
Abstract
The pyramidal neurons of the hippocampal CA1 region are essential for cognitive functions such as spatial learning and memory, and are selectively destroyed after cerebral ischemia. To analyze whether degenerated CA1 neurons are replaced by new neurons and whether such regeneration is associated with amelioration in learning and memory deficits, we have used a rat global ischemia model that provides an almost complete disappearance (to approximately 3% of control) of CA1 neurons associated with a robust impairment in spatial learning and memory at two weeks after ischemia. We found that transient cerebral ischemia can evoke a massive formation of new neurons in the CA1 region, reaching approximately 40% of the original number of neurons at 90 days after ischemia (DAI). Co-localization of the mature neuronal marker neuronal nuclei with 5-bromo-2'-deoxyuridine in CA1 confirmed that neurogenesis indeed had occurred after the ischemic insult. Furthermore, we found increased numbers of cells expressing the immature neuron marker polysialic acid neuronal cell adhesion molecule in the adjacent lateral periventricular region, suggesting that the newly formed neurons derive from this region. The reappearance of CA1 neurons was associated with a recovery of ischemia-induced impairments in spatial learning and memory at 90 DAI, suggesting that the newly formed CA1 neurons restore hippocampal CA1 function. In conclusion, these results show that the brain has an endogenous capacity to form new nerve cells after injury, which correlates with a restoration of cognitive functions of the brain.
Introduction
Brain neurons have a high demand for oxygen and only little or no endogenous reserves of glycogen. Thus, adult neurons are vulnerable to ischemic conditions that occur, for example, after cardiac arrest, stroke, brain trauma or subarachnoid hemorrhage (Bendel et al, 2005a). Hippocampal CA1 neurons are particularly sensitive and undergo selective, delayed degeneration in response to global ischemia (Kirino, 1982; Pulsinelli et al, 1982). These pyramidal neurons are critically involved in spatial learning and memory, and degeneration of these neurons results in deficiencies in such functions (Morris et al, 1982).
Formation of new neurons from neuronal stem or progenitor cells, neurogenesis, continues throughout life in discrete parts of the brain of vertebrate species including humans (Altman and Das, 1965; Kaplan and Hinds, 1977; Kuhn et al, 1996; van Praag et al, 2002). Under physiological conditions neuronal proliferation occurs in two regions; the subventricular zone, which lines the lateral ventricles and produces neuronal progenitor cells that migrate to the olfactory bulb, and the subgranular zone of the dentate gyrus, from where neuronal progenitors migrate to the dentate gyrus. Stem cells have also been reported to be located in the posterior periventricle adjacent to the hippocampus (Nakatomi et al, 2002).
Ischemia is a powerful stimulus to enhance cell proliferation in the subgranular zone, leading to an increase of newborn granule cells in the dentate gyrus (Choi et al, 2003; Liu et al, 1998), although the enhancement is independent of the ischemic injury (Arvidsson et al, 2001; Liu et al, 1998). Ischemia also increases generation of neurons in the subventricular zone. These newborn cells migrate into the striatum to replace a minute fraction of the destroyed medium spiny neurons (Arvidsson et al, 2002; Parent et al, 2002). Even in other structures that normally do not show signs of neuronal turnover, such as the hippocampal CA1 region (Nakatomi et al, 2002; Schmidt and Reymann, 2002) and the cerebral cortex (Gu et al, 2000; Jiang et al, 2001; Jin et al, 2003), ischemia induces sparse numbers of new pyramidal neurons.
Whether the newly generated neurons have a role in restoring ischemia-induced behavioral impairments remains to be proven. The only evidence for functional neuronal replacement after ischemia derives from a study by Nakatomi et al (2002). They showed that intraventricular infusions of FGF-2 and EGF stimulated the regeneration of CA1 neurons, and that such enhanced regeneration was associated with restoration of deficiencies in learning and memory. However, it cannot be excluded that growth factors contributed to the enhanced functional improvements through other mechanisms.
Therefore, we have investigated whether endogenous neurogenesis occurs in the CA1 region in response to ischemic injury without any stimulatory treatment and whether reappearance of CA1 neurons is associated with recovery of deficient hippocampal-dependent spatial learning and memory.
Materials and methods
Induction of Transient Global Ischemia
In all, 97 male Sprague–Dawley rats (B&K Universal AB, Sollentuna, Sweden) weighing 250 to 330 g were used and kept under standardized conditions. All experiments were approved by the Animal Ethics Committee of Northern Stockholm. We used a recently developed transient global ischemia model (Bendel et al, 2005b) utilizing halothane to reduce systemic blood pressure (McBean et al, 1995). The rats were anaesthetized with halothane (Fluothane®, AstraZeneca, Södertälje, Sweden), intubated and ventilated. The carotid arteries were carefully exposed and the tail artery was catheterized to continuously monitor the mean arterial blood pressure (MABP). A laser-Doppler probe was attached to measure the cerebral blood flow (CBF) in the forebrain. The muscular tissue surrounding the common carotid arteries was infiltrated with bupivacain (Marcain®, AstraZeneca, Södertälje, Sweden) and the skin tissue of the tail and skull with lidocaine (Xylocain®, AstraZeneca, Södertälje, Sweden). Body temperature was maintained at 37.5° C. After the baseline of MABP and CBF had been established for 10 mins, the concentration of halothane was increased until the MABP was reduced to approximately 40 mm Hg. The common carotid arteries were subsequently occluded for 1 or 11 mins. During the ischemic period, adjustments in the halothane concentration were made to maintain the MABP at 40 to 45 mm Hg. Control rats did not undergo any anesthesia or surgery.
