
Abstract
Macrophage migration inhibitory factor (MIF) is involved in various immune-mediated pathologies and regulates both innate and adaptive immune reactions, thus being related to several acute and chronic inflammatory diseases such as rheumatoid arthritis, septic shock, and atherosclerosis. Its role in acute and chronic brain pathologies, such as stroke and neurodegenerative diseases, has attracted increasing attention in recent years. In response to stimuli like hypoxia, inflammation or infection, different cell types can rapidly release MIF, including immune cells, endothelial cells, and neuron cells. Notably, clinical data from past decades also suggested a possible link between serum MIF levels and the severity of stroke and the evolving of neurodegenerative diseases. In this review, we summarize the major and recent findings focusing on the mechanisms of MIF modulating functions in brain injury and neurodegenerative diseases, which may provide important therapeutic targets meriting further investigation.
Keywords
Introduction
Macrophage migration inhibitory factor (MIF), a 12.5 kD protein that inhibits macrophage migration, has been primarily recognized as a product of T lymphocytes. 1 Later, rapidly growing evidence revealed that MIF can be broadly expressed in various cell types, including macrophages, smooth muscle cells, endothelial cells, and neuron cells.2–4 The physiopathological roles of MIF have come to light subsequently. It’s an inflammatory cytokine with chemokine-like functions that is involved in several immune-mediated pathologies and is responsible for inflammatory reactions throughout the central nervous system.5–9 The role of MIF in pathological conditions appears to be diverse with both protective and detrimental effects. For example, MIF defective animals showed decreased neuronal death and improved recovery from transient middle cerebral artery occlusion (tMCAo) in the brain, 10 suggesting its detrimental role in acute cerebrovascular diseases.11,12 However, both ischemic and hemorrhagic models could benefit from MIF overexpression by attenuating oxidative stress.13,14 Furthermore, evidence from both clinical studies and basic research have suggested that MIF could be the biomarker to predict the outcome of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). The purpose of this review is to provide an overview of the research on MIF, with a particular focus on the context of brain injury and neurodegenerative diseases.
Regulatory mechanisms of MIF expression under different pathological contexts
The MIF gene is located on human chromosome 22 (22q11.2) and 10, 15 where functional polymorphisms -794 CATT 5-8 microsatellite repeat (rs5844572) and -173 G > C single nucleotide polymorphism (SNP) (rs755622) control MIF promoter activity and MIF expression. 16 It is expressed at a high level in various parts of the rat brain, including the cortex, hypothalamus, and hippocampus. 16 MIF’s expression can be increased in vivo following physiological and pathological stimuli like hormones, glucose, cytokines, and microbial products. 17 For instance, after lipopolysaccharide (LPS) injection, both MIF mRNA and protein levels were increased and quickly released into the cerebrospinal fluid of the rat, where cytokines such as IL-1β and IL-6 could be detected. 16 A similar elevation of gene expression was also found in a LPS treated marine invertebrate model. 18 What’s more, Rice et al. demonstrated that angiotensin II stimulated MIF mRNA and protein production, which induced a rapid release of 50% of MIF protein within 20 minutes in the rat proximal tubular epithelial cells. 19 MIF, as previously reported, can boost transcription factor nuclear factor kappa B (NF-κB) expression, which therefore has a pro-inflammatory effect, and increasing NF-κB levels triggers MIF gene transcription, creating a feedback loop. However, these effects were context dependent, since NF-κB decreased MIF expression under hypoxia conditions, then diminished the neuronal protective effect of MIF in a cerebral ischemia mouse model. 13 Thus, the regulation of MIF expression is dependent on the different cellular background during physiological or pathological processes.
The intracellular downstream signals of MIF
The MIF homologous receptor, a cluster of differentiation 74 (CD74) was discovered in the early twentieth century. 20 It is a type II transmembrane protein required for MIF-induced mitogen-activated protein kinase (MAPK) cascade activation, cyclooxygenase 2 (COX2) and prostaglandin E2 (PGE2) production, as well as cell proliferation. 20 CD44 or C-X-C Motif containing chemokine receptor 2 (CXCR2), CXCR4, and CXCR7 are examples of co-receptors.21,22 MIF can activate the CD74/CD44 complex via tyrosine-protein kinase (SRC) and phosphorylate downstream MAPK family signals such as ERK1/2, PI3K, and protein kinase B (PKB), also known as AKT.20,23–25 Then, NF-κB and activator protein-1 (AP1) will be activated, 26 resulting in the release of pro-inflammatory cytokines like IL-6, IL-8, IL-10, and TNF-α. 27 Furthermore, MIF functions as a link between NF-κB and the tumor suppressor protein p53, as p53-dependent macrophage apoptosis can be inhibited by MIF, allowing the macrophage to retain its proinflammatory properties. 28 In addition, MIF binding to CXCR2 and CXCR4 is linked to the activation of mononuclear cell chemotaxis, whereas CXCR7 is linked to the ERK- and zeta-chain-associated protein kinase (ZAP)-70 signaling pathways, as well as B-lymphocyte migration.21,22 When CXCR7 and CXCR4 combine to form homo- and heterodimers, they are able to bind CXCL12 with a much more affinity than CXCR4 and subsequently mediate G-protein signaling.29,30 The significance of CXCL12/CXCR4/CXCR7 signaling in prostate cancer of obese mice has been clarified. 31 As a result, it can be assumed that high amounts of MIF may interfere with the CXCL12-CXCR4/CXCR7 axis by competing for binding sites especially after the stroke, 32 and it has been discovered that the MIF/CXCR7/AKT pathway promotes the development and metastasis of castration-resistant prostate cancer. 33
MIF upregulated the expression of toll-like receptor 4 (TLR-4) in response to LPS and Gram-negative bacteria, which was necessary for the production of proinflammatory cytokines such as TNF-α, IL-6, and IL-12.34–36 Moreover, MIF is involved in the inflammatory response of NLR family pyrin domain containing 3 (NLRP3) inflammasome. 37 Macrophages and dendritic cells (DCs) produce less IL-1α, IL-1β and IL-18 when NLRP3 inhibits MIF activation. 38 Hypoxia-inducible factor-1 (HIF-1) increases the transcriptional expression of MIF, and the elevated MIF level prevents HIF-1 degradation by interacting with constitutive photomorphogenesis 9 (COP9) signalosome subunit 5 (CSN5), which stabilizes HIF-1 by inhibiting its prolyl-564 hydroxylation.28,39 What’s more, MIF inhibits the anti-inflammatory and immunosuppressive effects of glucocorticoid (GC) via MAPK phosphatase.40,41 Figure 1 concludes the MIF-mediated immune response signaling transduction.
