
Abstract
Epilepsy is a common, chronic neurologic disorder characterized by recurrent unprovoked seizures. Experimental modeling and clinical neuroimaging of patients has shown that certain seizures are capable of causing neuronal death. Such brain injury may contribute to epileptogenesis, impairments in cognitive function or the epilepsy phenotype. Research into cell death after seizures has identified the induction of the molecular machinery of apoptosis. Here, the authors review the clinical and experimental evidence for apoptotic cell death pathway function in the wake of seizure activity. We summarize work showing intrinsic (mitochondrial) and extrinsic (death receptor) apoptotic pathway function after seizures, activation of the caspase and Bcl-2 families of cell death modulators and the acute and chronic neuropathologic impact of intervening in these molecular cascades. Finally, we describe evolving data on nonlethal roles for these proteins in neuronal restructuring and cell excitability that have implications for shaping the epilepsy phenotype. This review highlights the work to date on apoptosis pathway signaling during seizure-induced neuronal death and epileptogenesis, and speculates on how emerging roles in brain remodeling and excitability have enriched the number of therapeutic strategies for protection against seizure-damage and epileptogenesis.
Epilepsy and the Impact of Seizures on the Brain
Epilepsy is the most common neurologic disorder affecting people of all ages (Chang and Lowenstein, 2003; Meldrum and Bruton, 1992). Up to 3% of the population will suffer epilepsy at some point in their lives, and approximately 50 million people are affected worldwide. The incidence of epilepsy is highest in the first decade of life and after the age of 60 years, with rates in men usually higher (Meldrum and Bruton, 1992). Morbidity and mortality are significant problems for patients; severe limitations are often placed on working and driving, and the rate of sudden death in people with epilepsy is 24 times higher than the general population.
The defining characteristic of epilepsy is a tendency to recurrent, unprovoked seizures (Chang and Lowenstein, 2003). An epileptic seizure results from a temporary physiologic dysfunction of the brain caused by hypersynchronous discharge of neurons. The etiology of epilepsy is frequently unknown and referred to as cryptogenic (or idiopathic). Where a pathologic condition such as a malformation or tumor is found in the brain, epilepsy is symptomatic (or acquired). While there are at least 40 distinct epilepsy syndromes, they are classified under two general categories, generalized and partial; the former describing seizures beginning simultaneously in both cerebral hemispheres, while seizures in the latter originate in one or more localized foci, but can spread to involve the entire brain (termed secondary generalization). Temporal lobe epilepsy (TLE) is the most common and most intractable syndrome in adults, and about a third of TLE patients continue to experience seizures despite taking antiepileptic drugs (AEDs).
A link between epilepsy and brain damage is among the oldest known clinical–neuropathologic correlates, and as many as 70% of drug-refractory epilepsy patients have temporal lobe pathology. Experimental models and clinical studies have long-confirmed that a prolonged seizure or status epilepticus (SE) can cause neuronal death in the brain. Cell loss within Ammon's horn in the hippocampus remains the most commonly observed lesion, although damage to other limbic regions, the cerebellum and cerebral cortex, are also common (Meldrum and Bruton, 1992). It has been difficult in clinical practice to dissociate the consequences of epilepsy from its causes, from treatment of the disease or the sequelae of seizures themselves. However, the emergence of high-resolution neuroimaging of patients has revealed damage progression in epilepsy patients who continue to experience seizures (Briellmann et al, 2002; Fuerst et al, 2003; Jokeit et al, 1999; Kalviainen et al, 1998). Underlining the potential impact of such changes, patients who continue to experience seizures may have associated cognitive decline, particularly in memory function (Helmstaedter et al, 2003; Jokeit et al, 1999). As a result, there has been considerable interest in defining the molecular pathways involved in seizure-induced neuronal death. This may in turn lead to therapeutic strategies for not only mitigating damage and cognitive dysfunction in epilepsy but also for influencing epileptogenesis after a brain injury.
Apoptosis
Research on the mechanism by which neurons die after brain injuries such as stroke and SE has identified the involvement of apoptosis, a form of programmed cell death (Liou et al, 2003). Apoptosis is a physiologic process for killing cells that is critical for the normal development and function of multicellular organisms (Strasser et al, 2000). On the basis of ultrastructural analysis using electron microscopy, apoptosis was originally described as a series of well-defined and stereotyped morphologic changes in dying cells after certain stimuli, including growth factor withdrawal. Key features include aggregation of chromatin in large masses that abut the nuclear membrane, preservation of intracellular organelle integrity, and later dispersal of the cell contents in membrane-bound ‘apoptotic bodies’ (Wyllie et al, 1980). In contrast, the features of necrosis are gross mitochondrial matrix swelling, dilatation of the endoplasmic reticulum (ER), and rupture of intracellular membranes. While nuclear chromatin may initially be uniformly compacted during necrosis, chromatin masses later become evident before ultimate cell lyses leading to local tissue inflammation (Wyllie et al, 1980). Apoptosis is performed by a highly ordered molecular cascade that is typically energy-dependent and may involve new gene transcription. In a series of seminal papers, key genes were identified in the nematode worm Caenorhabditis elegans that promoted (ced-3, ced-4) or inhibited (ced-9) apoptosis (Ellis and Horvitz, 1986; Hengartner et al, 1992). Subsequently, mammalian homologues of these genes (Hengartner and Horvitz, 1994; Yuan et al, 1993) led to the characterization of two major families of genes regulating apoptosis—caspases and Bcl-2 family proteins.
Caspases
Caspases are a family of aspartate-specific cysteine proteases. Fourteen mammalian caspases have now been identified, which according to common classification schemes are divided into inflammatory/cytokine-processing caspases that includes 1, 5, and 11 and apoptosis-regulatory caspases that includes 2, 3, 6, 7, 8, 9, and 10 (Thornberry and Lazebnik, 1998). Caspases share a common four domain structure comprising p20 and p10 subunits separated by a linker domain flanked by aspartate residues to a prodomain of variable length. In general, the active conformation of the caspase is a (p20p10)2 tetramer, with the prodomain cleaved off during the activation step. Active caspases cleave after aspartic acid residues at P1, with substrate specificity between individual caspases dependent on amino acids in the P2–P4 position. The prodomain is long in initiator caspases (8, 9, and 10) and short in executioner caspases (3, 6, and 7). The intracellular localization of caspases is highly regulated in both zymogen and processed forms. Caspases 8 and 10 are typically cytoplasmic, caspases 2, 7, and 12 are associated with the microsomes and caspase-9 with the mitochondria (Krajewski et al, 1999; Mancini et al, 2000; Nakagawa et al, 2000; Zhivotovsky et al, 1999). A cell death-promoting stimulus typically activates an initiator caspase via a recruitment scaffold. In the case of caspases 8 and 10, this is thought to involve an induced proximity model in which death effector domains (DED) within the prodomain bind homologous regions on death receptor (DR) adaptor proteins such as Fas-associated death domain (FADD). On caspase-9, a caspase activation recruitment domain (CARD) is present, which mediates its recruitment and activation by apoptosis protease-activating factor 1 (APAF-1). Activated initiator caspases then process specific executioner caspases. The executioner caspases in turn cleave key structural and functional proteins within the cell such as actin (Mashima et al, 1997) and the inhibitor of caspase-activated DNase (ICAD) (Sakahira et al, 1998), as well as providing feedback loops for further processing of caspases (Thornberry and Lazebnik, 1998).