Experimental Groups
In all, 11 rats were excluded from further analysis because of surgical complications, early death or convulsions. The remaining rats were allowed to survive for 14 ± 1 days after ischemia (DAI) (mean ± s.d.), 21 DAI, or 90 ± 6 DAI. For the 14 DAI time point, 9 control, 8 1-min ischemia, and 8 11-min ischemia rats were investigated. At 21 DAI, 5 control and 5 11-min ischemia rats were assessed, and at 90 DAI, we used 12 controls, 9 1-min ischemia and 11 11-min ischemia animals. Four rats served as controls at 0 DAI. Data from these rats were included to provide initial values in the histological graphs, but were not included in the statistical analyses.
In a separate experiment, 6 control and 10 11-min ischemia rats were twice daily intraperitoneally injected with 50 mg/kg 5-bromo-2'-deoxyuridine (BrdU, Sigma-Aldrich, Stockholm, Sweden) during 6–12 DAI. These rats were allowed to survive until 14 DAI (3 controls and 4 11-min ischemia) or 90 DAI (3 controls and 6 11-min ischemia).
Learning and Memory
Spatial learning and memory were evaluated in an open-field water maze (Morris et al, 1982), which consisted of a circular pool, 180 cm in diameter (AB Lomma Plastpro-dukter, Lomma, Sweden). The animals were monitored by a video tracking system (Watermaze Software, Edinburgh, UK). The experimenter was blind to treatment groups.
Pretraining started 10 days before killing and consisted of four swim trials with a randomly positioned visible platform (10 cm in diameter) and no extra-maze cues. All rats were able to follow the experimenter's hand to the platform. After 2 days, the rats were trained for six consecutive days with 4 trials/day from any of the four pseudo-randomly selected starting points. The platform was submerged 1 cm below the water (22°C ± 1°C) surface in a fixed position. Several prominent extra-maze cues were present and kept constant throughout the experiment. The maximum trial duration was 90 secs, plus 30 secs on the platform at the end of each trial. At 24 h after the last training trial, memory retention was tested. The platform was removed and the animal was allowed to swim freely for 60 secs. The area where the platform had previously been located was defined as the platform area, and the circular zone (40 cm diameter) encompassing the platform zone was defined as the target zone (de Hoz et al, 2003).
Beam-Walk Performance
At 1 day before killing, beam-walk performance was tested using seven wooden planks of different widths, from 7.7 cm and narrowing 1 cm for each beam (von Euler et al, 1996). The rats were placed on successively narrower planks and the narrowest plank a rat could walk without foot slips within two trials was recorded.
Histology
After killing, the brains were rapidly removed, frozen in isopentane, and stored in −135°C until sectioning in 14-µm-thin coronal sections. Brain sections were thawed, rehydrated and post-fixated in 4% formaldehyde. Sections were stained with cresyl-violet or incubated in 0.1% (v/v) acetic acid containing 0.00002% (w/v) Fluoro-Jade (Schmeud et al, 1997).
For immunohistochemistry, sections were treated with 0.3% hydrogen peroxide after rehydration and fixation in 4% formaldehyde, and subsequently incubated with mouse anti-neuronal nuclei (NeuN, 1:1000, Chemicon, Temecula, CA, USA) or mouse anti-OX42 (1:1600, Harlan SERA-LAB, Loughborough, UK) in combination with the avidin–biotin blocking kit (Vector Laboratories, Burlingame, CA, USA). The sections were exposed to complementary biotinylated secondary antibodies (1:200; Vector Laboratories) and visualized with the DAB substrate kit. To examine co-localization of BrdU-immunoreactive (IR) and NeuN-IR, sections were first stained with the NeuN antibody as described above, except that the signal was visualized with the SG substrate kit (Vector Laboratories). Subsequently, the sections were treated with 2 × SCC buffer, 2N HCl, and 0.1 mol/L borate buffer (pH 8.5), and incubated with rat anti-BrdU (1:100, Harlan SERA-LAB, Loughborough, UK), in combination with the avidin–biotin blocking kit (Rao and Shetty, 2004). The sections were exposed to the complementary biotinylated secondary antibody (1:200; Vector Laboratories, Burlingame, CA, USA) and developed with the NovaRED substrate kit (BrdU; Vector Laboratories, Burlingame, CA, USA). To examine the expression of polysialic acid neuronal cell adhesion molecule (PSA-NCAM) or microtubule-associated protein 2 (MAP2), sections were rehydrated, post-fixated in 4% formaldehyde and incubated in 1% BSA buffer, followed by mouse anti-PSA-NCAM (1:1000, Chemicon) or by mouse anti-MAP2 (1:1000, Chemicon), and finally exposed to a donkey anti-mouse Cy3-conjugated antibody (1:300, Jackson Immunoresearch Lab, West Grove, PA, USA). Sections labeled with PSA-NCAM were counterstained with Hoechst 33342 (Molecular Probes, Eugene, OR, USA).
In the cresyl-violet- and NeuN-stained sections, the number of neurons was counted in 0.5-mm-wide sample areas (pyramidal layer approximately 0.1 mm thick) in the medial (towards the midline), central (indicated by the square in Figure 1A) and lateral (towards the CA3 layer) parts of the CA1, as well as in the CA3 region. Fluoro-Jade staining of CA1 pyramidal layer was analyzed with regard to area over a fixed threshold value, MAP2 and OX42 staining by determining specific gray values of the entire CA1 region using the Scion Image for Windows software (Scion Corporation, Frederick, MD, USA). Cell numbers and areas are presented as the average value of both hemispheres.
