion of the MCA was reduced by 81%±6.6%, 97%±1.3%, and 84%±7.1% at 1, 10, and 100 μg/kg per hour, respectively, when repinotan was administered as a 4-hour continuous intravenous infusion immediately after the onset of reperfusion (Figure 3). A similar reduction in infarct volume was observed when the start of infusion was delayed for 3 or 5 hours (Figure 4) when the repinotan dose was held constant at 10 μg/kg per hour.
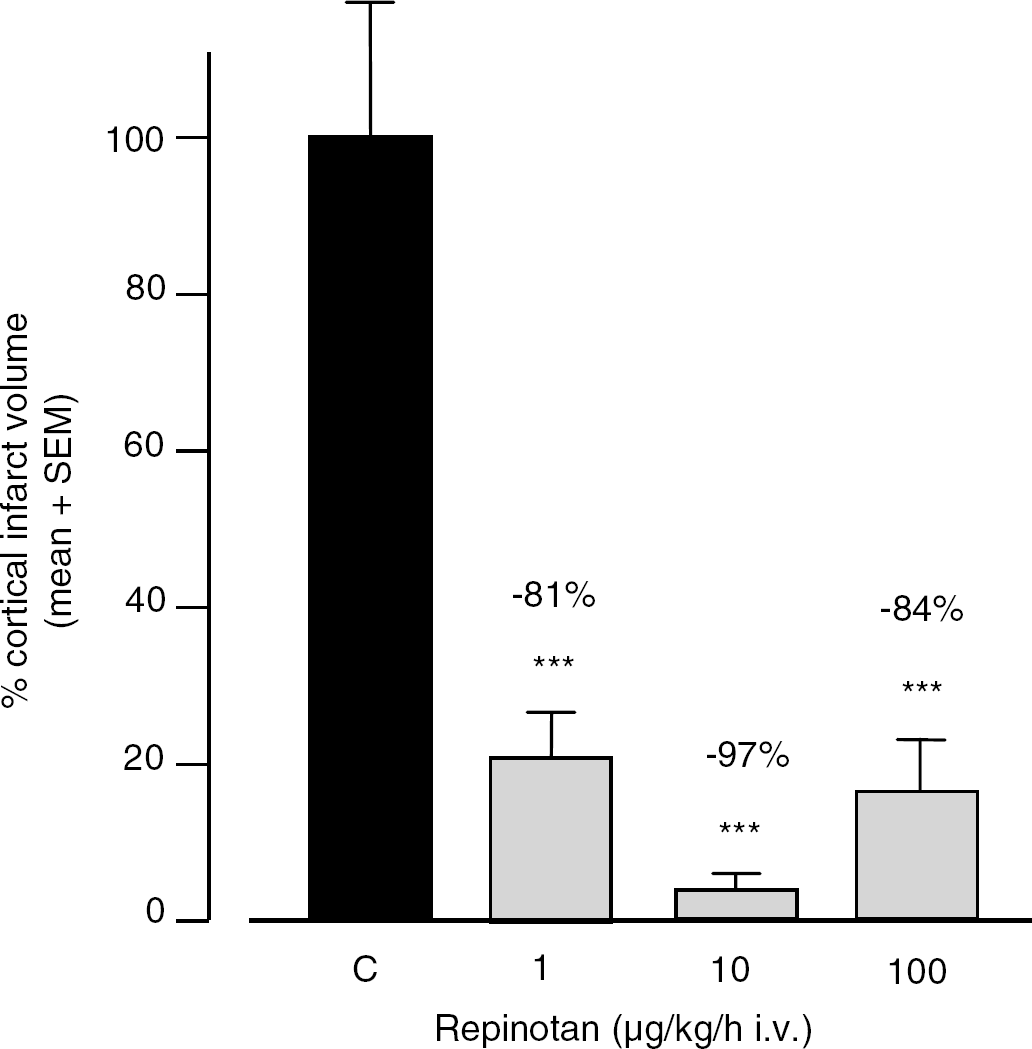
Neuroprotective efficacy of repinotan in the rat transient MCA-O model. Repinotan was administered as a 4-hour infusion, starting immediately after reopening of the 1-hour occlusion. Infarct volumes were determined 2 days after tMCA-O. Infarct volumes were calculated as the percentage of infarct volumes of the control group, which was set to 100%. C=control. *P⩽0.05, **P⩽0.01 (for (A) n=30 for C and 9–21 for all other groups, for (B) n=40 for C and 10 for treatment groups, ANOVA followed by post hoc LSD comparison).