Bcl-2 Family Proteins
The Bcl-2 gene family comprises more than 20 different members that either positively or negatively regulate apoptosis (Cory and Adams, 2002; Liou et al, 2003). Bcl-2 family proteins are typically small (~20 to 30kDa) and are defined according to the presence of one or more homology domains (BH domains), which are typically α helices. These range from four in Bax and Bcl-2 to one (BH3) in Bid, Bim, and Bad (Cory and Adams, 2002). A transmembrane domain is also present within the molecular structure of about half of the Bcl-2 family. Our understanding of the mechanism by which Bcl-2 family proteins regulate apoptosis continues to evolve. In normal cells, the ‘multidomain’ proapoptotic Bcl-2 proteins like Bax reside as monomers in the cytoplasm or loosely associated with the mitochondrial outer membrane. Their function requires an activation event, such as binding of a BH3-only member (e.g., truncated Bid). Likewise, BH3-only proteins require multidomain Bax or Bak to induce apoptosis (Wei et al, 2001). Interaction of BH3-only proteins with multidomain members triggers structural changes, formation of homooligo-merized multimers, insertion, and mitochondrial outer membrane permeabilization (Danial and Korsmeyer, 2004). However, BH3-only proteins also promote apoptosis by neutralizing antiapoptotic Bcl-2 family proteins, and this appears to be the basis for their relative potencies. For example, Bim and Puma, among the most potent proapoptotic BH3-only members, avidly bind all antiapoptotic Bcl-2 family proteins, while weaker members like Bad and Noxa interact with only a narrow subset (Chen et al, 2005). The mechanistic basis for the release of apoptogenic molecules from the mitochondria is probably multifaceted. It appears to involve either formation of the mitochondrial permeability transition, a large multiprotein complex pore, or is a result of direct functions of Bcl-2 family proteins as ion channels (Cory and Adams, 2002; Liou et al, 2003). Indeed, Bax is capable of forming pores in lipid membranes and during apoptosis undergoes a conformational change in which it embeds into the outer mitochondrial membrane. Antiapoptotic members of the Bcl-2 family like Bcl-2 and Bcl-xl can oligomerize and thereby neutralize proapoptotic Bcl-2 protein function, but may also confer protection through more direct actions on mitochondrial membrane integrity, Ca2+ mobilization, and perhaps antioxidant properties (Cory and Adams, 2002; Polster and Fiskum, 2004). Both pro- and anti-apoptotic Bcl-2 family proteins are associated with ER function, with roles identified in Ca2+ homeostasis, ER-to-mitochondrial Ca2+ signaling, and ER stress-induced apoptosis (Demaurex and Distelhorst, 2003; He et al, 1997; Nutt et al, 2001; Scorrano et al, 2003).
Extrinsic Pathway
Apoptosis can be initiated after activation of surface-expressed death receptors of the tumor necrosis factor (TNF) superfamily (extrinsic pathway), or after disruption of intracellular organelle function (intrinsic pathway) (Danial and Korsmeyer, 2004). The extrinsic pathway is triggered by binding of small molecular weight ligands to one or more membrane-expressed death receptors. Ligands for these receptors include TNFα, Fas ligand, and TNF receptor apoptosis-inducing ligand (TRAIL). To date, there are at least six members of this family including TNFR1, Fas (CD95), DR4 (TRAIL receptor 1), and DR5 (TRAIL receptor 2). Decoy receptors also exist for TRAIL, which lack an intracellular domain and therefore function to inhibit extrinsic pathway signaling (Ashkenazi and Dixit, 1999). Activation follows binding of the ligand to its receptor and oligomerization of the receptor. In the case of Fas, the cell death signal is propagated inside the cell by recruitment of FADD and an initiator caspase (e.g., caspase 8 or 10) to the intracellular side of the plasma membrane, resulting in formation of a death-inducing signaling complex (DISC). In contrast, activation of TNFR1 leads to direct association with TNF receptor-associated death domain (TRADD). Additional molecules including receptor interacting protein (RIP) and TNFR-activating factor 2 (TRAF2) may next be recruited to this complex, which can then modulate the nuclear factor κB pathway (Ashkenazi and Dixit, 1999; Strasser et al, 2000). TNFR1-mediated apoptosis is thought to require internalization of the complex, dissociation of TNFR1, and recruitment of FADD and caspase 8/10 (Micheau and Tschopp, 2003).
Intrinsic Pathway
The intrinsic apoptosis pathway is initiated after cell stressors that perturb intracellular organelle function (Danial and Korsmeyer, 2004). This can include raised intracellular Ca2+, proapoptotic Bcl-2 protein activation, or reactive oxygen species (Orrenius et al, 2003; Polster and Fiskum, 2004). Cytochrome c release from the mitochondrial inter-membrane space serves as the trigger for the mitochondria-originated intrinsic pathway (Liu et al, 1996). Cytosolic cytochrome c binds APAF-1 in the presence of dATP and recruits an initiator caspase (caspase-9) (Li et al, 1997; Qin et al, 1999). This results in a functional ‘apoptosome’. Progression of this pathway can be interrupted by the inhibitors of apoptosis protein (IAP) family (Salvesen and Duckett, 2002), a molecular brake that is in turn alleviated by release of a second mitochondrial protein (Smac/DIABLO) that binds IAPs (Du et al, 2000; Verhagen et al, 2000).
The ER regulates protein synthesis and folding, protein trafficking, responses to stress, and intracellular Ca2+ levels, and is the second major site for intrinsic apoptosis pathway initiation (Demaurex and Distelhorst, 2003; Paschen, 2003; Rao et al, 2001). Endoplasmic reticulum stressors may trigger apoptosis via several mechanisms. Impairment of protein movement triggers the unfolded protein response (UPR) that activates a series of transcription factors and membrane-associated signaling molecules that culminate in either restoration of ER function or apoptosis (Matsuzawa et al, 2002; Yoneda et al, 2001). Inositol (1,4,5) tris phosphate/ryanodine receptor activation, or blockade of the sarcoplasmic/endoplasmic Ca2+ ATPase (SERCA), triggers Ca2+ disturbances and/or release that can trigger Ca2+-dependent protease activation and apoptosis (Nakagawa and Yuan, 2000). The ER is also intimately connected with the mitochondria both physically and biochemically whereby ER stress can lead directly to mitochondrial Ca2+ accumulation and dysfunction (Rizzuto et al, 1998; Scorrano et al, 2003).
