
Abstract
In the adult brain, neurogenesis occurs in the subgranular zone of the dentate gyrus (DG), where high levels of vesicular zinc are localized in the presynaptic terminals. To determine whether zinc has a role in modulating hippocampal neurogenesis under normal or pathologic conditions, we manipulated the level of vesicular zinc experimentally. To reduce hippocampal vesicular zinc, rats were either fed a zinc-deficient diet or treated with a zinc chelator, clioquinol (CQ). The number of progenitor cells and immature neurons was decreased significantly in the DG after 6 weeks of dietary zinc deprivation. Conversely, the number of progenitor cells and immature neurons was restored after a 2-week reversal to a normal zinc-containing diet. Similarly, a 1-week treatment with the zinc chelator, CQ, reduced the number of progenitor cells. The results of our previous study showed that hypoglycemia increased hippocampal neurogenesis. This study shows that zinc chelation reduced hypoglycemia-induced progenitor cell proliferation and neurogenesis. Finally, the role of vesicular zinc on neurogenesis was further assessed in zinc transporter 3 (ZnT3) gene deleted mice. Zinc transporter 3 knockout (KO) mice had significantly fewer proliferating progenitor cells and immature neurons after hypoglycemia. Our data provide converging evidence in support of the essential role zinc has in modulating hippocampal neurogenesis.
Introduction
Divalent cationic zinc is the second most abundant transition metal in the brain following iron. Chelatable (loosely bound) zinc is highly localized in the synaptic vesicle of mossy fiber (MF) terminals of the dentate gyrus (DG) and olfactory bulb (Perez-Clausell and Danscher, 1985), sites where neurogenesis and neural migration are most active in the adult brain (Ming and Song, 2005). Zinc has long been recognized as a biologically essential element for both brain and systemic physiology. It is a component of more than 300 enzymes, and is thus involved in the regulation of a wide variety of cellular processes, including cell division and DNA synthesis (MacDonald, 2000). Zinc also influences hormonal regulation of cell division, specifically, those cells regulated by pituitary growth hormone—insulin-like growth factor-I (MacDonald, 2000) or nerve growth factor (Stewart et al, 1984). The division and migration of cerebellar granular cells is delayed after severe zinc deficiency (Dvergsten et al, 1983; Sandstead et al, 2000). Golub et al (1994) showed that zinc deficiency impaired performance in short-term memory tasks. Thus, the evidence described above suggests that zinc is an essential element required in cell division, proliferation, migration, and development, and further suggests that this element may have a critical role in neurogenesis and cognitive function.
Hypoglycemia occurs frequently in diabetic patients who attempt tight control of their blood glucose levels (Hershey et al, 1999; Rovet and Ehrlich, 1999). The results of our previous study showed that hypoglycemic brain insult transiently increases the number of proliferating progenitor cells and immature neurons in the DG of rats (Suh et al, 2005), followed by a sustained decrease in progenitor cell proliferation and immature neurons weeks later. The mechanism underlying the rise and decline in the hippocampal progenitor cell proliferation is unclear. Our results suggest that the high amount of vesicular zinc in the MF terminals of the dentate granule neurons is released after hypoglycemia, lasting from several hours to several days (Suh et al, 2004). Although zinc release and translocation have been implicated as the upstream events of hypoglycemia-induced neuronal death (Suh et al, 2004, 2008), the relationship between the zinc released from the MF terminals during acute hypoglycemia and neurogenesis has not been established (Stewart et al, 1984). Zinc homeostasis in the brain can be regulated by dietary zinc intake. It has previously been shown that zinc concentration in the presynaptic vesicle of the hippocampus was decreased in rats fed a zinc-deficient (ZD) diet for 4 weeks. In addition, zinc deficiency by dietary zinc deprivation is associated with a decreased learning ability (Golub et al, 1994; Takeda et al, 2000). However, the precise mechanism of the learning impairment in ZD animals is still unclear. As learning and memory are linked to hippocampal neurogenesis, it is possible that a ZD diet may affect neurogenesis in the adult animal. This study sought to determine the role of vesicular zinc in modulating hippocampal neurogenesis in the normal brain after hypoglycemia. In addition to dietary zinc deprivation, we used two more experimental approaches, namely pharmacological chelation of zinc with a cell-permeable zinc chelator, (5-chloro-7-iodo-8-hydroxyquinoline; clioquinol, CQ) and genetic vesicular zinc depletion by targeted gene deletion of the zinc transporter 3 (ZnT3 knockout (KO)), to study the effect of reducing vesicular zinc on neurogenesis. Hippocampal progenitor cell proliferation and neurogenesis was determined in each experimental condition, and the hippocampal MF zinc level was evaluated by autometallography or fluorescence zinc staining to verify the efficacy of zinc depletion.
Materials and methods
Zinc Deficiency Diet Feeding
To determine the effect of dietary zinc deficiency on basal progenitor cell proliferation, rats were subjected to a ZD diet. We have previously established that a 4-week dietary zinc deprivation regiment has significantly depressed vesicular zinc level and triggered less zinc release into the extracellular space (Takeda et al, 2003). In this study, we extended the period of dietary zinc deprivation to 6 weeks to ensure the depletion of brain vesicular zinc. Zinc-deficient diet experiments were conducted at the School of Pharmaceutical Sciences in the University of Shizuoka, Japan. All experiments were carried out in accordance with the Japanese Pharmacological Society guide for the care and use of laboratory animals. Normal diet (ND; 44mg Zn per kg) and ZD (2.7 mg Zn per kg) diet were purchased from Oriental Yeast Co. Ltd. (Yokohama, Japan). Normal diet and ZD diet differ only in zinc concentration. Oriental MF diet composition was described in a reference paper (Mori et al, 1987) and in Online Supporting Material 1. Male Sprague—Dawley rats (3 weeks of age) were purchased from Japan SLC (Hamamatsu, Japan). Animals were housed in room temperature at 23°C ± 1°C and humidity of 55% ± 5% and had free access to tap water and food. Room lights were automatically turned on at 0800 and off at 2000 hours. The ZD diet was begun at 4 weeks of age, and the rats were fed the diet for 4 weeks. Thereafter, ZD and control rats were injected intraperitoneally with 5-bromo-2-deoxyuridine (BrdU) (50 mg/kg) twice a day for 4 days. After the start of BrdU injections, one group of ZD rats were fed the same ZD diet for 2 more weeks, whereas another group of ZD rats were fed the normal zinc-containing diet (ZD + ND) for 2 weeks after the 4 weeks of ZD diet. The control group was fed the normal zinc-containing diet (ND) for 6 weeks. After these 6 weeks, all three groups of rats were deeply anesthetized, and then perfused intracardially with 4% (w/v) paraformaldehyde in 0.1 mol/L of sodium phosphate buffer (pH 7.4). The rat's body weight was measured at the time of killing. The body weight data are available in the Online Supporting Material 2.