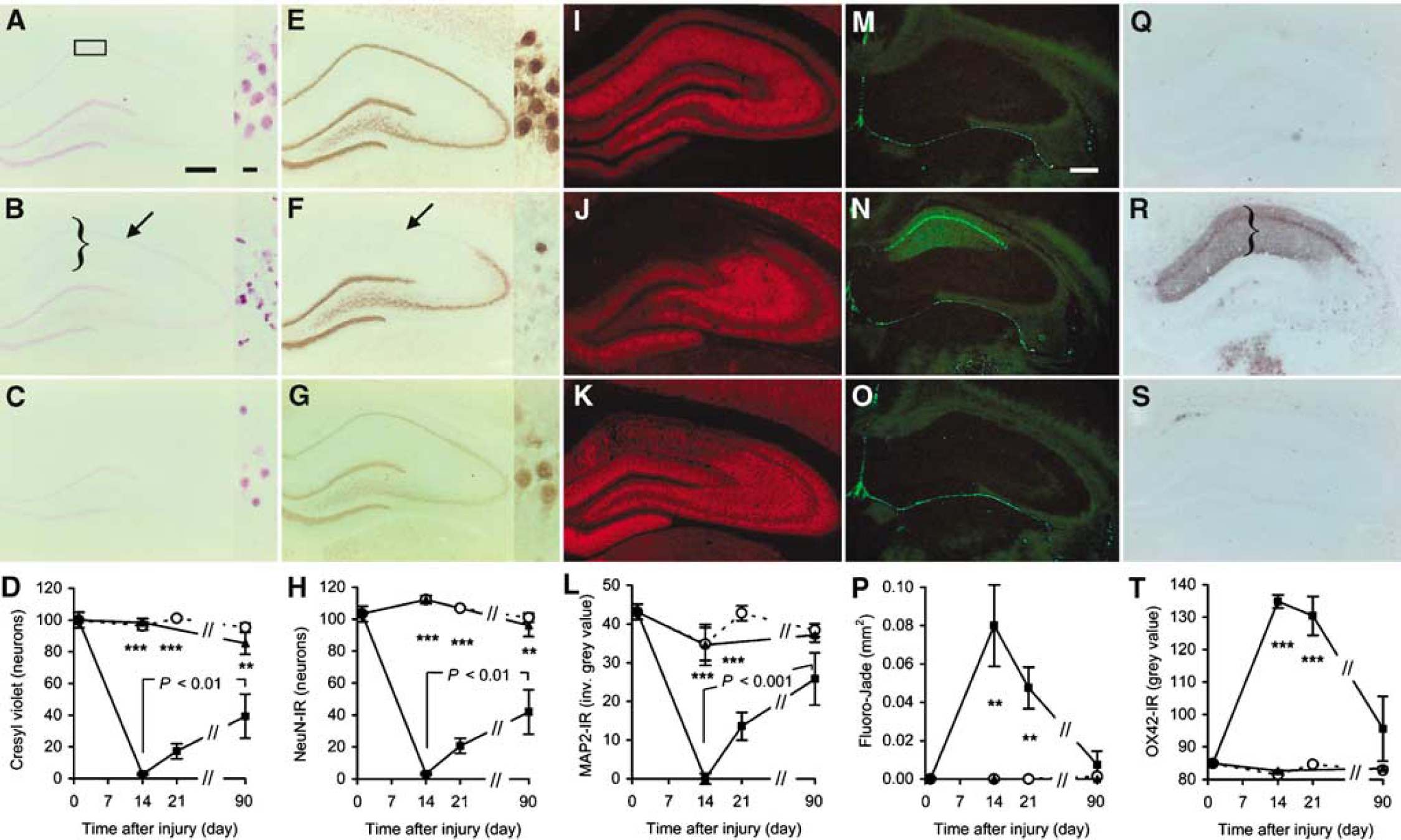
Degeneration and reappearance of CA1 neurons after 11 mins of transient global ischemia. (
Statistical Analysis
Histological data were analyzed by one-way ANOVA followed by Fisher's PLSD, both to investigate the differences between 11-min ischemia, 1-min ischemia, and controls at discrete time points, and to investigate time-related changes within the 11-min ischemia groups. Learning test data were analyzed by repeated-measures two-way ANOVA followed by Fisher's PLSD, and memory test data were analyzed by one-way ANOVA followed by Fisher's PLSD. Beam walk data were analyzed by Kruskal–Wallis test.