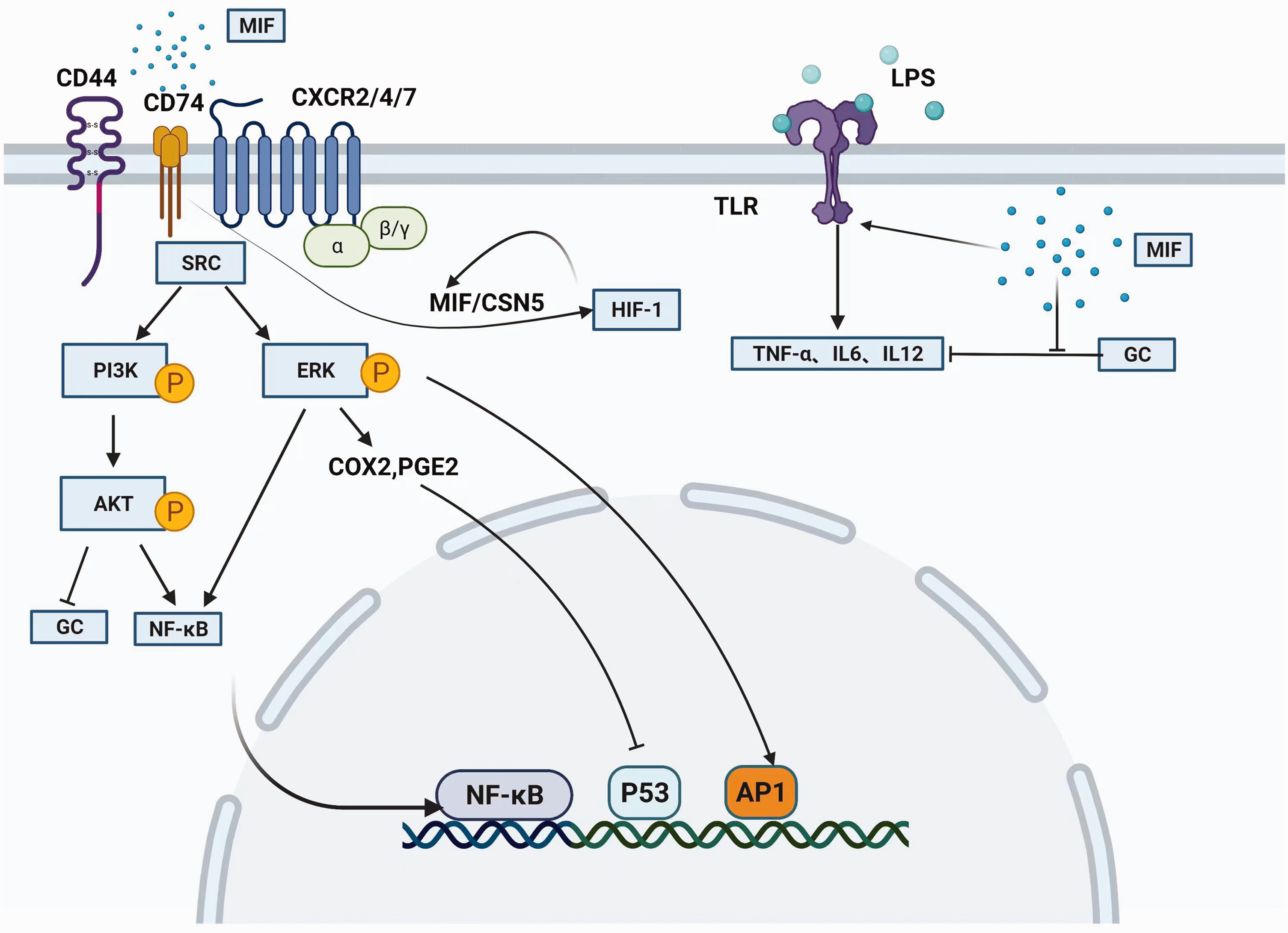
The overview of MIF-mediated cellular signaling transduction. MIF binds the ligand-binding protein CD74 and activates MAPK cascade, COX2, and PGE2. MIF activates the CD74/CD44 complex via SRC and phosphorylates downstream ERK1/2, PI3K, and protein kinase B. NF-κB and AP1 then be activated to induce the release of IL-6, IL-8, IL-10, and TNF-α, while the tumor suppressor protein p53 is inhibited by NF-κB. HIF-1 increases the expression of MIF, and the elevated MIF stabilizes HIF-1 in turn by preventing its degradation through CSN5. MIF suppresses GC immunosuppressive effects via MAPK phosphatase and upregulates TLR-4 expression in response to LPS stimuli, then induces the production of downstream proinflammatory cytokines such as TNF-α, IL-6, and IL-12. MIF: macrophage migration inhibitory factor; MAPK: mitogen-activated protein kinase; COX2: cyclooxygenases 2; PGE2: prostaglandin E2; SRC: tyrosine-protein kinase; ERK: extracellular regulated protein kinases; PI3K: Phosphoinositide 3-kinase; AP1: activator protein-1; IL: interleukin; TNF: tumor necrosis factor; HIF-1: Hypoxia-inducible factor-1; CSN5: constitutive photomorphogenesis 9 signalosome subunit 5; GC: glucocorticoid; TLR-4: toll-like receptor 4. The figure were created with BioRender.com.