Surgery and Hypoglycemia Induction
To investigate the role of zinc chelation on hypoglycemia-induced progenitor cell proliferation, rat or mouse hypoglycemia surgery was performed. This study was approved by the San Francisco Veterans Affairs Medical Center Animal Studies Committee. Animals were housed two per cage under conditions of constant room temperature (18°C to 20°C) and humidity (50% to 55%), and had free access to tap water and food. Room lights were automatically turned on at 0600 and off at 1800 hours. In this study, we used both rats and mice that were fed a normal zinc-containing diet (Harlan TEKLAD rodent diet 7956, San Francisco, CA, USA) for the entire experiment. Two-month-old male Sprague—Dawley rats (230 to 250 g; Charles River, CA, USA) and 3-month-old male C57/B16 mice (20 to 25 g; Charles River) were housed and cared for according to federal and institutional guidelines. Hypoglycemia surgery for animals was performed as previously described (Auer et al, 1984; Suh et al, 2003). Hypoglycemia was induced by an intraperitoneal injection of 10 U/kg (in rats) or 5 U/kg (in mice) of regular insulin (Novolin-R, Novo Nordisk, NC, USA). Isoelectric electroencephalograph was maintained for 30 mins in rats and for 45 mins in mice. Blood glucose was measured using an YSI 2700 glucose analyzer (YSI, Yellow Spring, OH, USA) at 30-min intervals during the induction of hypoglycemia and at 60-min intervals during recovery from hypoglycemia. Mean arterial blood pressure was maintained between 160 and 200 mmHg during the entire isoelectric period by adjusting isoflurane concentration, and bradycardia during hypoglycemia was prevented with atropine (1 mg/kg; American Pharmaceutical Partners, Los Angeles, CA, USA). Hypoglycemia in the rats was terminated by an intravenous injection infusion of 1:1 solution of 50% glucose and Krebs—Henseleit buffer (1.5 mL/h for 3 h). Hypoglycemia in the mice was terminated by an intraperitoneal injection of 100 μL of 25% glucose. Animals were monitored under anesthesia for 3 h after the onset of glucose infusion. Anesthesia was discontinued after removal of the arterial and venous catheters, and the animals were returned to their cages when fully awake and ambulatory. One rat and two mice that showed seizure activity were discarded from this study.
Zinc Chelator, Clioquinol, Injection
To depress vesicular zinc level or to chelate extracellular zinc, a zinc chelator, CQ, was used. Three-month-old male rats were injected with CQ (30 mg/kg, intraperitoneally) twice a day (0900 to 1000 hours and 1700 to 1800 hours) for 1 week with or without hypoglycemia. Clioquinol was dissolved with DMSO (dimethyl sulfoxide) (30 mg per 300 μL; Sigma, St Louis, MO, USA) and then injected intraperitoneally. In the rats that were subjected to hypoglycemia, CQ injection was started immediately after 30 mins of isoelectric electroencephalography Control rats were injected with the same volume of DMSO. The nonhypoglycemia group also had CQ/DMSO or DMSO vehicle only. For this experiment, adult male rats were fed a ND.
5-Bromo-2-Deoxyuridine Labeling
To test the effects of ZD diet on neurogenesis, BrdU was injected twice daily for four consecutive days starting 4 weeks after the ZD diet. The thymidine analog, BrdU, was administered intraperitoneally (50 mg/kg; Sigma) to investigate the progenitor cell proliferation. The rats were killed 10 days later. To test the effects of zinc refreshment after zinc deficiency, the rats were fed a ND and simultaneously injected with BrdU starting 4 weeks after the ZD diet. To test the zinc chelation effects on neurogenesis after hypoglycemia, the rats received BrdU injections twice daily for four consecutive days from the third day after hypoglycemia, and were killed on day 7.
Immunohistochemistry and Immunofluorescence Staining
Rats and mice were anesthetized with isoflurane and then transcardially perfused with 4% paraformaldehyde. Their brains were removed, postfixed, and then cryoprotected by 20% sucrose. Free-floating coronal sections of 40-μm thickness were immunostained as described (Suh et al, 2005) using the following reagents: mouse anti-BrdU (Roche, Indianapolis, IN, USA); rat anti-BrdU (Accurate Chemicals, Westbury, NY, USA); rabbit anti-Ki67 (recognizing nuclear antigen expressed during all proliferative stages of the cell cycle, except GO (Kee et al, 2002); Novocastra, Newcastle UK); goat anti-doublecortin (DCX) (recognizing immature neurons) (Nacher et al, 2001); Santa Cruz Biotechnology, Santa Cruz, CA, USA); and ABC solution (Vector laboratories, Burlingame, CA, USA).