Activation of Apoptotic Pathways by Seizures
Apoptosis and its biochemical machinery emerged as a focus of research on seizure-induced brain injury in the mid-1990s. In the earliest work, researchers detected in situ ‘apoptotic’ DNA fragmentation (detected by terminal deoxynucleotidyl dUTP nick end labelling; TUNEL) and DNA laddering in tissue samples from the rat brain after prolonged seizures (Filipkowski et al, 1994; Pollard et al, 1994). Cell phenotype analysis suggests neurons comprise >90% of such cells (Henshall et al, 2001b,c). The small numbers of cells that die after brief seizures also exhibit ‘apoptotic’ DNA fragmentation (Bengzon et al, 1997; Zhang et al, 1998b). However, in rat models relatively few such degenerated neurons exhibit classical apoptotic morphology (Fujikawa et al, 2000a,b; Henshall et al, 2000c, 2002b). These data suggest that the end-stage processes of cell demise after seizures, at least in rats, does not incorporate all features of apoptosis. However, as many as a third of degenerating, TUNEL-positive cells in the hippocampus of mice exhibit nuclear features of apoptosis after seizures (Shinoda et al, 2004a).
Extrinsic Pathway Activation After Seizures
A diagram highlighting key molecular regulators of the intrinsic and extrinsic pathways in the setting of seizure-induced neuronal death is shown in Figure 1. Of the three caspases associated with the extrinsic, death receptor linked pathway, evidence for caspase-2 and caspase-8 has been provided in seizure models. The presence and/or significance of caspase-10 in the brain is yet to be widely recognized. Caspases 2 and 8 are both constitutively expressed in adult brain and immunoblotting and pseudosubstrate assays have established that both caspases are activated very shortly after induction of seizures, suggesting they lie at the apex of the caspase cascade (Henshall et al, 2001b, c ). Studies have also confirmed that caspase-8 processing occurs during seizure-induced neuronal death in mice (Shinoda et al, 2004a).
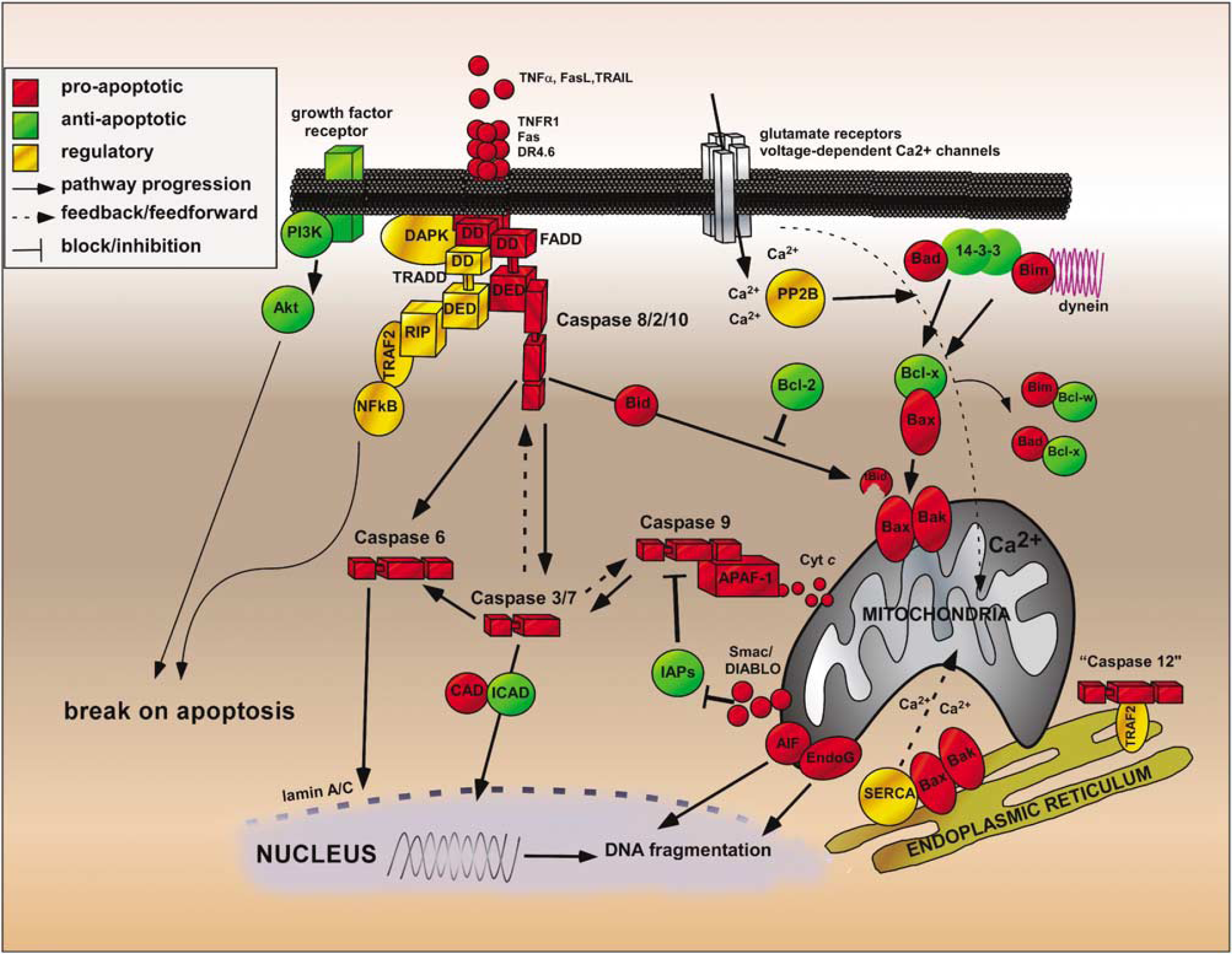
Major apoptosis pathways activated by seizures. Cell death is most likely triggered after prolonged glutamate receptor activation and/or release of death receptor ligands. Raised intracellular calcium (Ca2+) may directly trigger mitochondrial dysfunction or activate calcium phosphatases (e.g., PP2B, calcineurin), which releases Bad from 14-3-3 leading to Bax activation in the mitochondria. In turn, cytochrome c is released which binds Apaf-1 and triggers caspase-9 activation. Alternatively, death receptor activation leads to formation of a death-inducing signaling complex (DISC) and caspase-8 processing. Both pathways converge to executioner caspases, which target key substrates (e.g., inhibitor of caspase-activated DNase—ICAD) and structural proteins as well as providing feedback loops for further caspase activation. These pathways may be blocked at several steps premitochondrially by Akt and antiapoptotic Bcl-2 proteins or postmitochondrially by the inhibitors of apoptosis proteins (IAPs).