Divergent pathological mechanisms of MIF in different brain resident cells
Double side effects on neuronal cells
In addition to serving as an immune mediator, MIF has a direct influence on neuronal cells. The protective effects of MIF on neuronal cells via blocking microglial activation were discovered at the end of last century. 42 Sumners et al. then conducted a series of studies using MIF to investigate its function on neurons. 43 They found that MIF, via its intrinsic thiol-protein oxidoreductase (TPOR), acted as an intracellular negative regulator of neuronal activity after the stimulation of Angiotensin II (Ang II), the hormone responsible for increased blood pressure.44–46 This negative regulation was insufficient in spontaneously hypertensive rat neurons, because increased Ang II level did not elevate the expression of MIF and led to the chronotropic effect of Ang II. 47
Zis et al. discovered that the MIF content in cerebral blood vessel endothelial cells was significantly increased in ischemic stroke patients, and that the protective property was also observed in cultured cortical neuron model of oxygen-glucose deprivation (OGD) to mimic ischemia, treatment of MIF for 12 hours prevented OGD-induced cell death. 48 MIF depletion via shRNA in vitro or genetically modified animals was used to examine the effect of MIF on neuron cell death. The absence of MIF increased neuronal cell apoptotic caspase 3/7 activity,13,49 and its presence rescued neurons from death by shifting the pro-/anti-apoptosis pathway balance to the survival side. 50 MIF treatment improved the proliferation and survival of neural stem/progenitor cells via many pathways, including PKB and extracellular regulated protein kinases (Erk), thereby extending these benefits to neural stem/progenitor cells.49,51
In contrast to the positive effects, the detrimental aspects of MIF were also significant. Eric et al. found that after a single injection of 200 ng exogenous MIF into the mouse hippocampus, the pharmacological kinetics of induced N-methyl-D-aspartic acid receptor (NMDA) responses in the apical dendrites of hippocampal CA1 pyramidal neurons were altered for more than two weeks, which may be responsible for sequelae after CNS injury. 52 In another rat diffuse axonal injury model, cortex MIF levels were considerably elevated 3 hours after injury and were primarily localized in neurons, intracerebroventricular injection of the MIF antagonist ISO-1 reduced neuronal apoptosis and axonal injury. 53 The similar neuronal cell death promotional effects of MIF were found to be involved in other studies, including the traumatic brain injury (TBI) model, which led to more severe neurodegeneration and neurologic deficits.10,54
MIF accumulates in astrocytes and regulates its activity
The inhibitory effects of MIF on the reactivity of astrocytes were discovered as early as 1996. 55 In addition, MIF was found to be accumulated in astrocytes at the blood brain barrier (BBB) in the Borna disease virus (BDV) infected rat brain, where it modulates inflammation during virus-induced encephalitis. However, during BDV-induced inflammation, MIF in astrocytes was not produced at the gene level since MIF mRNA was absent in the experimental groups. 56 Recently, Okazaki and colleagues discovered that antipsychotics increased MIF expression in astrocytes via increasing the acetylation of H3K27 in the MIF promoter, indicating that MIF may play a role in the pathophysiology of schizophrenia disease and the working mechanisms of antipsychotics. 57
The promoting effects of MIF on astrocyte activity were investigated at the cellular level as well. Svenningsen et al. found that MIF and Serine protease high-temperature requirement A1 (HTRA1), a tumor suppressor implicated in cell growth and cerebral small vessel disease, 58 were both expressed in cultured mouse astrocytes. 59 HTRA1 can reduce astrocyte migration, whereas MIF inhibits this influence, which has potential benefits for CNS development and disease treatment. 59 The enhanced astrocyte proliferation phenomenon was also observed in a TBI mouse model, MIF concentrations were increased after traumatic brain injury and associated with poor outcome, and the pre-administration of MIF antagonist might prevent TBI induced astrocytosis. 60
MIF produced by microglia and involved in its inflammatory response
Previous studies have suggested that spinal microglia, not invading immune cells, create MIF in inflammatory hyperalgesia.61,62 In primary cultured spinal microglia cells, MIF administration boosted COX2 expression and PGE2 production, while CD74 deletion completely reversed these effects. 63
Overall, the interactions of MIF with microglia are detrimental. The inhibition of MIF tautomerase activity downregulated LPS-induced nitro oxide (NO) and TNF-α production in microglial cells, indicating that MIF is involved in microglia-mediated neuron inflammatory pathology. 64 Zheng et al. found that Z-312, a novel MIF inhibitor, reduced proinflammatory factors produced by LPS-stimulated microglial cells in another LPS-induced PD mouse model. 65 Not surprisingly, MIF induced neuroinflammation and Parkinson's disease-like symptoms when microglial autophagy function was inhibited. 66 In addition to PD, the MIF level in glioblastoma patients was significantly elevated, and its expression in malignant gliomas exceeded that of normal brain tissue. The suppression of the MIF/CD74 pathway promoted interferon (IFN)- secretion in microglia, resulting in the death of tumor cells and a change from M2 to M1 in microglia associated with glioma. 67 Infusion of MIF into the animal hippocampus decreased microglia activation and prevented the production of microglia-derived tissue plasminogen activator, thereby protecting against excitotoxic neuronal cell death. 42