Cell Counting
For Ki67 and DCX immunohistochemistry, every twelfth (for rats) or sixth (for mice) coronal section spanning the septal hippocampus was collected. The number of Ki67-and DCX-immunopositive cells was determined in the subgranular zone and granule cell layer using optical fractionators probe unbiased stereology investigation (Stereo Investigator, MicroBrightField, Williston, VT, USA) (Gundersen et al, 1988). Counting frames (20 times 20 times 20 μm for rats; 15 times 15 times 20 μm for mice) were placed at the intersection of a matrix (100 times 100 μ for rats; 40 times 40 μ for mice) randomly superimposed onto the region of interest by the program. Cells were counted using a × 63 oil objective. For BrdU immunohistochemistry, the number of BrdU-immunopositive cells in the subgranular zone and in the granule cell layer was counted under a Zeiss microscope (Carl Zeiss Microimaging, Inc, Thornwood, NY, USA) in every twelfth (for rats) or sixth (for mice) coronal section spanning the septal hippocampus.
Fluorescence Zn2+ Staining (TSQ Method)
Vesicular free zinc was imaged using the N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) method (Frederickson et al, 1987). Rats were euthanized 3 h after CQ (30 mg/kg) treatment and the fresh frozen brains were sectioned coronally. Five evenly spaced sections were collected through the hippocampal region of each brain and immersed in a solution of 4.5 μmol/L TSQ (Molecular Probes, Eugene, OR, USA) for 60 secs, then rinsed for 60 secs in 0.9% saline. The TSQ—zinc binding was imaged and photographed using a fluorescence microscope with 360 nm of ultraviolet light and a 500-nm long-pass filter. The mean fluorescence intensity within the MF terminal area was measured and expressed as arbitrary intensity after subtraction of background fluorescence as measured in the lateral ventricle. Measurements from the five sections were averaged for each observation.
Autometallography Zn2+ Staining (AMG Method)
Four weeks after hypoglycemia or sham operation, rat brains were stained with zinc-selenium autometallography for evaluating MF zinc contents. Rats were injected with sodium selenide (10 mg/kg, intraperitoneally) dissolved in phosphate-buffered saline under isoflurane anesthesia. One and a half hours later, the rats were anesthetized and then killed by transcardial perfusion with 3% glutaraldehyde solution in phosphate-buffered saline. The brains were cryoprotected in 20% sucrose followed by freezing with dry ice, cut into 20 μm-thick sections on a cryostat, and placed on rinsed slides with Farmer's solution (Danscher and Stoltenberg, 2005). The AMG silver enhancement was performed with the original silver lactate developer (Danscher, 1982). The stained sections were dehydrated in alcohol, cleared in xylene, coverslipped with mounting media, and then examined under light microscope. After AMG staining, sections were mounted on the microscope, transilluminated with a fixed intensity of white light (tungsten), and individual images were captured by camera, digitized, and stored. Every section that was obtained from an individual brain slice (typically 5 to 10) was used for quantification.
Neural Stem Cells Self-Renewal Assay
Embryonic day-14 forebrain tissue from wild-type mice was dissected and triturated into single cell suspension and cultured in the neurosphere culture medium (DMEM (Dulbecco's modified Eagle's medium)/Fl2 at 1:1 supplemented with 2% B27 and epidermal growth factor (EGF) at 20 ng/mL) to form primary floating neurospheres. One week later, 10 single primary neurospheres were mechanically dissociated into single cell suspension. A total of 400 viable cells per well were plated into a 6-well cell culture plate containing neurosphere culture medium in three to four replicates per mice. The number of secondary neurospheres generated in each well was counted 10 days after plating.
Statistical Analysis
All data were expressed as means ± s.d. The statistical significance of differences between means was calculated using SPSS (SPSS, Chicago, IL, USA). For statistical comparisons between data from ND, ZD diet, and ZD diet plus ND feeding rats in BrdU-, Ki67- and DCX-positive cells, significance was determined using one-way ANOVA (analysis of variance) followed by Bonferroni post hoc test. For statistical comparisons between data from all other experiments, significance was evaluated by two-tailed Student's t-test. P-values < 0.05 were considered significant.
Results
Dietary Zinc Deficiency Reduced Hippocampal Progenitor Cell Proliferation
After 6 weeks of consuming ZD diet, the number of Ki67-, BrdU-, and DCX-immunoreactive cells at the DG was lower than in the normal zinc-containing diet (ND) group (Figures 1A–1D, P < 0.05). In the 4-week ZD and then in the 2-week normal zinc diet (ZD—ND) group, the number of BrdU- (Figure 1A), Ki67- (Figure 1B), and DCX-immunoreactive cells (Figures 1C and 1D) was recovered 2 weeks after normal zinc-containing diet feeding.