The mechanism of extrinsic pathway activation after seizures has not been fully delineated. Perhaps unexpectedly, several death receptors of the extrinsic apoptotic pathway including TNFR1, Fas, and DR4 appear to be constitutively present in the brain (Dorr et al, 2002; Henshall et al, 2001b; Shinoda et al, 2003b). Assuming currently accepted models, the trigger for caspase-8 activation after seizures would be DISC formation downstream of DR ligand binding to surface-expressed receptors like TNFR1 and Fas. Because caspase-8 can be cleaved within 40 mins of seizure induction (Henshall et al, 2001b, c ) this would only be possible if mediated via constitutive TNFα, FasL, and/or TRAIL. This is possible; both TNFα and FasL are present in the adult rat hippocampus before and after seizures (Shinoda et al, 2004b, 2003b). However, TRAIL is not present in normal or epileptic brain (Dorr et al, 2002; Martin-Villalba et al, 1999). Modest neuroprotection achieved with neutralizing antibodies to TNFα would appear to support this as the mechanism of extrinsic pathway activation after seizures (Shinoda et al, 2003b). Functional evidence for seizures forming a death receptor molecular scaffold has also been provided by the elution of DISC components from the rat hippocampus after seizures (Henshall et al, 2003; Shinoda et al, 2003b). All major intracellular components of death receptor signaling are constitutively expressed in the rat hippocampus including TRADD and FADD, the latter of which is overexpressed after seizures (Henshall et al, 2001b). Eluted DISCs from the rat hippocampus have been shown to contain TNFR1 and TRADD, while TRADD coelutes with FADD and caspase-8 fragments are found to be associated with FADD (Henshall et al, 2003; Shinoda et al, 2003b). Additional signaling components such as RIP and TRAF2 are present in the rodent brain and they are also recruited to the DISC (Shinoda et al, 2003b).
Progression of death receptor signaling may be blocked by at least two proteins: FLIP (FADD-like interleukin converting enzyme inhibitory protein) and the silencer of death domains (SODD). While FLIP has yet to be investigated, expression of SODD within hippocampal CA3 neurons declines very rapidly after seizures commensurate with its cleavage (Shinoda et al, 2003b). Thus, it appears seizures activate extrinsic pathway-linked caspase-8 downstream from death receptors and a DISC. In light of recent evidence that interchain proteolysis without dimerization may be sufficient to trigger caspase-8 activation (Murphy et al, 2004), alternative activation mechanisms should not be ruled out.
Intrinsic Pathway Activation After Seizures
At least two mechanisms likely contribute to engaging the intrinsic mitochondrial apoptotic pathway after seizures. Mitochondrial calcium loading has long been known to follow seizure activity (Griffiths et al, 1984). However, Bax particle clustering has also been visualized at the outer membrane of the mitochondria within 2 h of a seizure (Henshall et al, 2002a), and this coincides with the timing of cytochrome c release (Henshall et al, 2000a). A third mechanism involving calpain-mediated release of apoptosis-inducing factor (AIF) might also be particularly relevant (Polster et al, 2004). The released cytochrome c has been shown to bind APAF-1 in the hippocampus after seizures (Henshall et al, 2001a), followed by caspase-9 processing and increased proteolytic (LEHDase) activity (Henshall et al, 2001a, b ; Li et al, 2004).
There is evidence that the IAP family may be important in regulating intrinsic pathway progression during seizure-induced neuronal death. Loss of XIAP has been shown in degenerating neurons after seizures (Korhonen et al, 2001). While seizures in rats do not decrease expression of cIAP2, they trigger its neutralization by binding to Bcl10 (Shinoda et al, 2003b).
While the mitochondria have been the major focus of intrinsic pathway study, there is evidence that the ER is important in seizure-induced neuronal death. Several epilepsy-relevant ER stressors are known including oxidative stress, disruption of intracellular Ca2+ homeostasis, and TNFR1 activation. A general ER stress response occurs after seizures and TRAF2 and apoptosis signal-regulating kinase 1, which can be ER-associated proteins, are activated (Jang et al, 2004; Shinoda et al, 2003b). Second, 150-kDa oxygen-regulated protein, an ER-specific molecular chaperone, is protective against seizure-induced neuronal death (Kitao et al, 2001). Third, ER Ca2+ store inhibitors reduce seizure-induced cell death in vitro (Pelletier et al, 1999). Finally, ceramide, which can promote apoptosis via an ER-facilitated Ca2+ release mechanism (Pinton et al, 2001), is generated during seizures (Mikati et al, 2003). However, studies have yet to specifically address whether ER stress is a primary trigger for seizure-induced activation of the apoptosis machinery.
While the temporal ordering of intrinsic and extrinsic pathways after seizures has yet to be undertaken in a single study, findings to date suggest the extrinsic pathway precedes the intrinsic. Caspase-8 cleavage precedes the mitochondrial dysfunction and caspase-9 activation after seizures (Henshall et al, 2001a, b ). Additional indirect support for caspase-8 lying upstream includes: (a) caspase-8 inhibition confers somewhat greater protection on the hippocampus than does caspase-9 inhibition (Henshall et al, 2001a, b ); (b) caspase-8 inhibition reduces cytochrome c release and caspase-9 cleavage (Henshall et al, 2001b); and (c) caspase-9 inhibition has little effect on caspase-3 activity while caspase-8 inhibition reduces both caspase-3 and caspase-9-like protease activity (Henshall et al, 2001a, b).
Executioner Caspase Activation After Seizures
Caspase-3 is among the most studied regulators of apoptosis in the setting of seizure-induced neuronal death. Induction of caspase-3 mRNA and protein occurs within the hippocampus and extrahippocampal regions after seizures (Akbar et al, 2001, 2003; Ferrer et al, 2000; Gillardon et al, 1997; Narkilahti et al, 2003b). Caspase-3 activation in the form of increased DEVDase activity and/or detection of cleaved caspase-3 has also been described in the hippocampus after seizures and extrahippocampal regions (Araki et al, 2002; Faherty et al, 1999; Gervais et al, 1999; Gillardon et al, 1997; Henshall et al, 2000a; Kondratyev and Gale, 2000; Narkilahti et al, 2003b; Tan et al, 2002; Troy et al, 2002; Weise et al, 2005). Further indirect evidence for activated caspase-3 after seizures includes degradation of ICAD and spectrin breakdown products (Kondratyev and Gale, 2004; Kondratyev et al, 2002). However, some studies have not reported any processing of caspase-3 after seizures (Ananth et al, 2001; Fujikawa et al, 2002).
Caspase-7 is expressed in the adult brain, albeit at low levels (Henshall et al, 2002b), but activation does not appear to occur after seizures in rats, at least when evoked by intraamygdala kainate (Henshall et al, 2002b). However, elevated caspase-7 expression after seizures in both the hippocampus and temporal cortex suggests that there may yet be roles for caspase-7 (Henshall et al, 2002b). The explanation for the lack of caspase-7 processing is unclear, but may reside with the cell type in which it is constitutively expressed or its subcellular location, neither of which have been well described. Ongoing work in our lab suggests that caspase-7 processing occurs after seizures in mice (unpublished observation).