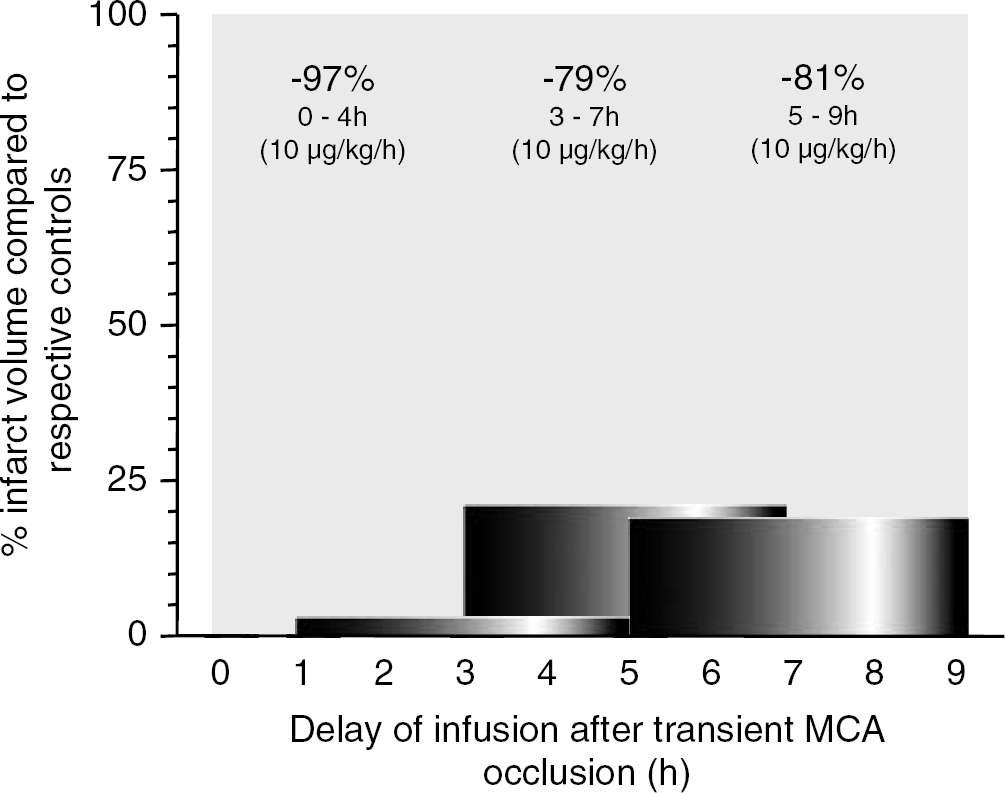
Therapeutic time window for intervention with repinotan after intravenous infusion in the rat model of tMCA-O. Repinotan was administered as a continuous 4-hour infusion either immediately, 3, or 5 hours after transient occlusion of the MCA. Dose was 10 μg/kg per hour.