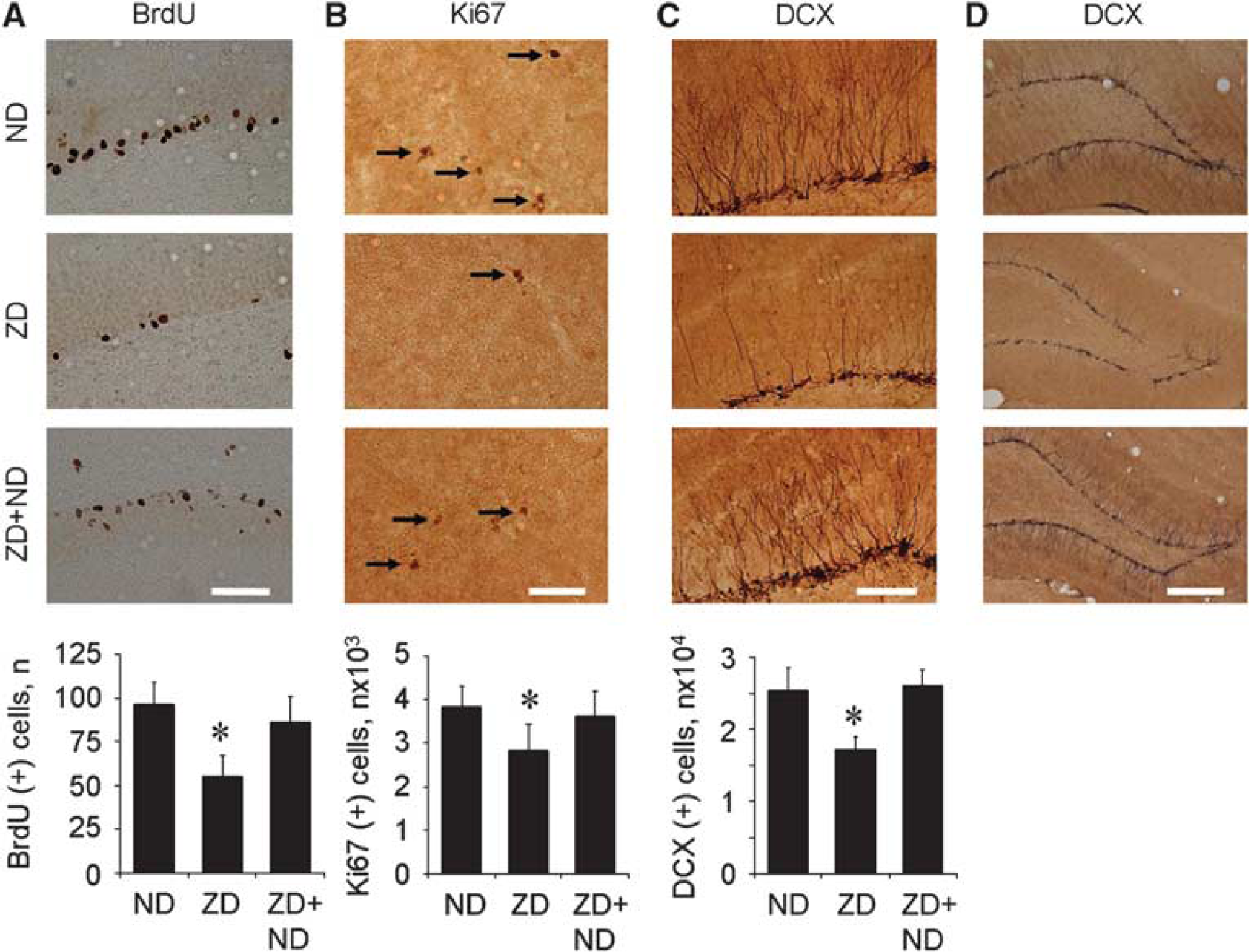
Zinc-deficient (ZD) diet decreased progenitor cell proliferation and neuroblast production in rats. AZD diet decreased BrdU-(
Clioquinol Decreased Vesicular Zinc and Hippocampal Progenitor Cell Proliferation
Consistent with a previous observation (Nitzan et al, 2003), 3 h after CQ treatment (30 mg/kg), the intensity of MF zinc in the rat hippocampus was 43.0% ± 6.8% lower than in vehicle-treated controls (Figure 2A). There were fewer BrdU-, Ki67-, or DCX-labeled cells in the CQ-injected rats compared with that in the vehicle-treated rats (Figures 2B–2D).
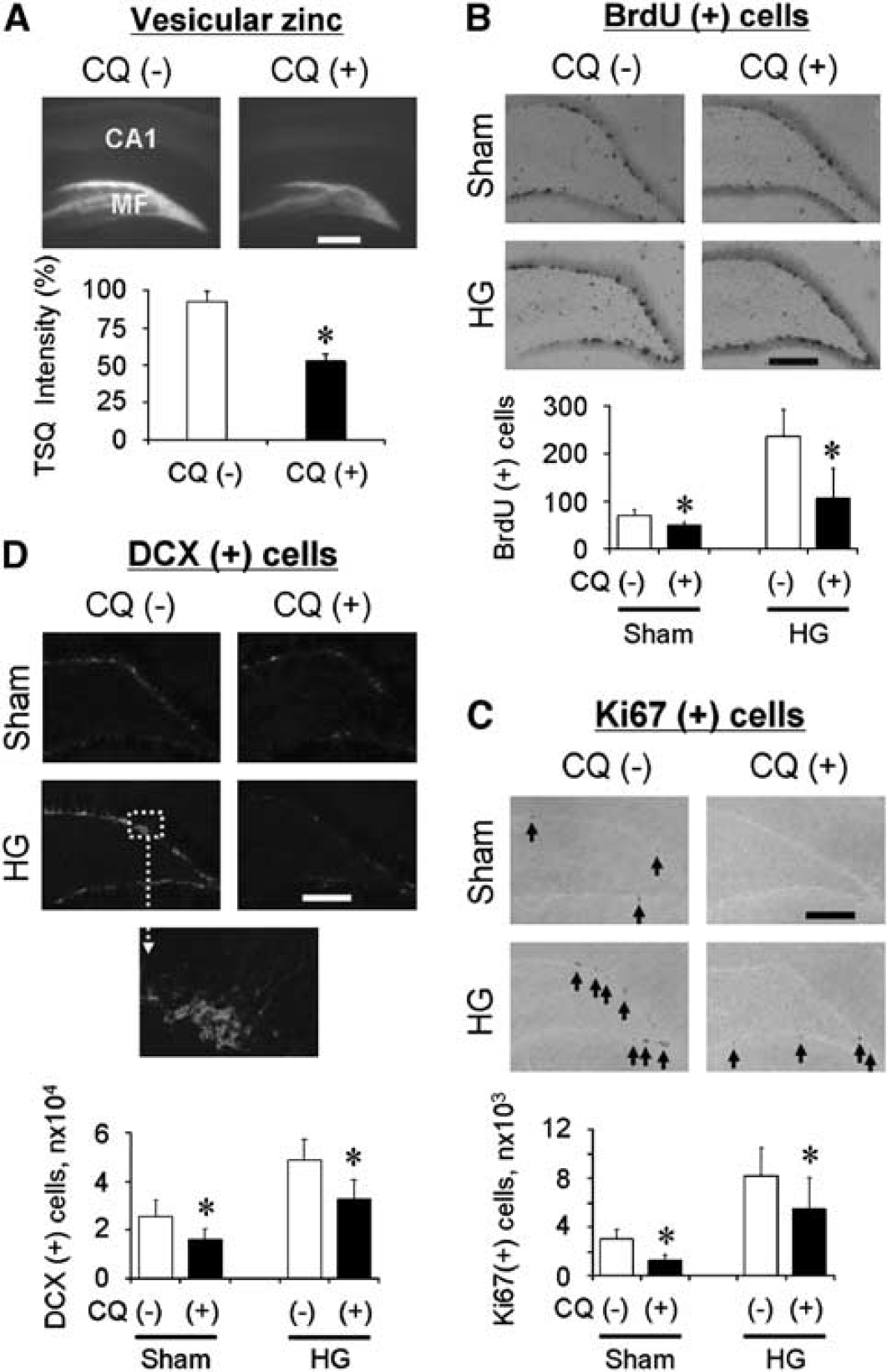
Zinc chelator decreased vesicular zinc, and basal or hypoglycemia (HG)-induced progenitor cell proliferation, and neuroblast production in rats. (
Zinc Chelation Reduced Hypoglycemia-Induced Progenitor Cell Proliferation
After 1 week, a group of rats that were subjected to hypoglycemia showed increased progenitor cell proliferation and the number of immature neurons as detected by immunohistochemistry against BrdU, Ki67, and DCX (Figures 2B–2D). However, a group of 1-week CQ-treated rats showed lower number of BrdU-, Ki67-, or DCX-immunoreactive cells in the hippocampus after hypoglycemia (Figures 2B–2D).