While caspase-3 is known to have the largest number of substrates, several lines of evidence implicate caspase-6 as an important mediator of neuronal death. First, caspase-6 may be constitutively expressed at higher levels in the brain than caspase-3 and −7 (Henshall et al, 2000b, 2002b; LeBlanc et al, 1999; Narkilahti and Pitkanen, 2005). Second, caspase-6 may lie upstream of caspase-3 in the setting of neuronal death (Allsopp et al, 2000; Henshall et al, 2002b). Third, neurons may be particularly vulnerable to caspase-6, perhaps because IAPs do not bind active caspase-6 (Roy et al, 1997). This may explain in part why injected active caspase-6 is more effective at triggering apoptosis in human neurons than is caspase-3 or −7 (Zhang et al, 2000). Several studies have now reported hippocampal caspase-6 activation after seizures in rat models (Henshall et al, 2002b; Narkilahti and Pitkanen, 2005; Troy et al, 2002). Of particular interest, active caspase-6 strongly stains dendritic elements within the seizure-damaged hippocampus, supporting a role for caspase-6 in restructuring as well as cell death. The role of caspase-6 has yet to be addressed in a mouse seizure model.
A diagram highlighting the mechanism of caspase activation in the setting of seizure-induced neuronal death is shown in Figure 2.
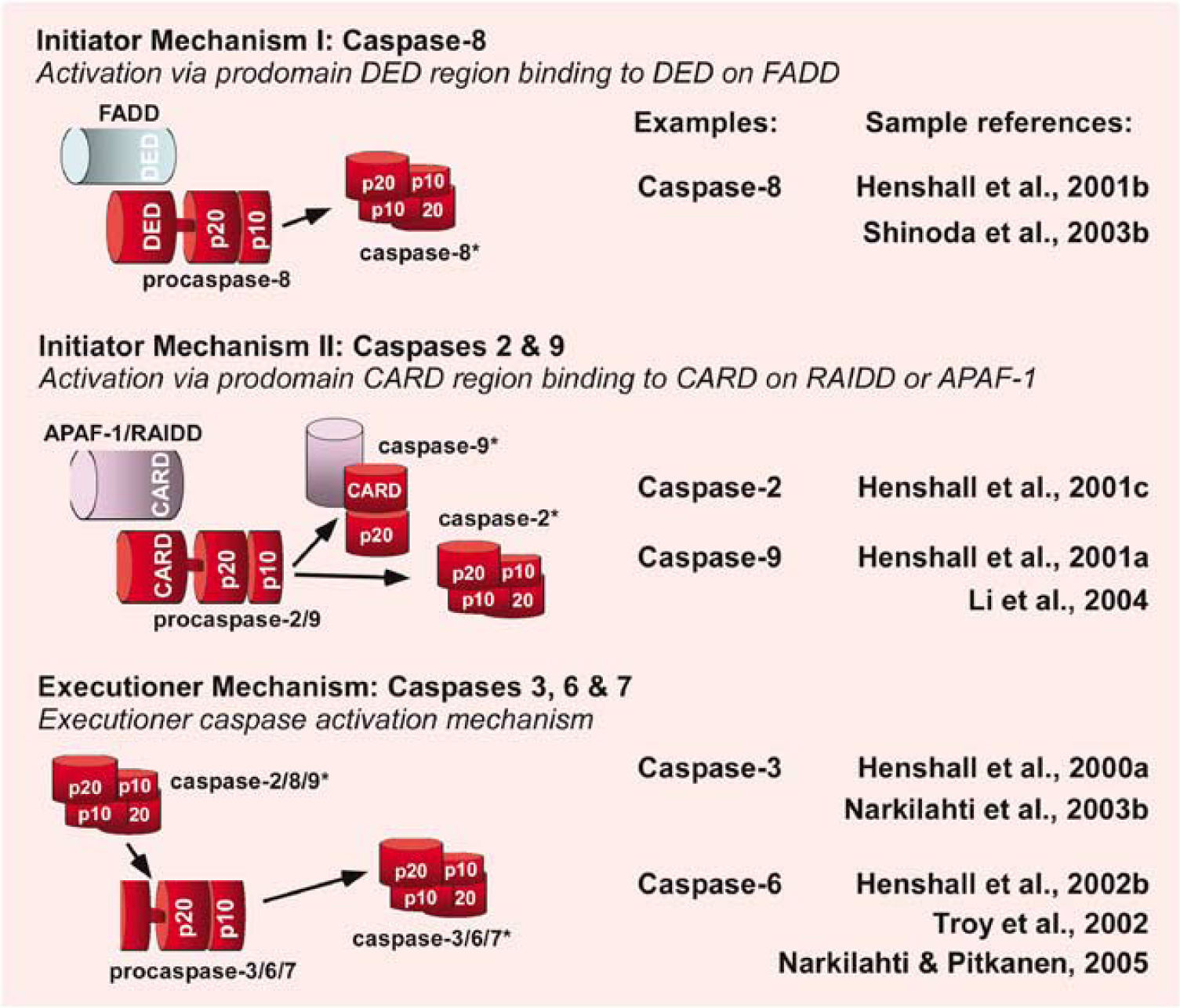
Caspase activation mechanisms. Caspases are proteolyzed from inactive zymogens to their active form; typically a (p20:p10)2 tetramer. They function either as initiators or executioners. Initiator caspases have long prodomains (~20 kDa), which function in their activation mechanism by enabling interaction with scaffold components: for example, caspase activation recruitment domains (CARD) on apoptosis protease-activating factor 1 (Apaf-1), death effector domains (DED) on Fas-associated death domain (FADD). Executioners have short prodomains (~5 kDa) that are cleaved off by initiator caspases. This figure highlights these mechanisms and the evidence for each in the setting of seizure-induced neuronal death.
Bcl-2 Family Proteins in Seizure-Induced Neuronal Death
Bcl-2 family proteins, like caspases, are involved in regulating seizure-induced neuronal death. Of the proapoptotic regulators, the major focus has been on the expression of Bax, which typically increases after seizures (Ananth et al, 2001; Gillardon et al, 1995; Korhonen et al, 2003; Lopez et al, 1999). Bcl-xs is also upregulated by seizures (Kondratyev et al, 2001), a response prevented by nondamaging electroshock exposure. Of the BH3-only proteins studied, changes of Bid expression have not been noted, although Bid is cleaved to its active, p15 conformation after seizures (Henshall et al, 2001b; Schindler et al, 2004). Increased expression of Bim but not Bad is seen after seizures (Henshall et al, 2002a; Schindler et al, 2004; Shinoda et al, 2004b). In this setting, the site of their function is highly specific, with Bad cytoplasmic while Bax and Bid are found in both cytoplasmic and mitochondrial compartments (Schindler et al, 2004). However, mixed reports on expressional regulation of proapoptotic Bcl-2 family proteins after seizures suggest the extent of involvement of individual members may be model dependent (Ferrer et al, 2002; Korhonen et al, 2003; Puig and Ferrer, 2002; Schindler et al, 2004).