Results
Degeneration and Reappearance of CA1 Neurons After Ischemia
Transient global ischemia for 11 mins caused a nearly complete disappearance of cresyl-violet-stained neurons with a normal neuronal morphology in the anterior CA1 at 14 DAI (Figures 1A–D). The numbers of neurons in the medial, central, and lateral sample areas of the CA1 were 1.9% ± 0.3% (mean ± s.e.m.), 2.9% ± 0.4%, and 4.0% ± 0.8% of control values, respectively. In contrast, the neurons in the CA3 and dentate gyrus were spared. At 21 DAI there was a tendency to an increased number of neurons, and at 90 DAI the number of medial, central, and lateral CA1 neurons reached 45% ± 13% (mean ± s.e.m.), 41% ± 14%, and 45% ± 13% of control values (all areas P < 0.01 against respective 14 DAI values, by one-way ANOVA followed by Fisher's PLSD). Subsequently, we analyzed the number of neurons IR to the neuronal nuclei marker NeuN (Figures 1E–H). Similar to the results obtained with cresyl-violet staining, the NeuN-IR neurons in anterior CA1 disappeared at 14 DAI, and reappeared at 21 and 90 DAI. The numbers of NeuN-IR neurons in the medial, central, and lateral sample areas of CA1 were 2.8% ± 0.7% (mean ± s.e.m.), 2.8% ± 0.6%, and 3.8% ± 0.2% of control values, respectively, at 14 DAI, and 36% ± 13%, 41% ± 14%, and 40% ± 12% of control values at 90 DAI (all areas P < 0.01 against respective 14 DAI values by one-way ANOVA followed by Fisher's PLSD). We used an antibody against microtubule-associated protein 2 (MAP2), which is stringently expressed in the neuronal perikarya and dendrites, to visualize the dendrites in the hippocampal formation. Consistently, MAP2-IR in the entire CA1 region was abolished to 0.6% ± 3.4% (mean ± s.e.m.) of control values at 14 DAI (Figures 1I–L). MAP2-IR increased to 59.8% ± 15.6% of control values at 90 DAI (P < 0.001 against 14 DAI value by one-way ANOVA followed by Fisher's PLSD). Also, MAP2-IR in the hilus was diminished to 41.3% ± 13.5% relative to control, which was reversed to control levels at 21 and 90 DAI (P < 0.05 against 14 DAI value by oneway ANOVA followed by Fisher's PLSD). The neuron-specific death marker Fluoro-Jade (Schmeud et al, 1997) confirmed the massive degeneration of neurons in the anterior CA1 at 14 DAI (Figures 1M–P). Fluoro-Jade staining decreased at 21 DAI and returned to control values at 90 DAI. The caudal part of CA1 did not show any Fluoro-Jade staining, suggesting that this part is spared after the ischemic insult. We found a marked increase in the reactive microglia marker OX42-IR in the stratum oriens, pyramidale, radiatum, and lacunosum of the CA1 region at 14 and 21 DAI, with little or no OX42-IR appearing at 90 DAI (Figures 1Q–T). This coincides with an infiltration of the small and densely stained cresyl-violet-positive cells (Figure 1B) indicating the presence of a substantial inflammatory response. Transient global ischemia for only 1 min did not affect the number of cresyl-violet- and NeuN-labeled neurons, MAP2-IR, Fluoro-Jade staining, or OX42-IR at any time (Figures 1D, 1H, 1L, 1P, 1T). This shows that the operative procedures per se did not cause neuronal degeneration.
Ischemia Induces Neurogenesis in the CA1