ZnT3 Knockout Lowered Hypoglycemia-Induced Progenitor Cell Proliferation and Neurogenesis
To assess the role of vesicular zinc in mediating hippocampus neurogenesis after acute hypoglycemia, we determined the level of neurogenesis in wild-type and in ZnT3 KO mice. The TSQ-stained section from ZnT3 KO mice showed completely devoid fluorescence intensity in the hippocampus either after sham operation or after hypoglycemia (Figure 3A). In this study, we found that sham-operated wild-type and ZnT3 KO mice showed a similar number of progenitor cells (Figure 3B) and immature neurons (Figure 3C), suggesting that ZnT3 deletion did not affect either progenitor cell proliferation or immature neuron production in mouse hippocampus without hypoglycemia insult. This result is different with pharmacological zinc chelation by CQ. Following 1 week after hypoglycemia (Suh et al, 2007a), we detected a robust increase in hippocampal neurogenesis in the wild-type mice similar to that observed in the rats. However, the increase of Ki67- and DCX-labeled cells after hypoglycemia was less pronounced in the ZnT3 KO mice (Figures 3B and 3C). In support of the results obtained with pharmacological zinc chelation on reduction of synaptic zinc, targeted genetic depletion of ZnT3 also attenuated hypoglycemia-induced neurogenesis in the adult rodent brains.
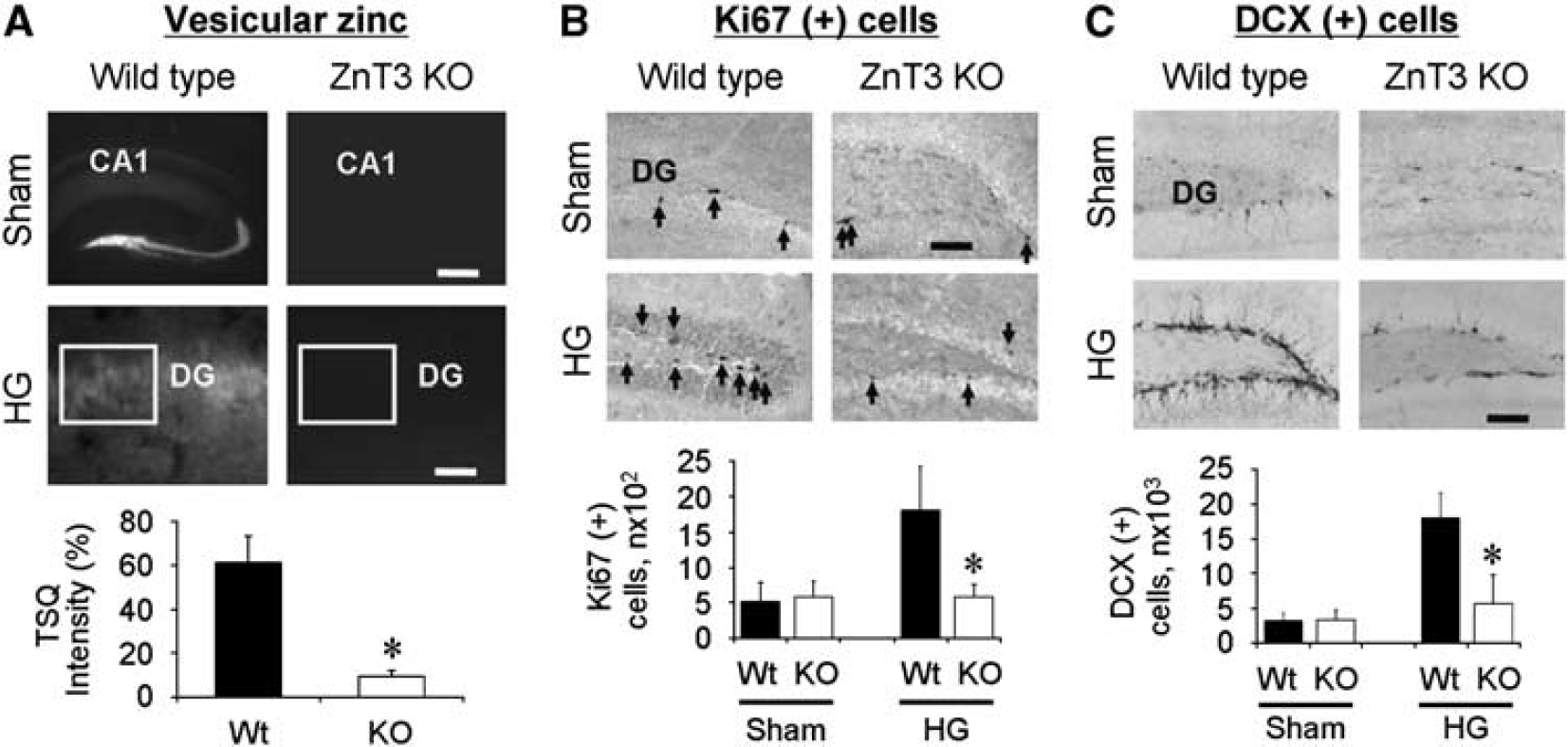
ZnT3 KO mice showed the absence of vesicular zinc, and decreased hypoglycemia-induced progenitor cell and neuroblast production. (
Progenitor Cell Proliferation is Associated with Vesicular Zinc at Day 4 or 4 Weeks After Hypoglycemia