MIF involved in cerebrovascular structural cells dysfunction
Endothelial cells can release and respond to MIF when proatherogenic stimuli are present because they play a crucial structural function in blood vessel walls. Consequently, they can contribute to a number of vascular disorders, including atherosclerosis, retinopathy, and pulmonary hypertension.68–70
MIF was expressed in the cerebral micro-vessels in the peri-infarct area of 10 acute stroke patients, in contrast to negative findings in normal brain. 48 The elevated expression of MIF in both the MCAo and OGD tests led to the discovery that MIF may stimulate angiogenesis via the AKT and ERK signaling pathways in rat brain microvascular endothelial cells. 71 MIF levels in the blood of 39 stroke patients were also shown to be elevated; the subsequent investigation indicated that MIF injection disrupted tight junction in rat OGD-treated brain endothelial cells (ARBECs) and compromised BBB integrity in a tMCAo mice model. 72 In experimental cerebral malaria produced by Plasmodium berghei ANKA, brain endothelial cells that displayed CD74 signaling contributed to the cross-presentation of antigens to CD8+ T lymphocytes, and pharmacologic Plasmodium MIF antagonism protected against cerebral malaria. 73
The mural cells of brain microvasculature is composed of pericytes and vascular smooth muscle cells (VSMCs).74,75 Previous studies demonstrated that human placental NG2+ pericytes are the primary source of MIF in response to pathogenic activation, and these pericytes also offer migratory pathways for extravasated leukocytes in rat mesentery tissue and in vitro EC-PC barrier model.76–78 Human VSMCs are likewise the secretory source of MIF under hypoxic conditions via HIF-1 activation, 79 with vascular remodeling properties through VSMC differentiation and proliferation interference in carotid and pulmonary arteries.80,81 Nevertheless, the interaction of MIF with cerebrovascular pericytes and VSMCs remains limited. Greater comprehension of this topic will allow us to prevent BBB damage and hasten the recovery of the neurovascular unit after a stroke. The interaction of MIF with brain resident cells in experimental stroke were concluded in Table 2.
The function of MIF on brain injury
Dualistic roles of MIF on ischemic stroke
Although MIF depletion had no effect on cytokine levels in the brain or serum of mice one week after tMCAo, 82 the detrimental effect of MIF on stroke recovery was found elsewhere. 10 All the underlying mechanisms, including pro-inflammation, enhance immune reaction and accelerate neuron cell death, inhibit the recovery of post-stroke physiological function.10,83–85 MIF levels were found to be considerably higher in acute ischemic stroke patients and in the experimental tMCAo model. The serum MIF levels of patients were positively correlated with infarct volume and long-term outcomes, while treatment of MIF increased BBB permeability in animal models following tMCAo.72,86 Acute ischemic stroke patients with higher serum MIF concentrations at admission were more likely to have poor clinical outcome, post-stroke depression (PSD) and recurrent stroke in the long term,87–89 and its major receptor CD74 was also found to have a similar predictive role in ischemic stroke patients. 90 In a different mouse focal ischemia model, knocking down MIF or inhibiting its nuclease activity decreased infarct volume and improved behavioral performance. 91 Based on the nuclease property, the most recent work utilizing a photothrombotic ischemia model demonstrated that MIF acetylation on the K78 residue decreased the translocation and nuclear intensity of MIF, therefore providing a therapeutic target for protecting neurons in ischemic stroke. 92
MIF, on the other hand, has a neuroprotective effect in ischemic stroke by inhibiting neuronal cell death and inducing brain-derived neurotrophic factor (BDNF) expression.12,50,93 It was found that 60 ng/mL MIF administration was most effective for BDNF expression and inhibiting neuronal apoptosis. 94 MIF promoted angiogenesis after ischemic stroke, this process could be inhibited by miR-493, which was previously thought to be a tumor angiogenesis suppressor. 71 Furthermore, the deletion of MIF gene accelerated neuronal loss after stroke because MIF shielded neurons from oxidative stress and ischemia-reperfusion (I/R) induced apoptosis by reducing caspase-3 activation. 13 HIF-1 activation boosted MIF promoter activity and dramatically reduced OGD-induced neuron cell death from a genetic perspective. 48 Finally, in the MCAo model, sex-specific protective benefits were discovered; MIF deletion was detrimental to female mice due to its involvement in regulating inflammation and cell death. 95
When examining particular genetically engineered ischemia models, we see that MIF-KO mice displayed smaller infarct volume at day 7 after 45 min tMCAo, 10 with improved behavioral scores at day 1, 3, and 7 in the same surgical model, as reported by Wang's innovative study. 91 However, another research found that MIF-KO mice had a larger infarct volume 10 hours after 2 hours of tMCAo, with no significant difference at 4 and 22 hours after ischemia. 13 Similar outcomes were seen in MIF-KO mice administered 90 minutes of tMCAo on day 3, with the exception that females had bigger infarcts than WT females but males did not. 95 MIF appears to serve a protective effect at an early stage following prolonged ischemia, but no conclusion has been reached so far. Different observation timepoints, the length of occlusion, or even the origin of genetically engineered animals may account for contradictory MIF function results in ischemic stroke. Further research is required to evaluate the potential bias caused by the model discrepancies noted previously.