To detect newly formed cells, rats were injected with BrdU. In the CA1, BrdU was only found in nonneuronal cells at 14 DAI (Figure 2A), showing that BrdU was not incorporated in the few neurons that remained after 11 mins of ischemia at this time. In contrast, at 90 DAI, BrdU was found in some of the CA1 neurons, supporting that these neurons indeed were formed subsequent to the ischemic injury (Figure 2B). In agreement with previous studies (Choi et al, 2003; Liu et al, 1998; Sharp et al, 2002), the dentate gyrus showed BrdU-positive neurons at both 14 and 90 DAI (Figures 2C and 2D). In non-ischemic animals, we observed BrdU incorporation in the dentate gyrus, but not in CA1 neurons (not shown).
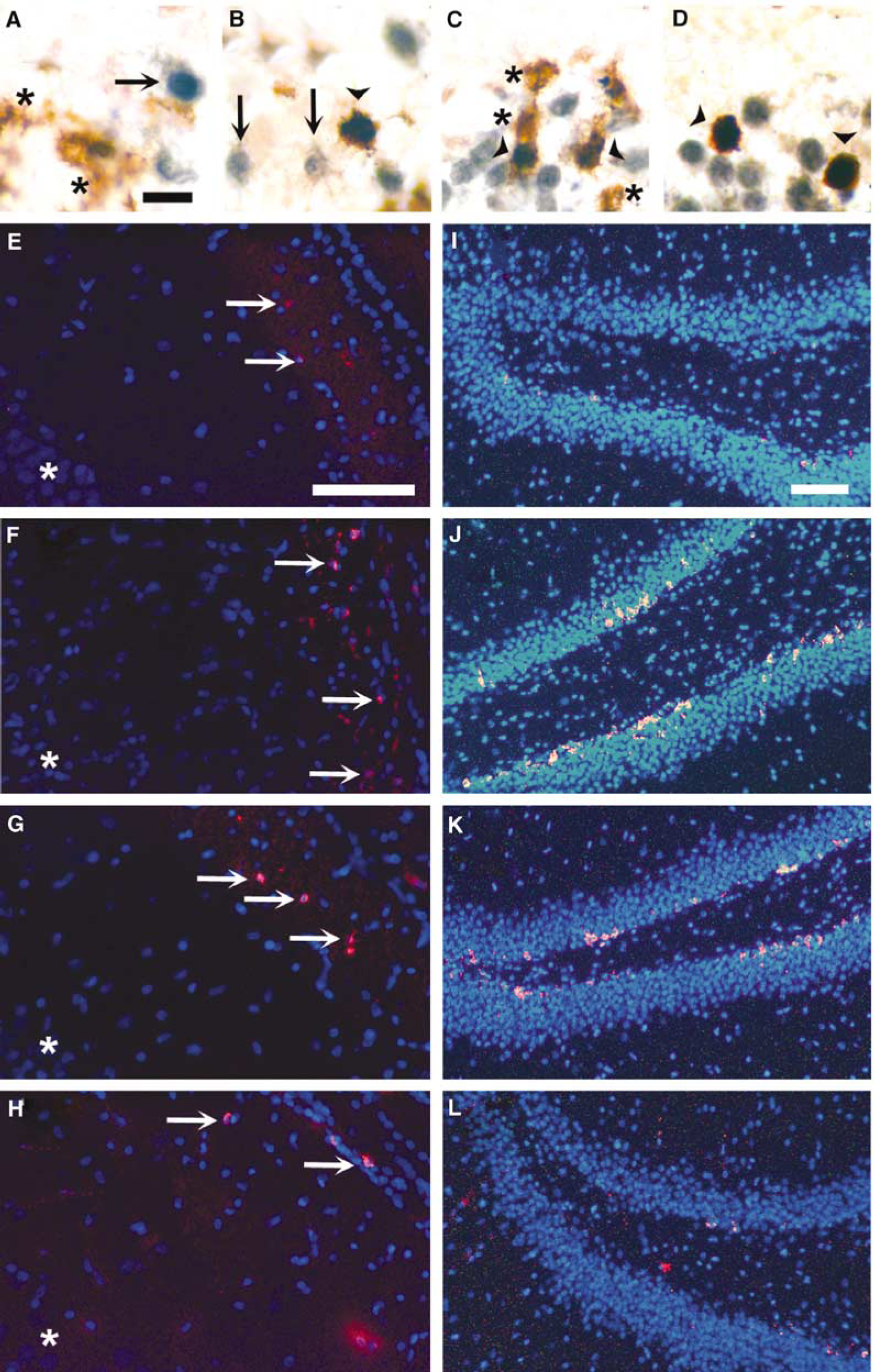
Neurogenesis in CA1 and dentate gyrus after transient global ischemia. (
Immature Neuronal Markers are Induced in the Posterior Lateral Periventricle
Recent work suggests that newly formed neurons migrate from the posterior lateral periventricle towards the CA1 after transient global ischemia (Nakatomi et al, 2002) or neonatal hypoxia (Daval et al, 2004). In agreement, we found increased numbers of cells expressing the immature neuron marker PSA-NCAM (Iwai et al, 2001) in the adjacent lateral periventricular region at 14 and 21 DAI (Figures 2E–H). In addition, we observed increased numbers of PSA-NCAM-IR cells in the subgranular zone adjacent to the inner blade of the dentate gyrus (Figures 2I–L), supporting that ischemia-enhanced neurogenesis takes place in this region.
Ischemia-Induced CA1 Degeneration Correlates with Learning and Memory Deficits
To see whether the formation of new CA1 neurons was associated with any functional recovery, we investigated hippocampal-dependent spatial learning and memory performance at 14 and 90 DAI. In the pretraining sessions, we found similar swim maze performances between the groups (data not shown). At 14 DAI, the 11-min ischemia rats showed longer escape latencies than control and 1-min ischemia rats (both P < 0.001 by repeated-measures two-way ANOVA; Figure 3A), indicating a marked deficiency in spatial learning. The learning deficiency persisted throughout the entire training period. During the first trials, control and 1-min ischemia rats showed a rapid learning followed by overnight forgetting (trials 4 and 5; Figure 3B). This pattern was not observed in the 11-min ischemia rats (Figure 3B), resulting in longer average escape latencies already at day 1. The 11-min ischemia rats swam longer distances than control and 1-min ischemia rats (both P < 0.001 by repeated-measures two-way ANOVA; Figure 3C). In fact, the relative difference in swim distance was even greater than the difference in escape latency, due to slightly higher swim speeds of the 11-min ischemic rats (P < 0.001, P < 0.05 to 0.001 at each day, against both controls and 1-min ischemia rats, by repeated-measures two-way ANOVA followed by Fisher's PLSD; there was no difference in swim speed between days in any group). The average swim speed during the training days 1 to 6 was 24 ± 1 cm/s (mean ± s.e.m.) for controls, 25 ± 1 cm/s for 1-min ischemia rats, and 30 ± 1 cm/s for 11-min ischemia rats. The increased swim speed of the 11-min ischemic rats could possibly be related to a slightly increased stress level due to failure of finding the escape platform and/or due to a different search strategy.