Acute Subdural Hematoma
A reduction in cortical damage was achieved after repeated postinjury intravenous bolus injections of repinotan, immediately and 2 and 4 hours after surgery. Infarct volumes were reduced by 60%±15.5% and 64%±15% at repinotan doses of 10 and 100 μg/kg, respectively (Figure 5A). When repinotan was administered as a 4-hour continuous intravenous infusion immediately after the induction of SDH, infarct volumes were significantly (P<0.05) reduced by 76%±7.2%, 69%±7.8%, 63%±10%, and 43%±15% at repinotan doses of 1, 10, 100, and 300 μg/kg per hour, respectively (Figure 5B). At lower (0.1 μg/kg per hour) or higher doses (1000 μg/kg per hour) the efficacy of repinotan decreased, resulting in nonsignificant reductions of infarct volume of 40 and 15%, respectively. Thus, continuous intravenous infusion of repinotan 3 μg/kg per hour resulted in infarct volume reductions of 74 and 54% after a treatment delay of 3 and 5 hours, respectively (Figure 6).
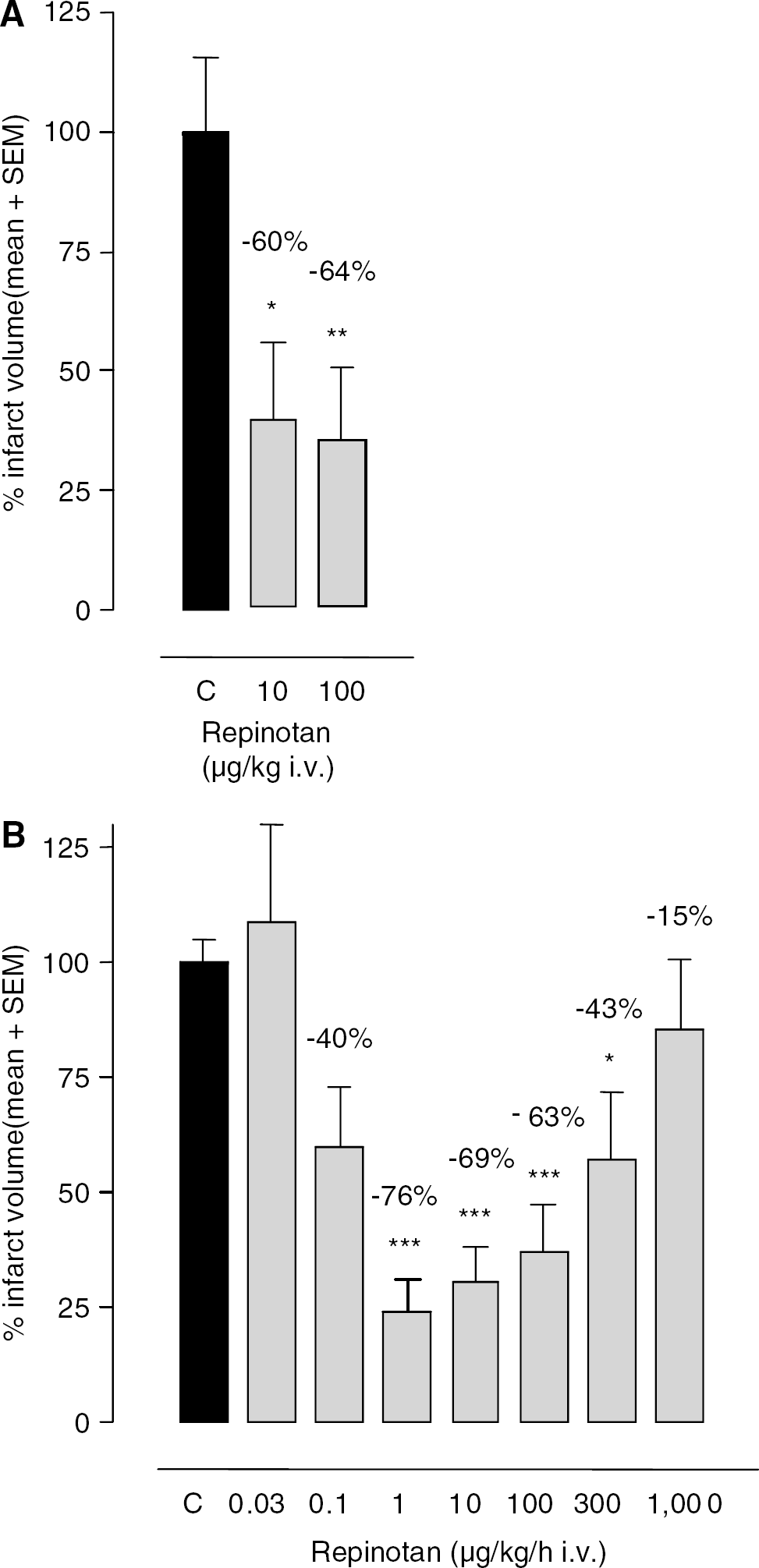
Neuroprotective efficacy of repinotan in the rat SDH model. Repinotan was administered (
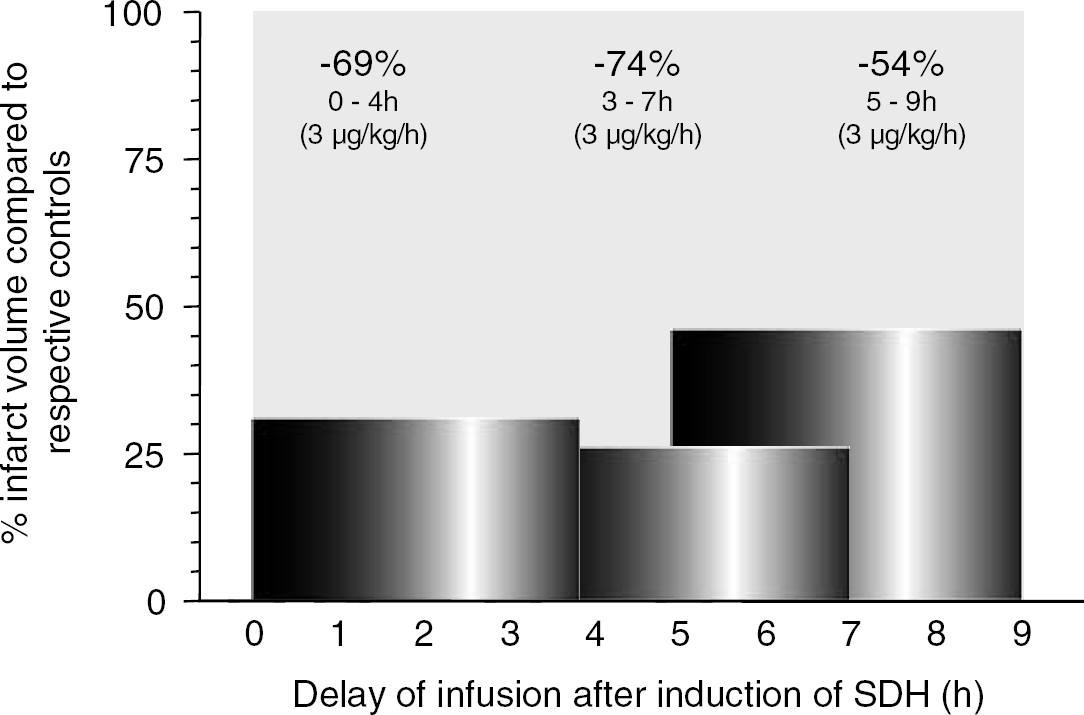
Therapeutic time window for intervention with repinotan after intravenous infusion in the rat model of TBI. Repinotan was administered as a continuous 4-hour infusion either immediately, 3, or 5 hours after transient occlusion of the MCA. Dose used was 3 μg/kg per hour.