The double-faced role of MIF in hemorrhagic stroke
Blood MIF level was reported to be an ideal predictor of delayed cerebral ischemia (DCI) after acute stage of aneurysmal subarachnoid hemorrhage (aSAH), as reported by a single center, prospective, observational cohort study which enrolled 201 patients. 96 This positive correlation between serum MIF concentration and DCI after aSAH was postulated due to the inflammatory nature of MIF, which can exacerbate brain injury and stimulate the immune response during subarachnoid hemorrhage. 96 The difference between the two investigations was that the MIF concentration was measured in either cerebrospinal fluid (CSF) or normal pressure hydrocephalus. 97 These results were comparable to those of an earlier clinical trial using a small sample size. In the study, the MIF concentration in CSF was also identified as a biomarker for aSAH patients who suffered cerebral vasospasm (CV) and DCI sequelae. To obtain more convincing results, a multicenter study with a larger sample size of patients is still required. In patients with intracerebral hemorrhage (ICH), serum MIF concentration rose immediately after disease onset and was strongly correlated with plasma C-reactive protein level, hematoma size, and NIHSS score. 15 Furthermore, it accurately reflected injury severity and can be used as an independent predictor of 6-month unfavorable outcome after acute ICH. 98
Interestingly, compared to clinical data, injecting 2 days before or 2 hours after bacterial collagenase induced experimental intracerebral hemorrhage, MIF alleviated ICH injury by inhibiting microglial activation and macrophage infiltration, resulting in lower reactive oxygen species (ROS) production and neuronal degeneration.14,99
Evidence of MIF functions on traumatic brain injury
Possessing a similar predictive function as stroke, severe blunt trauma patients were also found had higher serum MIF concentrations and related to the clinical outcome. 100 Data from an observational study enrolled 116 severe TBI patients revealed that serum MIF concentrations were also a potential biomarker for identifying cerebrocardiac syndrome (CCS). 101 Another study involving 108 severe TBI found a correlation between the serum MIF level and the severity of TBI and clinical outcomes. 102 In sever blunt trauma patients, Chuang et al.
Wang et al. found that the total expression of MIF remained unchanged in a controlled cortical impact (CCI) induced TBI model. 54 However, MIF entered the nucleus of damaged neurons from the cytosol, causing neurodegeneration with a worsened neurological outcome in a poly ADP-ribose polymerase-1 (PARP-1) dependent manner. 54 Significantly, although MIF can promote neurodegeneration with the participation of CD74, 103 the treatment of MIF antagonist ISO1 failed to prevent neurodegeneration in the peri-injury cortex in a fluid percussion injury (FPI) model of TBI, despite the astrocyte suppression property of MIF during this process. 60
MIF and neurodegenerative diseases
Pleiotropic effects of MIF on Alzheimer’s disease
MIF levels in cerebral spinal fluid and resident brain cells were found to be increased in AD patients and could serve as a biomarker. 104 When compared 52 cognitively healthy volunteers to 97 cognitively impaired or mild dementia patients, the latter had higher MIF levels in CSF, which were significantly correlated with tau and phosphorylated tau protein level. 105 Similarly, another study involving 31 patients with AD, 28 patients with mild cognitive impairment (MCI) and 19 control participants showed that MIF concentrations in CSF were increased in both the AD and MCI groups, with a link between MIF and TNF-α levels in the AD group. 106
MIF was proved to be involved in neuroinflammation and cell toxicity in animal models of AD and in vitro studies.107,108 Pharmacological MIF inhibition or knockout prevented tau protein hyperphosphorylation in C57BL/6 mice, along with better memory improvement in the same streptozotocin induced sporadic AD model.107,109 After treatment with streptozotocin, extracellular MIF levels were considerably increased in cultured astrocytes and microglia, and the MIF antagonist ISO-1 decreased cytokine production in vitro. 107
In the Aβ1-40-stimulated PC12 cell line AD model, treatment with diabetes pathogenic compounds advanced glycation end products (AGEs) led to an increase in MIF expression and a decrease in PC12 cell viability. 110 Consequently, the use of ISO-1 reversed AGEs induced cell deactivation and reduced inflammatory mediators such as IL-1β and TNF-α. 110 In the early stage of AD brains, before the formation of AGEs, fluorescent phenylboronate gel electrophoresis 111 identified glucose-modified MIF that lacked the ability to stimulate enzymes and glial cells, suggesting that MIF participates in the entire progression of high glucose-induced AD-like changes. Using mass spectrometry-based imaging, Carlred et al. confirmed a high degree of co-localization of MIF and activated microglia to amyloid- (A) deposits in the hippocampal region, suggesting that MIF-associated neuroinflammation and glial cell reactivity played a crucial role in the AD pathology. 112 Notably, MIF deficiency impaired the spatial learning function of APP23 transgenic AD mice, suggesting that MIF can shield neuronal cells against A-induced cytotoxicity. The above evidence provides a more complete view of MIF in various AD models. 113
The multifaced role of MIF on Parkinson’s disease
Prior to the year of 2000, the function of MIF on allogeneic fetal mesencephalic dopaminergic grafts was studied using the 6-hydroxydopamine rat model of PD. 114 The intracerebral administration of MIF decreased the activities of microglia and macrophages but didn’t improve the function and survival of grafts. 114 In 2011, Cheng et al. compared 92 PD patients with 87 matched controls and revealed a significant elevation of serum MIF levels in PD patients, proposing MIF levels as a diagnostic biomarker for PD. 115 All clinical studies mentioned in this review were summarized in Table 1.
Clinical investigations of the relationship between MIF and brain injury.