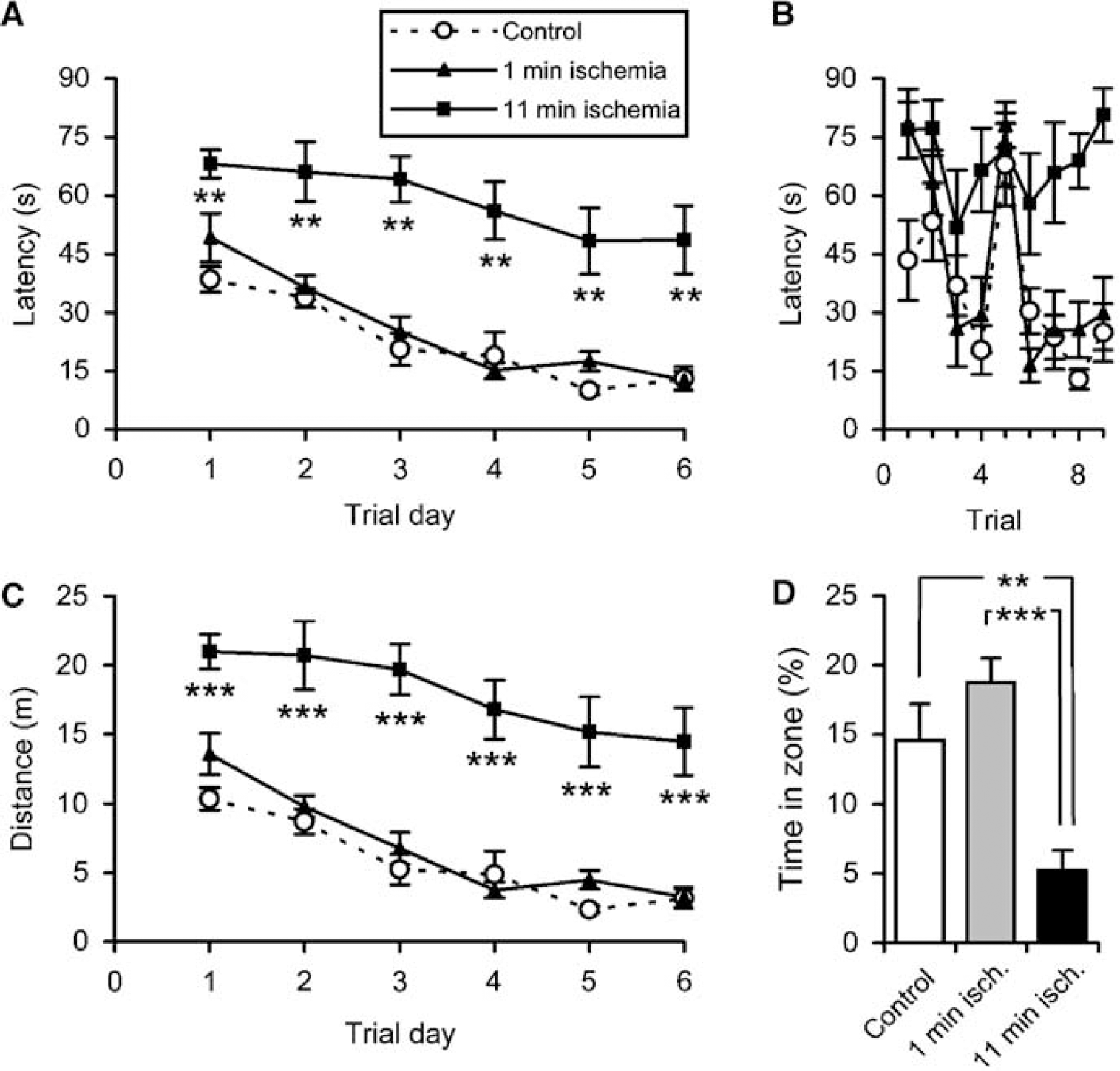
Effects of transient global ischemia on spatial learning and memory at 14 DAI. Data show mean ± s.e.m. (
In the subsequent memory test, the latencies to reach the target zone were longer in the 11-min ischemia rats (41 ± 7 secs; mean ± s.e.m.) than in the control (17 ± 5 secs; P < 0.01 by one-way ANOVA followed by Fisher's PLSD) and 1-min ischemia rats (20 ± 5 secs; P < 0.05). The 11-min ischemia rats spent significantly less time in the target zone as compared with controls and 1-min ischemia rats (Figure 3D). Likewise, the relative time spent in correct quadrant was shorter in the 11-min ischemia rats (23% ± 3%; mean ± s.e.m.) than in the control (36% ± 4%; P < 0.05 by one-way ANOVA followed by Fisher's PLSD) and 1-min ischemia rats (44% ± 3%; P < 0.001). Taken together, these results show that the 11-min ischemia rats have severely impaired spatial memory. No differences were found between the control and the 1-min ischemia groups in any measurement used.
To investigate whether the differences in escape latencies and time spent in the target zone were due to impairments in sensory, balance, or motor performance, the rats were subjected to beam-walk testing. No significant differences in beam-walk performance were observed, as analyzed by Kruskal–Wallis tests. The control, 1-min ischemia, and 11-min ischemia group beam widths were 1.7 (1.7 to 2.2), 2.2 (1.95 to 2.2), and 2.2 (1.7 to 2.2), respectively (medians and interquartiles). These results, together with the similar performance of the rats during the pretraining sessions and a lack of decreased swim speed, indicate that the longer escape latencies of the 11-min ischemia rats at 14 DAI are not due to deficiencies in sensory-motor performance or physiological state.
The Generation of New CA1 Neurons is Associated with a Restoration of Spatial Learning and Memory
In contrast to the 14 DAI rats, the 90 DAI rats showed no differences in escape latencies (P > 0.6; Figures 4A and 4B) or swim distance (P > 0.3; Figure 4C), indicating that the deficiencies in spatial learning observed at 14 DAI had been restored.