The results of our previous study showed that neurogenesis after hypoglycemia was significantly depressed at 4 weeks after insult (Suh et al, 2005). To investigate the relationship between the amount of vesicular zinc and the state of hippocampal neurogenesis, brain sections were processed for TSQ (Frederickson et al, 1987) and AMG zinc staining (Danscher and Rytter Norgaard, 1985). Four days (day 4) after hypoglycemia, the intensity of zinc staining was increased by 143.80% ± 15.05% in the dentate granule cell layer (rectangular area in Figure 4A) compared with sham-operated rats. At this time point, the number of Ki67 (+) cells in the dentate granule cell layer was substantially increased as we saw before (Suh et al, 2005) (Figure 4B). Compared with the abundance of vesicular zinc in the sham-operated rats, the density of AMG staining was significantly decreased by 70.90% ± 1.16% in the MFs 4 weeks after hypoglycemia (Figure 4C). There was also a concurrent reduction of Ki67 immunoreactivity in the subgranular zone at 4 weeks after hypoglycemia compared with the sham-operated control rats (Figure 4D). Thus, these results support our hypothesis that higher zinc availability in the subgranular zone of the hippocampus may increase progenitor cell proliferation and, in contrast, loss of vesicular zinc caused by hypoglycemia-induced dentate granule cell death may depress neurogenesis or decrease newborn cell survival.
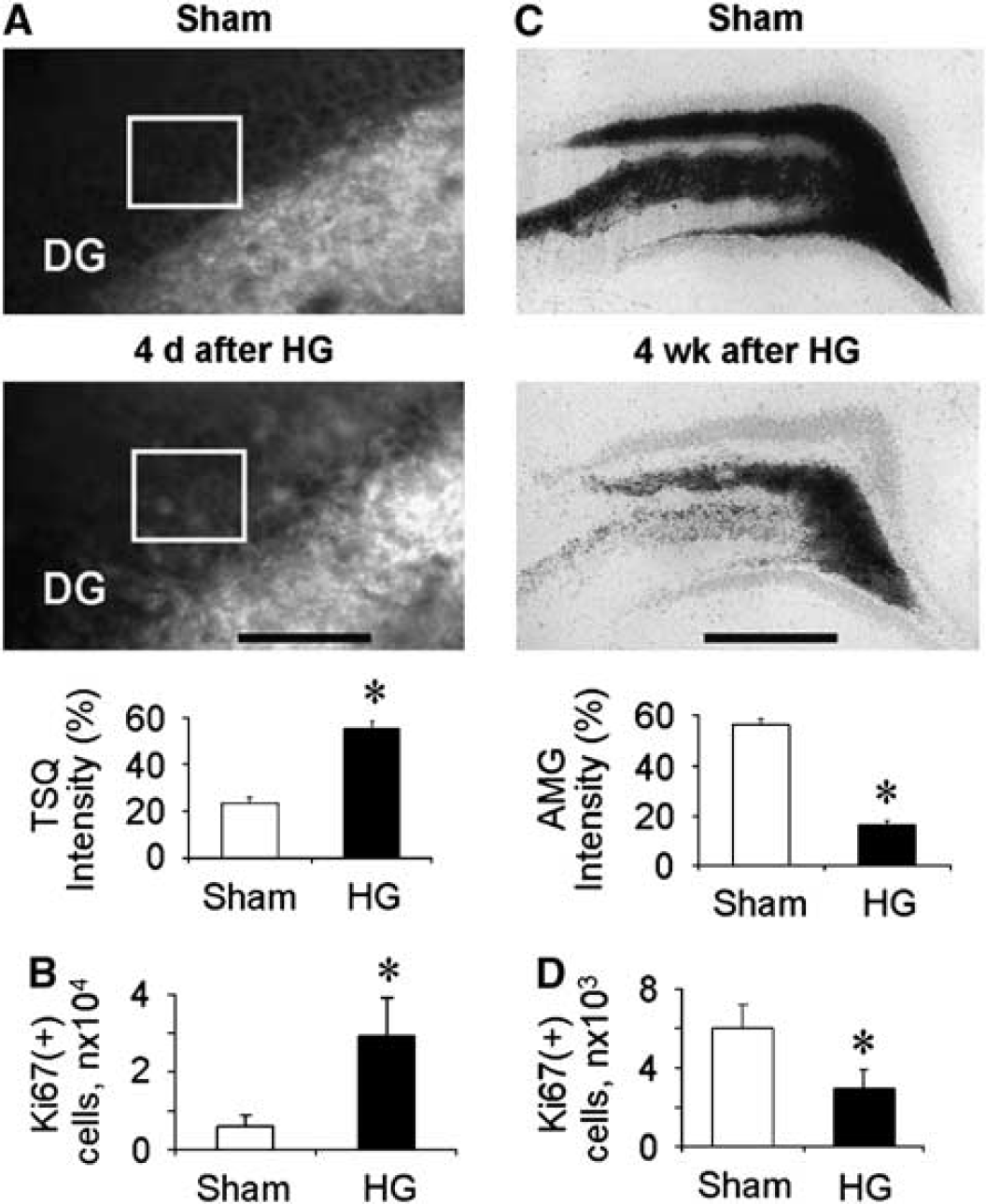
Decreased Ki67-labeled cells is related to lower vesicular zinc level in rat hippocampus. (
Zinc Chelation Reduced Secondary Neurosphere Formation
To investigate whether zinc chelation affects the self-renewal capacity of neural stem cells (NSCs) in vitro, single cells from primary neurospheres were cultured in the presence of the extracellular zinc chelator, calcium ethylenediaminetetraacetic acid (CaEDTA), or with the nonzinc chelator, ZnEDTA. The formation of the second neurosphere was 44.5% ± 6.5% reduced by 44.5% ± 6.5% with 100 μmol/L of CaEDTA treatment in the media. Contrary to the effects of CaEDTA, ZnEDTA showed little effects on NSC self-renewal (Figure 5).