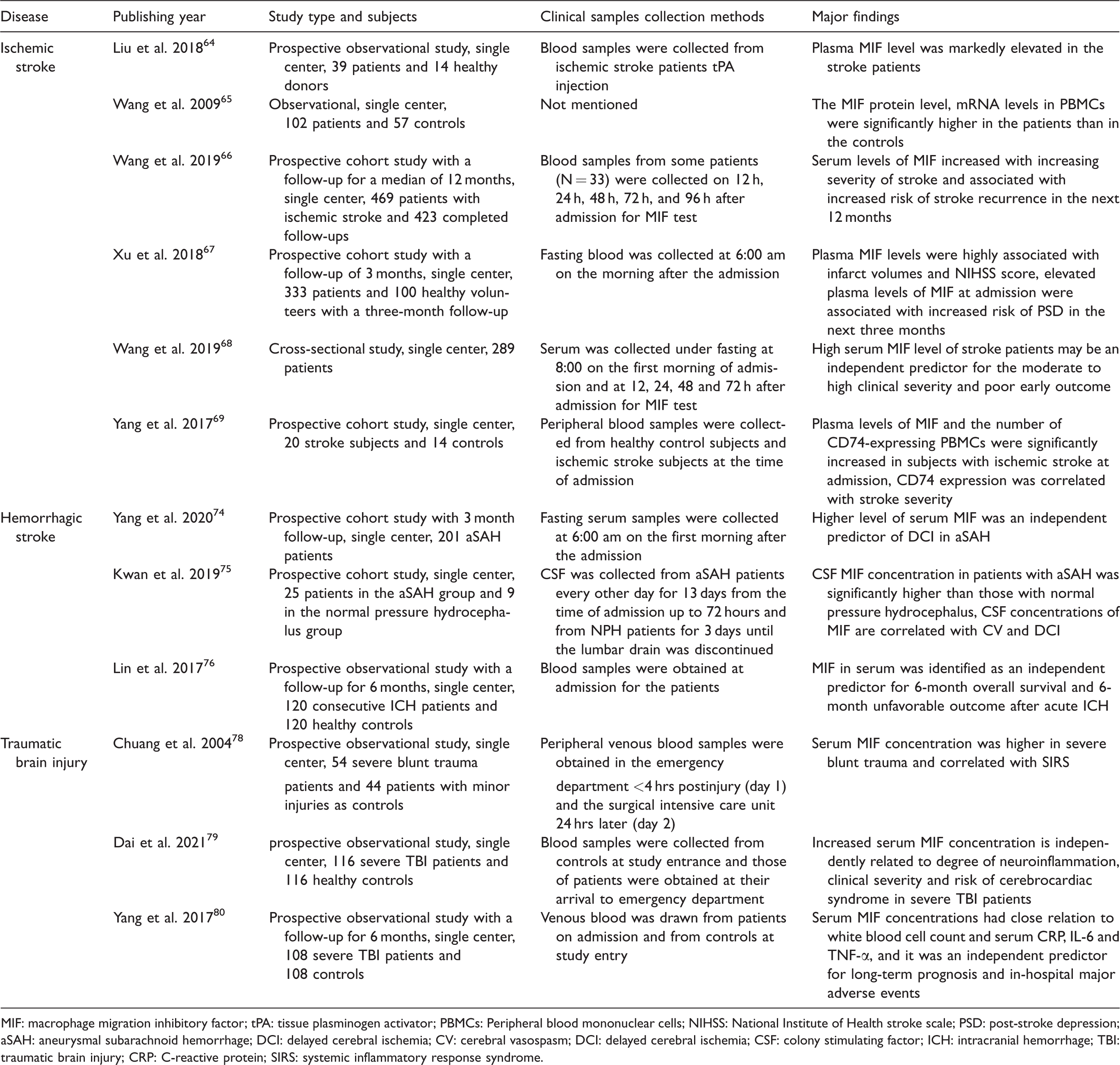
MIF: macrophage migration inhibitory factor; tPA: tissue plasminogen activator; PBMCs: Peripheral blood mononuclear cells; NIHSS: National Institute of Health stroke scale; PSD: post-stroke depression; aSAH: aneurysmal subarachnoid hemorrhage; DCI: delayed cerebral ischemia; CV: cerebral vasospasm; DCI: delayed cerebral ischemia; CSF: colony stimulating factor; ICH: intracranial hemorrhage; TBI: traumatic brain injury; CRP: C-reactive protein; SIRS: systemic inflammatory response syndrome.
The dualistic effects of MIF on brain resident cells in experimental stroke.