Altered expression of antiapoptotic proteins in this family has been showed after seizures in some but not all cases for Bcl-2, Bcl-xl, and Bcl-w (Ananth et al, 2001; Graham et al, 1996; Henshall et al, 2002a, 2001d; Schindler et al, 2004). As with proapoptotic members, subcellular location during seizures is specific. For example, Bcl-w appears to be mainly cytoplasmic in rat hippocampus while Bcl-2 is largely mitochondrial (Schindler et al, 2004). Of additional note, antiapoptotic Akt has been shown to block neuronal death in the cortex after seizures, likely through suppressing BH3-only Bad or Bim expression and/or function (Henshall et al, 2002a; Shinoda et al, 2004b).
Because many Bcl-2 family proteins are constitutively expressed in the brain, altered protein levels and even subcellular location are probably insufficient to determine ‘activation’. This requires assessment of changes in protein–protein interaction. Seizures cause such changes; Bad dissociates from its sequestered site with 14-3-3 and binds antiapoptotic Bcl-xl after seizures in vivo and in vitro (Henshall et al, 2002a; Meller et al, 2003). Bid in its truncated form also appears to bind 14-3-3 (Shinoda et al, 2003a). Bim normally resides sequestered to dynein but after seizures its interaction increases with Bcl-w (Shinoda et al, 2004b). Seizures also trigger increased binding of Bcl10 to cIAP2 (Shinoda et al, 2003b). In addition, functional inactivation of Bcl-2 has been showed after kainic acid-induced seizures in rats based on elevated phosphorylation levels (Korhonen et al, 2003). Thus, seizures induce a proapoptotic Bcl-2 family protein activation cascade that coincides, and in some cases precedes, intrinsic pathway activation. A diagram highlighting the various mechanisms of Bcl-2 protein function and evidence for their occurrence during seizure-induced neuronal death is shown in Figure 3.
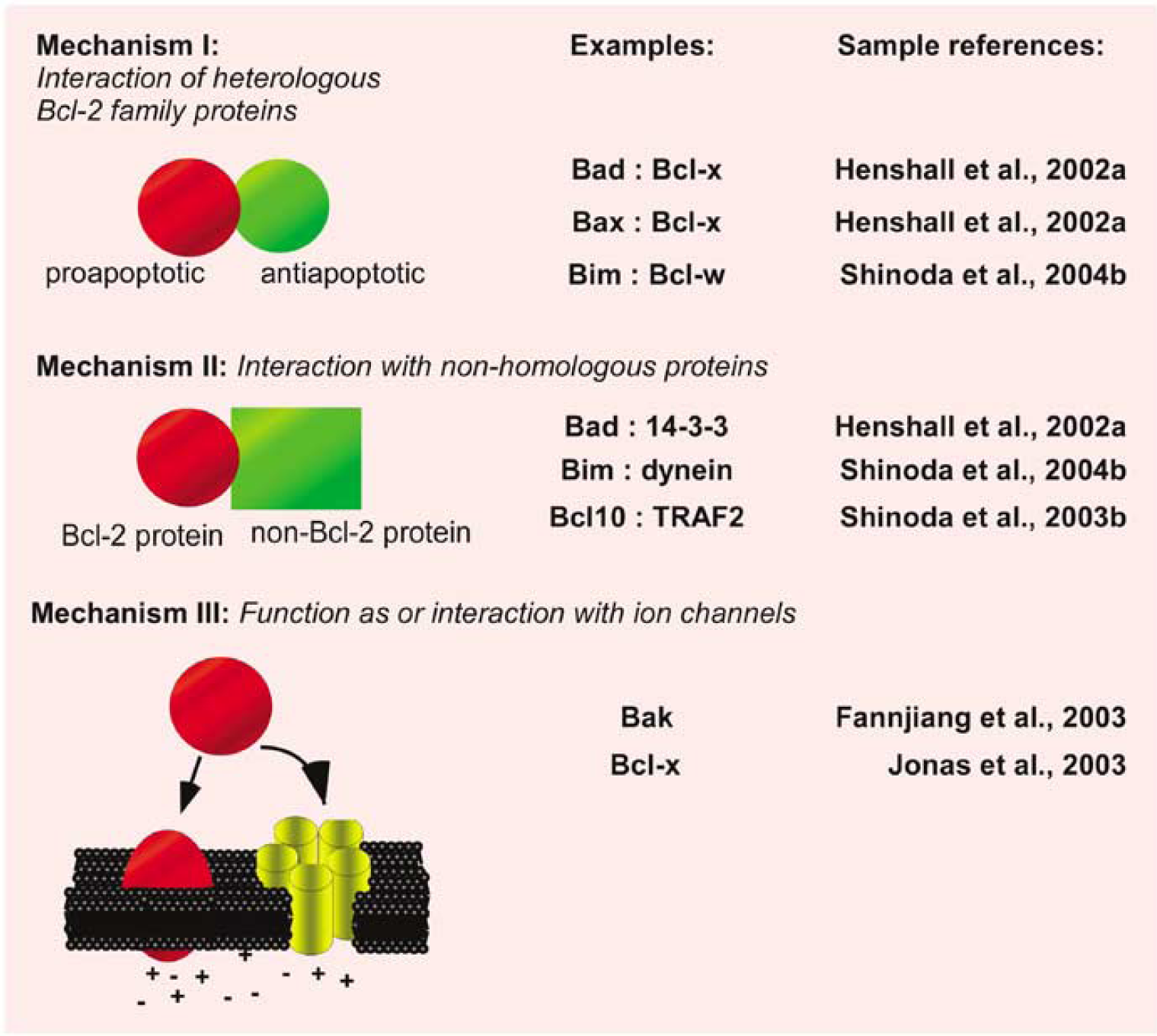
Mechanisms of Bcl-2 protein function. At least three mechanisms have been proposed to explain how Bcl-2 family proteins regulate apoptosis. This figure highlights these mechanisms and provides examples reported in the setting of seizure-induced neuronal death or relevant to epilepsy.
Additional Apoptosis Regulators
While most work on apoptosis in epilepsy has focused on caspases and Bcl-2 family proteins, genes in several other families known to regulate apoptosis may influence seizure-induced neuronal death. Among the most studied is the p53 tumor suppressor protein, which has been shown to be induced in response to seizures in several models and which is thought to promote apoptosis by induction of proteins including Bax (Liou et al, 2003). Additional modulators and pathways known to influence seizure-induced neuronal damage and epileptogenesis include the mitogen-activated protein kinase cascades (Liou et al, 2003; Shinoda et al, 2003b), cAMP response-element binding protein (Moore et al, 1996), death-associated protein (DAP) kinase (Henshall et al, 2003, 2004), Nuclear Factor κB (Albensi, 2001), cathepsins (Houseweart et al, 2003), and AIF (Cheung et al, 2005). Other cell death modulators that may have roles in seizure-induced neuronal death include inflammatory cytokines and calpains (Goll et al, 2003; Jankowsky and Patterson, 2001).
Therapeutic Insights
A range of interventions have been investigated in the setting of seizure-induced neuronal death that target apoptosis signaling pathways.