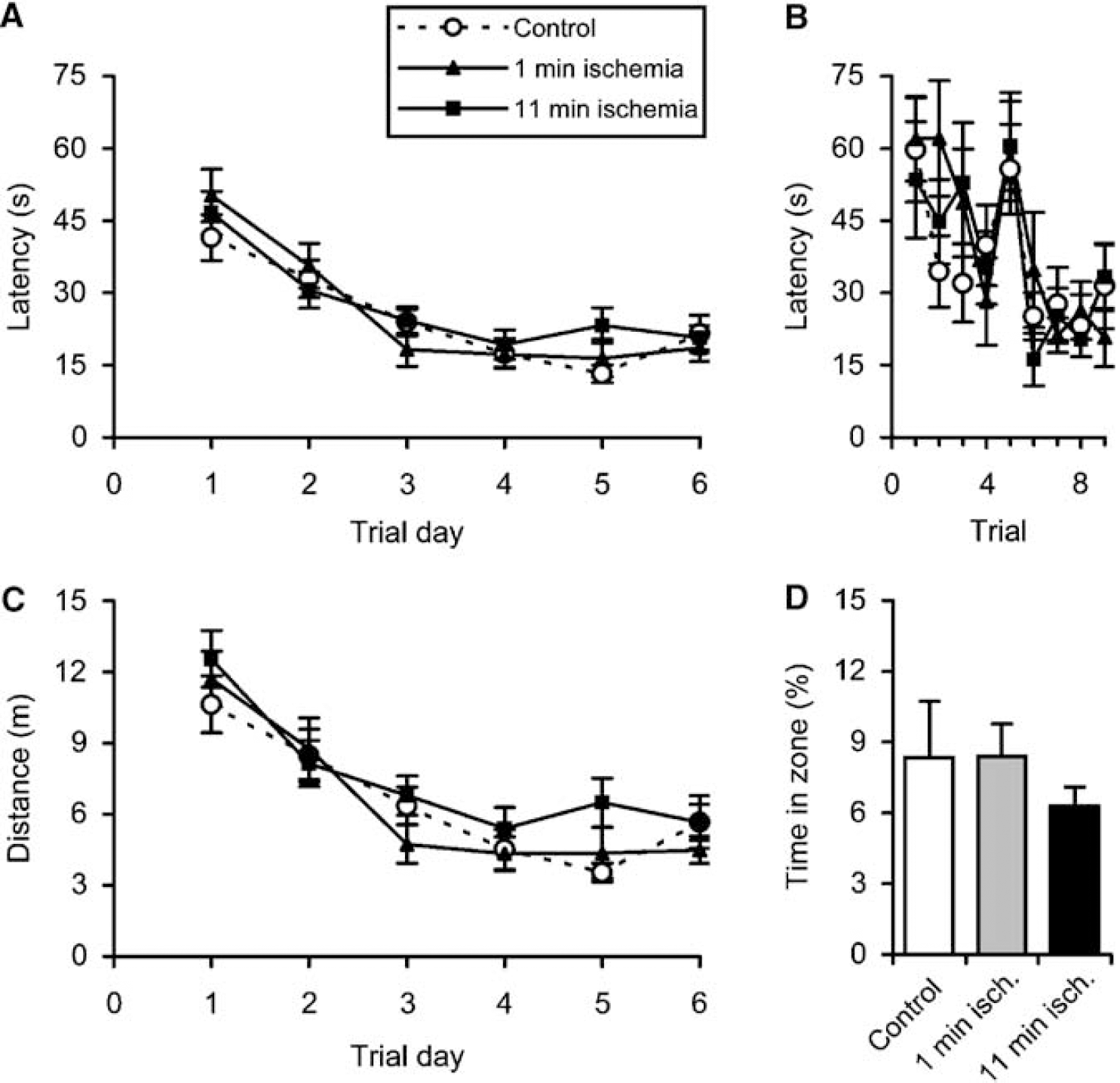
Effects of transient global ischemia on spatial learning and memory at 90 DAI. Data show mean ± s.e.m. (
The slightly enhanced swim speed observed in the 11-min ischemia rats at 14 DAI did not remain at 90 DAI (P > 0.1). The speeds remained the same throughout the testing period; the average values during the training days 1 to 6 were 25 ± 1cm/s (mean ± s.e.m.) for control, 24 ± 1 cm/s for 1-min ischemia rats, and 26 ± 1 cm/s for 11-min ischemia rats.
There were no differences in the latencies to reach platform area (P > 0.5); the latencies were 35 ± 8 secs (mean ± s.e.m.) for control, 26 ± 8 for the 1-min ischemia rats, and 38 ± 8 for the 11-min ischemia rats. Likewise, there were no differences in the time spent in target zone (P > 0.9; Figure 4D). Note, however, that the control rats at this age showed much less persistence than the younger control rats, making it difficult to detect possible deficiencies in spatial memory. This was even more evident for the parameter time in correct quadrant, which was similar (P > 0.8) and close to the 25% random value in all groups; 27% ± 3% (mean ± s.e.m.) for control, 28% ± 1% for the 1-min ischemia rats, and 26% ± 2% for the 11-min ischemia rats. Nevertheless, the short latency times observed at the ninth trial (i.e. the first trial at day 3; Figure 4C) indicate that spatial memory is indeed restored in the 11-min ischemic rats at 90 DAI. The beam-walk performances at 90 DAI did not differ between groups. The beam widths were 1.95 (1.575 to 2.2) for control, 2.2 (2.2 to 2.2) for 1-min ischemia rats, and 2.2 (1.7 to 2.2) for 11-min ischemia rats (medians and interquartiles).
Discussion
In the present study, we show that ischemia-induced degeneration of CA1 neurons is followed by a massive regeneration of CA1 neurons that, at least in part, is due to neurogenesis. These new nerve cells may originate from neuronal stem or progenitor cells residing in the posterior periventricle. The regeneration of CA1 neurons is associated with a restoration of CA1-dependent learning and memory tasks, indicating that the newly formed neurons are functionally incorporated.