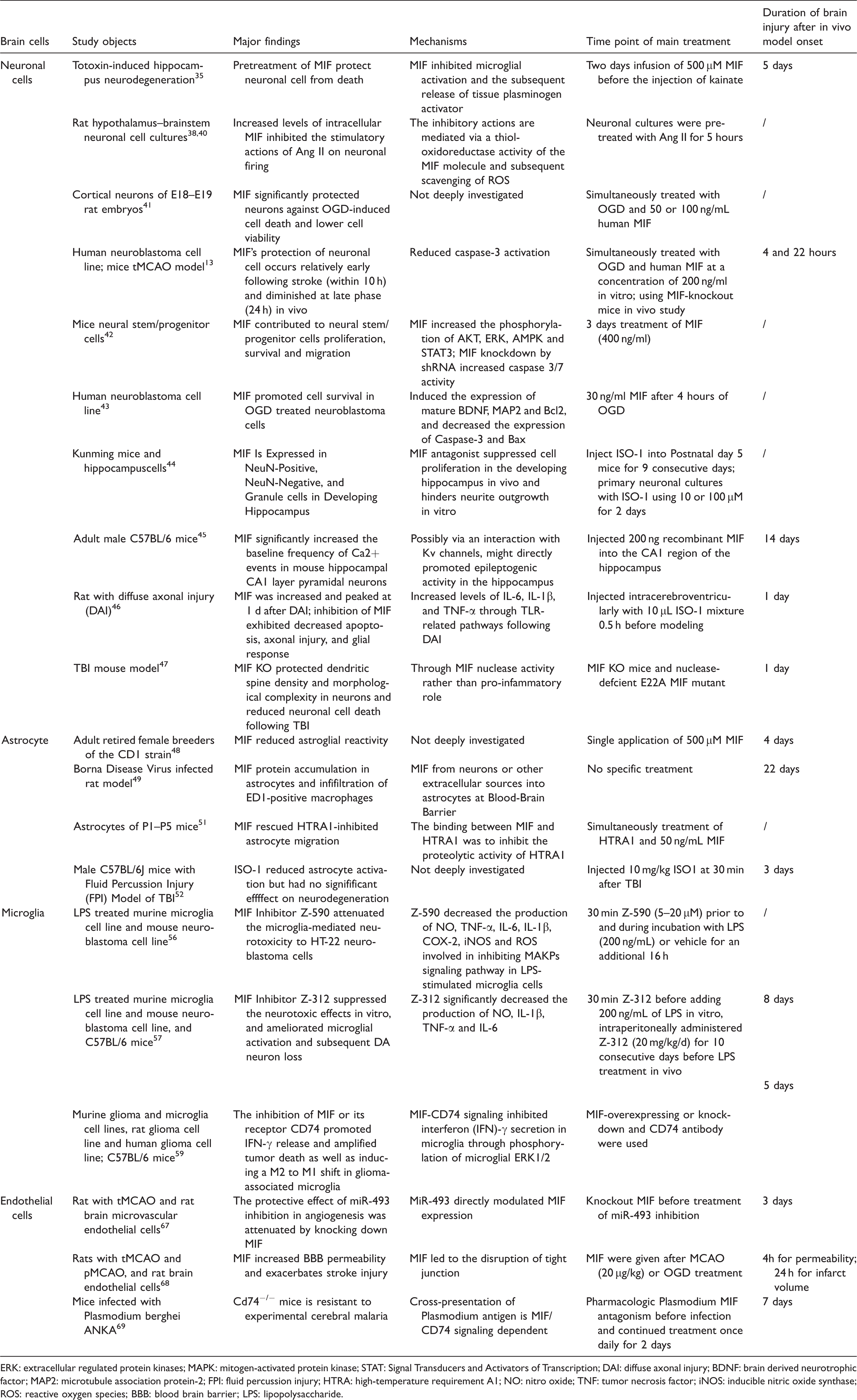
ERK: extracellular regulated protein kinases; MAPK: mitogen-activated protein kinase; STAT: Signal Transducers and Activators of Transcription; DAI: diffuse axonal injury; BDNF: brain derived neurotrophic factor; MAP2: microtubule association protein-2; FPI: fluid percussion injury; HTRA: high-temperature requirement A1; NO: nitro oxide; TNF: tumor necrosis factor; iNOS: inducible nitric oxide synthase; ROS: reactive oxygen species; BBB: blood brain barrier; LPS: lipopolysaccharide.
In Atg5 conditional knockout mice, the deficiency of microglial autophagy led to an increase of MIF in a NLRP3 inflammasome, causing neuroinflammation and PD-like symptoms. 66 The MAPKs and NF-κB pathways were implicated in the MIF related LPS-induced PD mouse model. 65 In co-culturing BV-2 microglia and HT22 neuroblastoma cells, the use of a MIF small-molecule inhibitor Z-312 attenuated LPS-induced neurotoxicity with decreased proinflammatory factors; this protective effect of Z-312 was also demonstrated in vivo by a decrease in LPS-induced dopaminergic neuronal loss. 65 For the α-synuclein (α-syn) preformed fibril (PFF) mouse model of sporadic PD, it was recently revealed that genetic and pharmacological inhibition of MIF nuclease activity can prevent neurodegeneration by decreasing PARP-1 activity. 116
Compared with the negative influence on PD pathology above, MIF’s expression was found to be positively correlated with IL-10 and to inhibit apoptosis in SH-SY5Y PD cells via decreasing the concentration of cleaved-PARP. Moreover, MIF enhanced autophagosome production in the SH-SY5Y PD cell model, which may be advantageous for neurodegenerative diseases such as PD. 117 Another neuroprotective effect of MIF was the ability to protect amino acid decarboxylase (AADC)-expressing Chinese hamster ovary (CHO) cells treated with MIF from intracellular but not extracellular dopamine toxicity. 118 Figure 2 shows how MIF interact with AD and PD.