Death Receptors and Extrinsic Pathway Caspases
In vivo inhibition of caspase-8 using the pseudosubstrate inhibitor IETDfmk has been shown to confer potent protection against hippocampal damage after seizures, reducing neuronal loss by ~50% (Henshall et al, 2001b). However, studies in which (presumably) upstream death receptor activation was prevented after seizures by neutralizing antibodies to death receptor ligands have produced less consistent neuroprotection (Shinoda et al, 2003b) and TNFR1 knockout mice exhibit increased damage in models of excitotoxicty (Gary et al, 1998). This disparity may lie with the pleiotropic actions of cytokines such as TNFα and death receptors, which may influence hippocampal excitability and serve pro- and antiapoptotic roles (Shohami et al, 1999; Micheau and Tschopp, 2003). Because death receptors and/or their intracellular adaptor proteins are expressed in human epilepsy brain (Dorr et al, 2002; Henshall et al, 2004), targeting them will require careful consideration of their duplicitous functions.
Caspase-2 inhibition does not appear to be neuroprotective after seizures despite biochemical evidence for its activation (Henshall et al, 2001c). This fits with other reports on the (lack of) functional significance of caspase-2 (Bergeron et al, 1998), including in neurons (O'Reilly et al, 2002). The function and specific substrates for caspase-2 remain rather obscure and concurrent caspase-8 activation may render caspase-2 function redundant (Henshall et al, 2001c). However, caspase-2 overexpression during the epileptogenesis phase (A Pitkanen, personal communication) suggests that more work on this caspase is required.
Intrinsic Pathway and Executioner Caspases
Caspase-3 inhibitors have proven efficacious against neuronal loss in several models of brain injury known to trigger epileptogenesis, including traumatic brain injury (Liou et al, 2003), and caspase-3 has received the most attention for preventing caspase-dependent neuronal death after SE. Several studies have reported neuroprotection by pharmacological inhibition of caspase-3 with DEVDfmk within brain structures such as the hippocampus and cortex of the temporal lobe (Henshall et al, 2000a; Kondratyev and Gale, 2000; Narkilahti et al, 2003a). A trend to neuroprotection was also apparent in the study by Ebert et al (2002) in some affected regions. Limitations with the specificity of DEVDfmk as a caspase inhibitor in vivo are now understood, which may explain the protection afforded by this drug against seizure damage in brain regions in which caspase-3 activation is not apparent (Henshall et al, 2000a; Kondratyev and Gale, 2000). Further, DEVDfmk could block cell death via calpain (Knoblach et al, 2004). While DEVDfmk would likely block caspase-7 also, activation of this caspase was not detected after seizures in the rat (Henshall et al, 2002b). Alternative approaches to these pseudosubstrate inhibitors have also shown efficacy against seizure-induced neuronal death. For example, the baculovirus-derived broad-spectrum caspase inhibitor p35 reduces kainate-induced hippocampal damage in rats (Roy et al, 2002) and p35 transgenic mice are resistant to seizure damage (Viswanath et al, 2000).
Three studies have reported a significant neuroprotective effect of the caspase-9 inhibitor LEHDfmk on hippocampal damage after seizures (Henshall et al, 2001a; Li et al, 2004; Narkilahti et al, 2003a). However, in the only direct comparison, LEHDfmk was not as protective as DEVDfmk (Narkilahti et al, 2003a). While the explanation for this may lie with dosage or brain penetration, it may also point toward converging pathways and redundancy within the caspase cascade that is activated after seizures. Indeed, not all caspase inhibitors are neuroprotective after seizures (Henshall et al, 2001c; Tomioka et al, 2002).
Bcl-2 Family Proteins
Less is known of the neuroprotective potential of targeting Bcl-2 proteins because pharmacological inhibitors for use in vivo are not readily available. Inhibition of calcineurin, which may be responsible for Bad activation, confers protection on the hippocampus after seizures (Henshall et al, 2002a; Moriwaki et al, 1998). Blocking activation of FKHR and FKHRL-1 of the Forkhead box, class O subfamily of Forkhead transcriptional regulators of Bim expression with a phosphatase inhibitor is also protective against seizure damage (Shinoda et al, 2004b). In vitro, cell injury is reduced after seizures by the use of antisense oligonucleotides to Bim (Shinoda et al, 2004b). Overexpression of Bcl-2 using a viral vector approach has also yielded promising results, although function still appears to be compromised in surviving neurons (McLaughlin et al, 2000). Last, knockout of Bak has been shown to exacerbate seizures and damage in mice (Fannjiang et al, 2003). Further research, particularly attempts to target more than one protein/gene in a pathway or where genetic approaches are taken is now required to establish the most efficacious strategies.
Apoptosis Pathways and Epileptogenesis
Evidence has emerged that the activity of apoptotic pathways continues well beyond the period of major cell death after the initial precipitating injury (e.g., SE) into the time of epileptogenesis. Pitkanen's group has revealed executioner caspases-3 and −6 continue to be active during epileptogenesis (Narkilahti et al, 2003b; Narkilahti and Pitkanen, 2005). In particular, caspase-3 cleavage was most prominent 7 days after evoked, damaging seizures, during the period of epileptogenesis (Narkilahti et al, 2003b). Blocking caspase-3 function during this week did not ultimately lead to an antiepileptogenic effect however (Narkilahti et al, 2003b). Nevertheless, preliminary results in that study suggested that DEVDfmk might have reduced the numbers of rats with epilepsy and mossy fiber sprouting (Narkilahti et al, 2003b).
Caspase-6 activation is detected in the apical dendrites of the hippocampal pyramidal neurons during epileptogenesis and in epileptic rats (Narkilahti and Pitkanen, 2005), suggesting targeting caspase-6 may hold promise for influencing epileptogenesis. While the substrates of these activated caspases are not known, likely candidates include structural proteins or ion channels (Strasser et al, 2000). The identification of in vivo caspase substrates during epileptogenesis would no doubt provide a novel set of therapeutic targets. At present, it is unknown whether Bcl-2 family proteins are involved in epileptogenesis but because they regulate proximal apoptosis signaling events, their role should be addressed.
Apoptosis Pathways in Epilepsy Patients
There is considerable evidence for the regulation of apoptosis signaling pathways in human TLE. Analyses of the hippocampus and extra-hippocampal temporal cortex removed during surgery for intractable epilepsy has revealed modulation of caspases and Bcl-2 family proteins (see Table 1). Expression and cleavage of caspase-3 is significantly elevated in the temporal cortex of epilepsy patients compared with control (Henshall et al, 2000b). Caspase-1, an inflammatory caspase, is also activated in the epilepsy brain (Henshall et al, 2000b). No hippocampal data are yet available for either caspase. These data suggest that caspase activation is a feature of the chronic disease process in epilepsy. Because experimental modeling has showed that caspase inhibition reduces seizure-induced neuronal death, it is tempting to speculate that caspases may be contributing to damage progression in refractory epilepsy patients (Briellmann et al, 2002; Fuerst et al, 2003; Kalviainen et al, 1998; Tasch et al, 1999).