The very existence of CA1 neurogenesis in response to ischemia has been reported previously (Nakatomi et al, 2002; Schmidt and Reymann, 2002), at a much lower magnitude than what we have found here. Several reasons may explain why most other groups have observed no recovery of CA1 neurons. Subtle differences may exist in the methods employed to induce global ischemia. In our study, halothane was used to reduce systemic blood pressure during the time of ischemia (Bendel et al, 2005b; McBean et al, 1995). It can be hypothesized that halothane slows the progression of neuronal degeneration. In the used model, the CA1 neurons degenerate much slower (Bendel et al, 2005b) than in more conventional global ischemia models, where the maximal neuronal loss is obtained already at 3 DAI (Sugawara et al, 2000, 2002). The prolonged presence of CA1 neurons may provide increased stimulation of neurogenesis, especially with regard to initiation and proliferation. Ischemia induces abnormal changes in glutamatergic neurotransmission, including postsynaptic changes in NMDA receptor function (Crepel et al, 1998; Hori and Carpenter, 1994). Recently, it has been shown that stimulation of NMDA receptors potently enhances neurogenesis (Deisseroth et al, 2004), which in that way can adapt to changed physiological demands and to pathological events such as ischemia. Glutamatergic signaling may possibly be preserved longer in our experimental set-up than in most other models, which thus may result in a much higher number of newly produced neurons. If so, delaying neurodegeneration could be an alternative therapeutic approach after cerebral ischemia. The prolonged survival of CA1 neurons may also be favorable for the intactness of supporting glial cells that may create a more attractive environment for the new neurons, for example, by secretion of trophic factors such as FGF-2 and EGF, which have been shown to stimulate CA1 neurogenesis after ischemia (Nakatomi et al, 2002). However, the very presence of CA1 neurons themselves seems not to be of crucial importance, since the newly formed neurons appeared after the almost complete degeneration of the initial CA1 neurons. We did not find evidence that any newborn neurons were embedded and integrated among the slowly degenerating ischemic CA1 neurons before 14 DAI.
It can also be hypothesized that halothane, by exerting neuroprotective effects at more distant regions and cell populations (Gotts et al, 2000), underlies the reappearance of CA1 neurons. However, this is not supported by a recent study showing a similar remarkable increase of CA1 neurons at 90 DAI after 2-vessel occlusion in combination with blood withdrawal (Elsersy et al, 2004). They reported that the reappearance occurred both when the ischemia was induced in the presence of isofluorane and when it was induced in the presence of fentanyl, suggesting that the presence of volatile anesthetics cannot account for the massive reappearance of CA1 neurons.
That not all newly formed CA1 neurons at 90 days were labeled by BrdU might be due to several causes. 5-bromo-2'-deoxyuridine administration within the tolerable concentration range has been shown to label only a fraction of the total number of dividing cells (Iwai et al, 2001). Furthermore, some cell divisions might have taken place before or after the used BrdU administration period. Finally, incorporated BrdU might have been diluted to undetectable levels due to subsequent divisions after the administration period.
Thus far, limited evidence existed that new neurons generated in the hippocampal formation in response to ischemic insult actually repair ischemia-related brain injuries. We observed a decline in MAP2-IR in the hilus at 14 DAI, an area adjacent to the subgranular zone, which may reflect a degree of dendritic damage in this region, but we did not see any cell death in the dentate gyrus itself. Yet, there was an enhanced formation of new cells in the subgranular zone, which migrate into the granule cell layer of the dentate gyrus, in line with previous studies (Choi et al, 2003; Iwai et al, 2001; Takagi et al, 1999). Hence, the reparative role of neurogenesis in this brain region after ischemia remains unknown (Kokaia and Lindvall, 2003). In the CA1 area, recovery of ischemia-induced behavioral deficiencies has previously only been shown after growth factor treatment (Nakatomi et al, 2002). Our results show that such recovery can also be achieved without the administration of growth factors. The renewal of the CA1 pyramidal cell layer is correlated to a normalization of learning and memory functions, suggesting that the new CA1 neurons indeed restore disrupted neuronal circuits.
Other researchers have also found equal Morris water maze performance in ischemic and non-ischemic rats at 90 DAI, but without any shown initial deficiency in the ischemic rats (Elsersy et al, 2004). It should be pointed out that it is mandatory to show clear-cut short-term deficiencies in learning and memory for investigating correlations between the number of CA1 neurons and behavioral performance. Indeed, a general problem with previous two-vessel occlusion models has been to achieve a clear deficiency in learning and memory, which at least in rats may be due to a variable and/or incomplete degree of degeneration of the CA1 neurons (Block, 1999).
Clearly, also other brain structures and plastic mechanisms, apart from CA1 neurogenesis, may contribute to the recovery of performance. To elucidate the relative contribution of CA1 neurogenesis for the recovery of learning and memory impairments, one will have to be able to selectively inhibit the formation of new CA1 neurons. This will not be an easy task, since the region of origin of the new CA1 neurons is not unequivocally shown. A nonselective inhibition of neurogenesis by mitotic inhibitors or irradiation will not be sufficient to solve the issue, since such treatment is known to impair learning and memory also in nonischemic animals, possibly through inhibition of neurogenesis in the subgranular zone (Feng et al, 2001; Raber et al, 2004; Shors et al, 2001).
Taken together, we have found that the adult rodent brain has a substantial endogenous capacity to form new neurons after ischemia, which may originate from progenitor cells residing in the lateral periventricle. The reappearance of CA1 neurons is associated with a restoration of the ischemia-induced impairment of spatial learning and memory performance, suggesting that neurogenesis can provide functional restoration after injury.
Footnotes
Acknowledgements
We thank Kanar Alkass and Britt Meijer for excellent technical assistance.