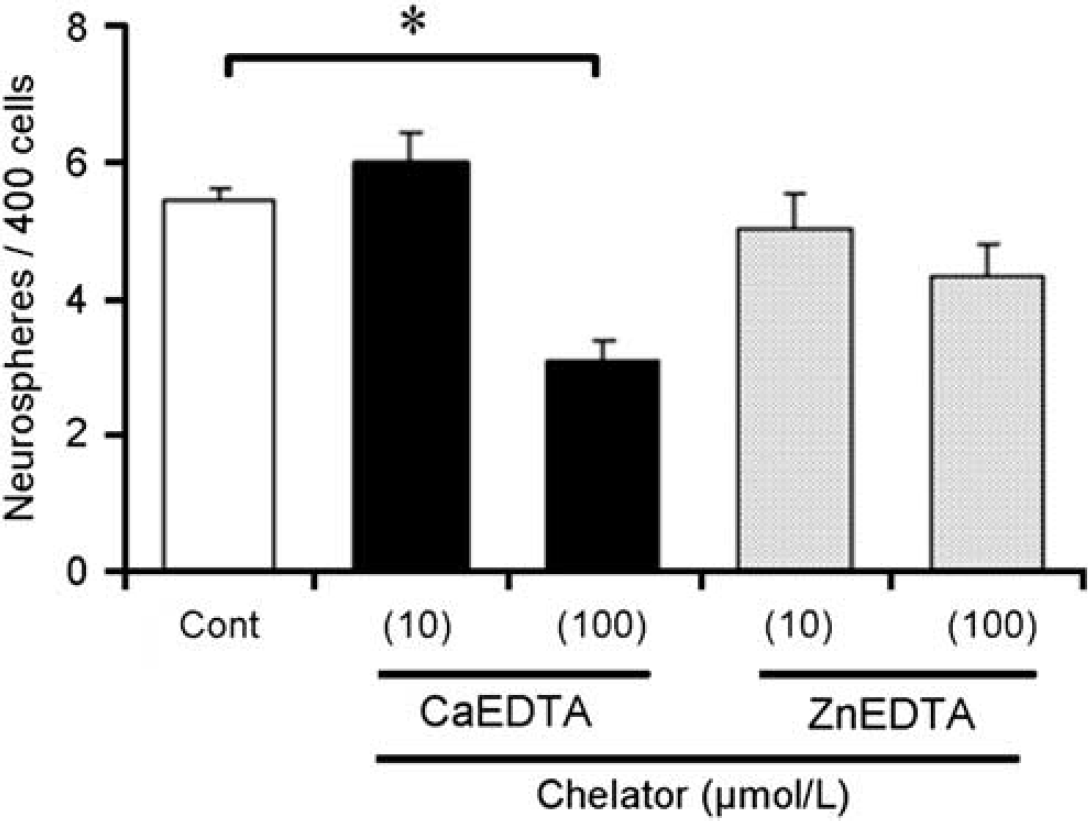
Extracellular zinc chelator, CaEDTA, inhibited secondary neurosphere formation. The number of secondary neurospheres was estimated under a microscope from 400 dissociated primary neurospheres 10 days after plating. Cont: neurosphere cell cultured in normal culturing medium. CaEDTA: neurosphere cells cultured in zinc chelator, CaEDTA (10 or 100 μmol/L). ZnEDTA; as a control, neurosphere cell cultured in nonzinc chelator, ZnEDTA (10 or 100 μmol/L). Values are means ± s.d., n = 5. *Different between vehicle-treated control (Cont) and CaEDTA-treated (100 μmol/L) neurosphere cells. P<0.05.
Discussion
The present evidence suggests that zinc in the brain might have a modulatory role in hippocampal neurogenesis either in the normal brain or in the hypoglycemia-insulted brain. This study provides validation of our hypothesis on the basis of converging evidences from four different approaches: first, dietary zinc deprivation decreased progenitor cell proliferation; second, pharmacological zinc chelation substantially prevented basal or hypoglycemia-induced progenitor cell proliferation; third, genetic vesicular zinc depletion decreased hypoglycemia-induced neuroblast generation; finally, reduction of the hippocampal MF zinc level is associated with a decline in progenitor cell proliferation. This study shows that vesicular zinc is an important mediator of neuronal regeneration in the hippocampus either under normal physiologic conditions or after brain insult.
Chelatable zinc is present in the MF of dentate granule cell of the hippocampus (Danscher and Rytter Norgaard, 1985; Frederickson, 1989). High levels of zinc accumulation in postsynaptic neurons contribute to direct neuronal death in several disease conditions, such as prolonged seizures (Frederickson et al, 1989; Suh et al, 2001), ischemia (Tonder et al, 1990; Koh et al, 1996), brain trauma (Suh et al, 2000, 2006), and hypoglycemia (Suh et al, 2004, 2007b). However, equally abundant studies have shown the beneficial or constitutive role of zinc (Sandstead et al, 2000). Zinc participates in the regulation of cell proliferation in several ways; it is essential to enzyme functions, which influence cell division and proliferation. Several studies have shown that zinc deficiency altered postnatal development of the brain (Halas et al, 1983, 1986). Thus, the evidences outlined above confirm that zinc is an essential transition element in cell division and proliferation, and further suggests that this element has a critical role in neurogenesis in the adult rodent brain.