Discussion
Previous studies of repinotan in the pMCA-O, tMCA-O, and SDH models in rats have demonstrated the neuroprotective potential of repinotan (De Vry et al, 1997; Horvath and Augstein, 1997; Horvath et al, 1997; Alessandri et al, 1999). This study focused on further elucidating the neuroprotective efficacy of repinotan using in vivo models of ischemic stroke and TBI. We have chosen three accepted models that mimic different aspects of brain injury: (i) permanent focal cerebral ischemia by occlusion of the middle cerebral artery (pMCA-O) because an infarct in the region of the MCA is the most common cause of stroke in humans (Mohr et al, 1986); (ii) reperfusion injury model by tMCA-O since many infarcts slowly reperfuse over time (Ringelstein et al, 1992) or recanalization is induced by thrombolytics, such as rtPA; (iii) a TBI model (SDH) in which other mechanisms such as reduction of regional cerebral blood flow in the neighboring cortical tissue because of direct compression of SDH and inflammatory processes (at least in the initial stage) (Holmin et al, 1995) play an important role.
Further, we have determined the therapeutic window for repinotan administration in each model. This is extremely important, because of the considerable delay that occurs from the onset of stroke to hospitalization, diagnosis, and first pharmacological intervention.
In general, repinotan displayed neuroprotective efficacy in the rat pMCA-O, tMCA-O, and SDH models. In some experiments, the dose–response curve suggested a wide U-shaped curve, which has previously been observed with repinotan (Horvath et al, 1997) and other compounds (Mauler et al, 2002).
In the pMCA-O and the SDH model, the neuroprotective efficacy of repinotan was independent of the dosage regimen. However, when administered as a 4-hour continuous infusion, repinotan appeared to be more potent in the pMCA-O model and exhibited a broader dose–response curve and larger dose variability. This is best explained by the assumption that brain levels of repinotan are maintained in the optimal range throughout the total infusion time, which is not the case with intravenous bolus administration. More importantly, data from the current studies suggest rapid penetration of repinotan into the brain. Because the technique used in the tMCA-O model is highly sophisticated, repinotan was investigated in this model only using the continuous infusion regimen. As seen in the other investigative models, repinotan was highly effective over a wide dose range (1–100 μg/kg per hour). Although we have not performed a complete dose–response analysis in the SDH model using intravenous bolus administration, the data obtained suggest similar differences in potency and efficacy in the SDH model as in the pMCA-O model.
Based on these data, we have used the continuous infusion regimen to investigate the therapeutic window. Repinotan displayed a therapeutic window of at least 5 hours in all models. At different time points, either the neuroprotective efficacy decreased when the administered dose remained the same (i.e. tMCA-O, SDH) or the dose had to be increased (i.e. pMCA-O) to yield neuroprotective effects comparable to those seen after immediate treatment. One can assume that this reflects emergent pathophysiological processes, such as Ca2+-independent glutamate release (Wahl et al, 1994) or glutamate liberation from non-neuronal and/or disrupted cells, which cannot be fully attenuated by repinotan.
It is difficult to view our results in the context of other published data, because the techniques, models, administration regimens, and results reported in the literature are highly diverse, as are the mechanisms of action of the tested drugs. Such compounds as ZK200775 (Turski et al, 1998), YM872 (Takahashi et al, 1998), NXY-059(Sydserff et al, 2002), FK506 (Furuichi et al, 2003), or SM-20220 (Horikawa et al, 2001) attenuate different mechanisms in the pathogenesis of stroke and exhibit therapeutic windows of 2 to 4 hours in either the pMCA-O or tMCA-O model, but were not evaluated in the SDH model. However, when administered as a continuous intravenous infusion, repinotan is the only compound that displays a therapeutic window of at least 5 hours in all models. In addition, as shown for the pMCA-O model, nonsignificant effects can be overcome by increasing the repinotan dose.
The broad dose–response curve obtained in investigations using immediate administration of repinotan suggests that the therapeutic window might be larger than 5 hours when the dose is increased in delayed administration models. These data also support the assumption that repinotan can penetrate the blood–brain barrier even at late time points when the blood–brain barrier might be closed, suggesting favorable pharmacokinetic properties of repinotan.