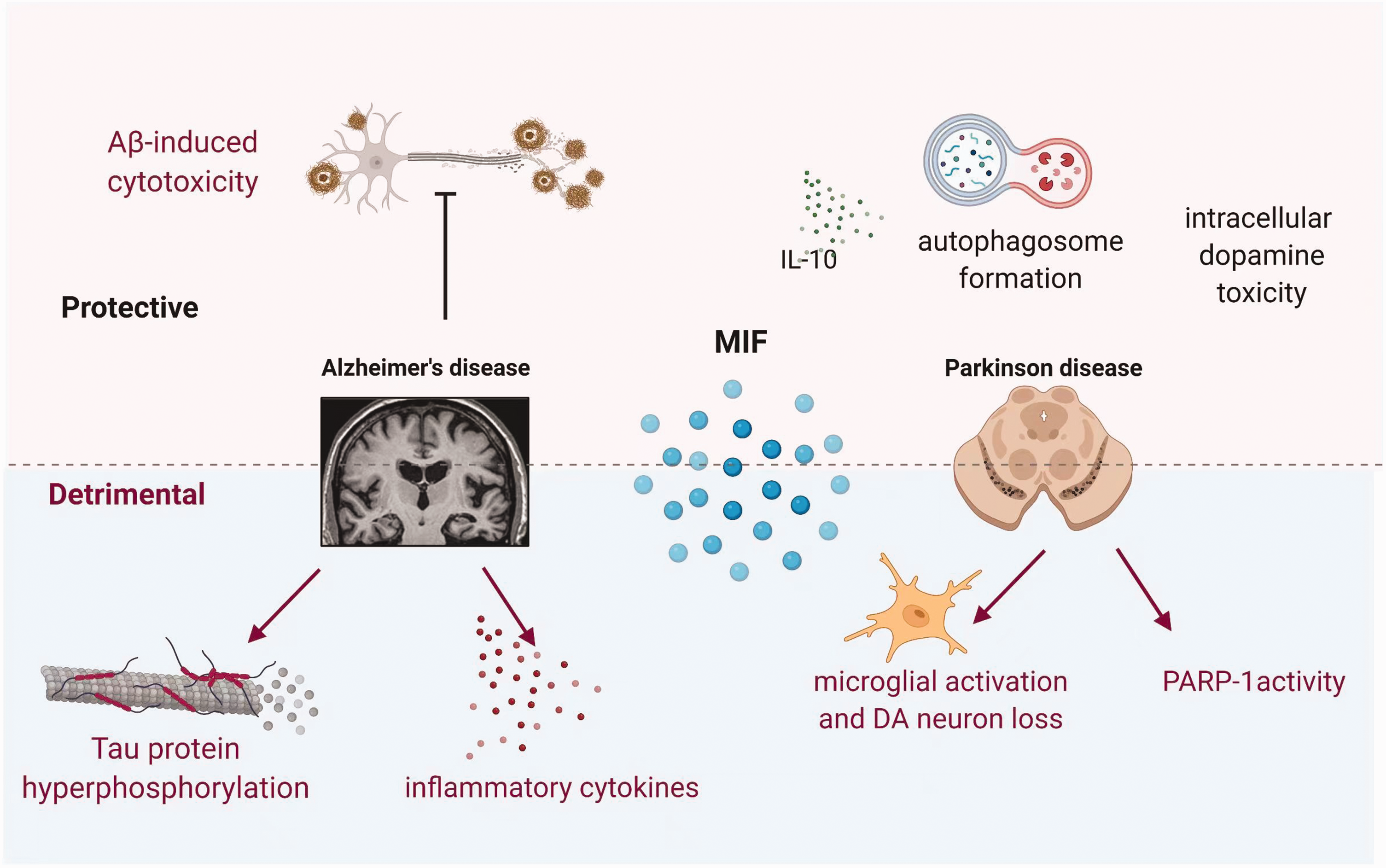
The double-sided effects of MIF on neurodegenerative disease models. MIF exerts protective effects by inhibiting Aβ-induced cytotoxicity, increasing anti-inflammatory cytokine IL-10 release, promoting autophagosome formation, and reducing intracellular dopamine toxicity in AD and PD models. In terms of the detrimental side, MIF is correlated to tau protein hyperphosphorylation, inflammatory cytokines production, microglial activation, and PARP-1 stimulation. Aβ: amyloid β-protein; AD: Alzheimer’s disease; PD: Parkinsons; PARP-1: poly ADP-ribose polymerase-1 (PARP-1); DA neuron: dopaminergic neuron. The figure were created with BioRender.com.
As previously stated, MIF is a diverse and contentious cytokine in both physiology and pathology. As illness progresses, MIF may display distinct roles in different cell contexts; hence, the impact of varied dosages of MIF on disease progression should not be overlooked.
Concluding remarks
Due to its proinflammatory characteristics, MIF was previously assumed to be deleterious in brain disorders. Recent research has demonstrated that MIF modulation effects are diverse, and that MIF may possibly have organ protective properties in brain damage and neurodegenerative disorders. However, the advantages of MIF in neuroinflammatory diseases were mainly found in basic research, and the specific type of disease model may also influence the explanation of the results. As the numerous activities of MIF in the development of brain damage and neurodegenerative disorders are elucidated, MIF has emerged as a novel and appealing therapeutic target. In MIF-based pharmacological methods, anti-MIF monoclonal antibody, antibodies directed against MIF receptors, and small-molecule inhibitors have been created.119,120 However, there are still limits to these pharmaceutical techniques. Synthesis inhibitors, for instance, may target an array of downstream pathways and lack selectivity. Therefore, MIF must be carefully targeted based on its varied functions.
In this review, we highlight the novel findings of MIF mechanistic insights in immune cells participating in acute and chronic brain disorders, which may shed light on MIF as a prospective therapeutic target in the future.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: P L. is supported by the National Natural Science Foundation of China (NSFC, 81722017, 91957111, 81971096, 82061130224), New Frontier Technology Joint Research (SHDC12019102) and Ward Building Project for Demonstration and Research sponsored by Shanghai Shenkang Hospital Development Center, Shanghai Municipal Education Commission-Gaofeng Clinical Medical Grant Support (20181805), Shuguang Program supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (20SG17), Shanghai Outstanding Academic Leaders’ Program from Shanghai Municipal Science and Technology Committee (20XD1422400), Newton Advanced Fellowship grant provided by the UK Academy of Medical Sciences (NAF\R11\1010), and Renji Clinical Research Funding (PYII20-03) the Innovative Research Team of High-level Local Universities in Shanghai (SHSMU-ZLCX20211602). W.Y. is supported by the Shanghai Pudong New Area Municipal Commission of Health and Family Planning Funding (PWZxq2017-06). Shanghai Municipal Key Clinical Specialty (shslczdzk03601 to Weifeng Yu.), Shanghai Engineering Research Center of Peri-operative Organ Support and Function Preservation (20DZ2254200). W.X. is supported by the National Natural Science Foundation of China (NSFC, 81901985).
Acknowledgements
Thank the authors of the references.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.