To test the hypothesis that zinc is an essential element for neurogenesis, first, rats were subjected to a ZD diet for 6 weeks. Previously, one of us showed that zinc concentration in the brain was significantly decreased during the 4-week ZD diet, but zinc was restored by feeding a zinc-adequate diet for 2 weeks (Takeda et al, 2003). We found that progenitor cell proliferation after 6 weeks of ZD diet significantly depressed the number of BrdU-, Ki67-, and DCX-positive cells in the hippocampus. This decrease in all the three markers was recovered by 2 weeks of adequate zinc-containing diet. This result shows that dietary zinc replenishment for 2 weeks can either prevent the degeneration of progenitor cells or increase de novo production. This study suggests that replenishment of zinc can have both roles, prevention of degeneration of progenitor cell and support of de novo synthesis. Recently, Corniola et al (2008) published that zinc deficiency impaired neurogenesis by the activation of proapoptotic gene induction. However, we cannot exclude the fact that ZD rats are not only ZD but also malnourished and hyperglycemic. All three factors can influence hippocampal neurogenesis. For this reason, in the next experiment, instead of inducing zinc deprivation through diet, we used the chemical zinc chelator, CQ, to see the zinc deprivation effects on hippocampal neurogenesis. Therefore, we tested our hypothesis that zinc chelation by CQ would reduce neurogenesis in the hippocampus of normal rats and in rats with a hypoglycemic insult. The results of our previous study showed a transient increase of progenitor cells after hypoglycemia until 2 weeks after insult (Suh et al, 2005). The reason for an increase in neurogenesis activity at early time points after hypoglycemia is uncertain. This study hypothesized that this transient increase in neurogenesis after hypoglycemia is related to the synaptic release of zinc and cytoplasmic liberation after dentate granule cell degeneration. Figure 4 represents several zinc accumulating neurons in cell bodies. Previously, our study suggested that those zinc-accumulated neurons degenerated after hypoglycemia. We believe that continuous free zinc liberation from the degenerating dentate granule cell may continuously stimulate progenitor cell proliferation and support the survival of neuroblast after hypoglycemia insult. Therefore, we tested the effects of zinc chelation on basal neurogenesis and on hypoglycemia-induced transient neurogenesis. Seven days of continuous treatment with CQ without hypoglycemia significantly decreased basal progenitor cell proliferation in the hippocampus compared with the vehicle-treated group, with a parallel reduction in the number of neuroblasts. Moreover, 7 days of consecutive treatment with CQ after hypoglycemia also substantially reduced progenitor cell proliferation in the hippocampus. These results suggest that zinc in the brain modulates neurogenesis after hypoglycemia. However, a major concern regarding the use of CQ is that this chelator is not entirely zinc specific, as it can also chelate other transitional metals in the brain, such as copper or iron (Nitzan et al, 2003). To verify our present finding that reduction of neurogenesis by CQ treatment is solely because of the depletion of extracellular zinc ion, we will need a more specific zinc chelator for the future study. Another concern is that CQ may not decrease the total amount of brain zinc. However, we speculate that CQ binds with chelatable (or free) zinc in the extracellular space and in the intracellular area, which depresses brain zinc availability to support neurogenesis.
Thus, to further verify our hypothesis, we used genetically mutated ZnT3 KO mice possessing no vesicular zinc in the presynaptic terminal (Palmiter et al, 1996; Cole et al, 1999; Linkous et al, 2008). The basal level of Ki67 and DCX (+) cells is similar between wild-type and ZnT3 KO mice possibly because of a floor effect, although the vesicular zinc level was completely absent in ZnT3 KO mice. Although, recent data by Frederickson et al have suggested that the ZnT3 KO mice have less amount of free zinc in the hippocampus (Linkous et al, 2008), it is possible that the basal amount of zinc for physiological function is supplied from blood, cerebrospinal fluid, or other unknown sources. Similar to rats, wild-type mice also showed a substantial increase in progenitor cell proliferation and neuroblast in the hippocampus at 1 week after hypoglycemia. However, ZnT3 KO mice showed fewer progenitor cells and neuroblasts after hypoglycemia. These results suggest that burst release of vesicular zinc from the synaptic vesicle is required for the transient increase in progenitor cell after hypoglycemia.
Other than zinc deprivation strategies stated above, we present here one more line of evidence that vesicular zinc is involved in neurogenesis. Our previous study found that neurogenesis is gradually reduced and declines below basal level at 4 weeks after hypoglycemia (Suh et al, 2005). The reason for this decline in progenitor cell proliferation after hypoglycemia is not clear. However, possible explanations may include a loss of progenitor cells as a result of direct injury, a reduced survival of newly formed neuroblasts resulting from an unfavorable microenvironment, or suboptimal DNA synthesis for stem cell production after hypoglycemia. As several studies have shown that hypoglycemia leads to dentate granule cell damage, it is not surprising that their vesicular zinc depletion from their synaptic terminal can be a consequent event at several weeks after hypoglycemia. In this study, we found that the content of vesicular zinc is significantly depressed at 4 weeks after hypoglycemia, which may be related to depressed progenitor cell proliferation at this time point. Thus, on the basis of the above findings, we propose that there is a close association between the depletion of vesicular zinc and reduced neurogenesis.
In lieu of converging in vivo evidences supporting the crucial role of zinc in neurogenesis, we found that zinc chelation significantly reduced the ability of embryonic NSC in forming secondary neurospheres. The reduction of NSC self-renewal by CaEDTA, but not by ZnEDTA, suggests that zinc is an essential element for NSC self-renewal. Furthermore, concentrations of CaEDTA or ZnEDTA (10 to 100 μmol/L) used in this study are not cytotoxic according to previous studies (Koh et al, 1996).
Taken together, our study shows that vesicular zinc in the hippocampus modulates neurogenesis in the adult brain under physiological and pathologic conditions. The mechanisms involved in the zinc-mediated hippocampal neurogenesis warrant further investigation.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.