Our data provide evidence that repinotan triggers a single mechanism that could attenuate the pathophysiological processes over a long time period or, more likely, that the 5-HT1A receptor triggers several processes that attenuate infarct formation at varying time points. As mentioned, activation of presynaptic 5-HT1A receptors located on glutaminergic terminals mediates cell hyperpolarization and inhibition of glutamate release in vitro and in vivo (Raiteri et al, 1991; Matsuyama et al, 1996; De Vry et al, 1997; Dong et al, 1998; Casanovas et al, 2000; Mauler et al, 2001). The ensuing neuronal hyperpolarization results in activation of G protein-coupled inwardly rectifying K+ channels (Andrade, 1992). Additionally, a recent study reported that activation of 5-HT1A receptors also may provide neuroprotection by directly interacting with voltage-gated Na+ channels to reduce Na+ influx (Melena et al, 2000). These mechanisms could explain the neuroprotective efficacy of repinotan per se, but not necessarily the efficacy observed when administration is delayed.
Thus, it is likely that other processes involved in the pathogenesis of brain injury are also attenuated by repinotan. It has been shown that activation of 5-HT1A receptors by repinotan attenuates several pathways, which are involved in either neuroprotection or recovery of neuronal function. When the neuroprotective potency of repinotan was investigated in vitro against glutamate-induced degeneration of hippocampal slices, repinotan reduced the number of damaged neurons and preserved cell morphology and integrity of the neuronal network (Semkova et al, 1998). Moreover, repinotan inhibited or reduced apoptosis induced by serum deprivation in cultured neurons (Ahlemeyer et al, 1999), staurosporine-induced apoptosis in primary cultures of hippocampal and cortical neurons (Suchanek et al, 1998; Ahlemeyer et al, 2000), and apoptosis of hippocampal and striatal neurons after transient forebrain ischemia in the rat (Schaper et al, 2000). All of these effects could be blocked by the specific 5-HT1A receptor antagonist WAY 100635, again showing a specific 5-HT1A receptor-mediated effect. Further, repinotan also attenuated the levels of death-inhibiting protein BCL-2 in rat brains during ischemia/reperfusion (Kukley et al, 2001) and levels of S-100β, a serotonergic glial growth factor that has been reported to protect cultured neurons against glutamate-induced damage (Ahlemeyer et al, 2000). Both of these mechanisms may contribute to the neuroprotective efficacy of repinotan. Recently, it was shown that repinotan also suppresses caspase-3 through MAPK and protein kinase Cα (Adayev et al, 2003), which is assumed to be a new pathway leading to neuroprotection. However, so far, the detailed mechanisms leading to neuroprotection after delayed administration of repinotan cannot be ruled out.
The main purpose of a neuroprotective therapy must be to reduce the severity of initial damage and improve neurological outcome weeks and months after a cerebral ischemic event. In fact, repinotan significantly improved and accelerated the recovery of abnormal hindlimb grasping reflex and forelimb flexion compared with vehicle-treated controls (Gerlach et al, 1998) for up to 42 days after brain injury. Supportive data show that repinotan attenuates NMDA-induced delayed neuronal death in rat magnocellular nucleus basalis when administration is delayed for 2 or 3 days after injury (Harkany et al, 2001). Extended therapeutic efficacy of repinotan has been observed in a contusion model of experimental brain injury. In this study, it was shown that repinotan conferred neuroprotection and attenuated spatial learning deficits after controlled cortical impact injury (Kline et al, 2001).
The current experiments establish the efficacy and define the therapeutic time window of repinotan in animal models of acute stroke and TBI. In addition, the results of the current and previous studies show that the neuroprotective efficacy of repinotan is related to a 5-HT1A receptor-mediated attenuation of ischemia in the first phase after cerebral infarction. Further, results suggest that repinotan also triggers additional 5-HT1A-mediated mechanisms that become effective at a later time point, as shown by investigations focusing on long-term beneficial effects. These in vivo results indicate that repinotan is a promising candidate for neuroprotective therapy of brain-injured patients suffering from acute ischemic stroke.