Findings on caspases and Bcl-2 family proteins in human TLE
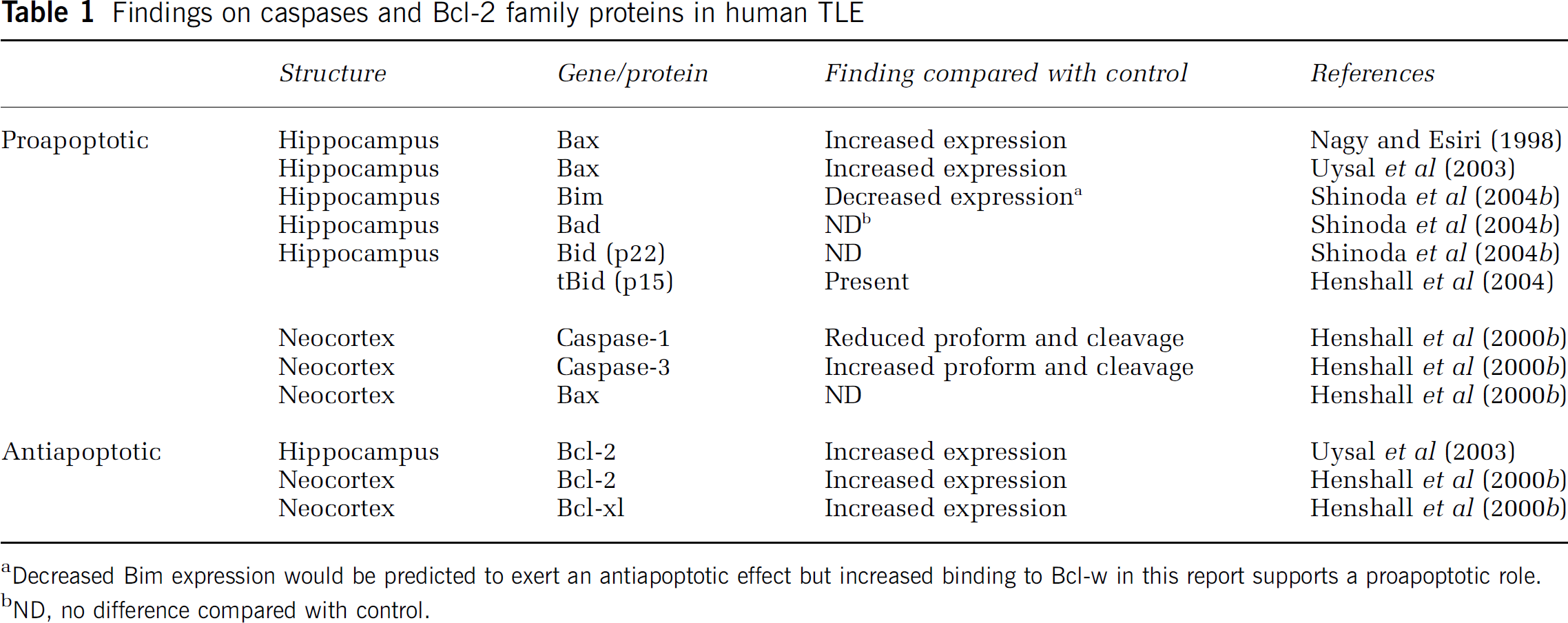
Decreased Bim expression would be predicted to exert an antiapoptotic effect but increased binding to Bcl-w in this report supports a proapoptotic role.
ND, no difference compared with control.
There are several descriptions of altered Bcl-2 family protein expression in the human epilepsy brain (Table 1). Qualitative descriptions of increased Bax expression have been reported for the hippocampus (Nagy and Esiri, 1998; Uysal et al, 2003), but not the neocortex (Henshall et al, 2000b). Bid cleavage is detected in the hippocampus (Henshall et al, 2004) but expression of Bid (p22) and Bad is unaltered (Shinoda et al, 2004b). Surprisingly, expression of Bim is significantly lower in patient brain than control (Shinoda et al, 2004b). Qualitative (Uysal et al, 2003) and quantitative (Henshall et al, 2000b) evidence for increased Bcl-2 expression has been provided, and Bcl-xl is also elevated in the temporal neocortex (Henshall et al, 2000b). Insight into the functional significance of these Bcl-2 family changes of course remains difficult but the increased interaction of Bim with antiapoptotic Bcl-w in human epilepsy brain could be interpreted as a cell death-promoting conformation (Shinoda et al, 2004b).
These data establish apoptotic pathways are active in patients with epilepsy. They also suggest that there is an ongoing balance of adaptive protective responses, but that BH3-only Bcl-2 family proteins may overcome these changes and may promote progressive damage via a caspase-dependent mechanism. Insight has also emerged on the role of other apoptosis-regulatory genes in the human TLE brain. Antiapoptotic Akt is phosphorylated (activated) in the human TLE brain compared with control (Shinoda et al, 2004b). In the same samples, elevated phosphorylation of the Akt target FKHR was found suggesting Akt may be functionally active in epilepsy brain (Shinoda et al, 2004b). While mitochondrial pathways have been the major focus so far, significant elevations in ER levels of DAP kinase and its E3 ligase DAP kinase interacting protein 1 are found in the human epilepsy brain (Henshall et al, 2004).
Issues and Confusions
As detailed above, the therapeutic potential of targeting apoptosis to improve epilepsy treatment falls into either approaches to neuroprotection or antiepileptogenesis. But while correlations between neuronal loss, epilepsy phenotype, and cognitive performance deficits have been shown (Kotloski et al, 2002; Zhang et al, 2002), epilepsy can arise after brain manipulations that do not result in appreciable neuronal loss (Zhang et al, 2002) or despite profound neuroprotection (Brandt et al, 2003). Furthermore, the extent to which caspase/Bcl-2-dependent pathways contribute to seizure-induced neuronal death and epileptogenesis requires further assessment. While consensus is emerging with regard to the neuroprotective benefits of targeting caspases after seizures, a consistent profile of their activation and functional importance has yet to emerge.
It is fair to say that not all approaches to studying apoptosis pathways in epilepsy carry equal interpretive value. Western blotting can provide direct and quantitative evidence for the formation of a processed and active caspase but lacks spatial resolution and its sensitivity may be lower than that of pseudosubstrate assays (DEVDase) (Narkilahti et al, 2003b). However, caspase assays lack specificity, particularly when concurrent caspases are activated or where calpain activity is likely (Knoblach et al, 2004). Immunohistochemistry does not lend itself well to quantitative analysis, and zymogen forms of caspases are rarely detected despite constitutive expression, while ‘active’ caspase antibodies may reveal staining, which only loosely correlates with immunoblotting results (Narkilahti et al, 2003b; Narkilahti and Pitkanen, 2005).
As the only study to date to have examined how caspase inhibition influences epileptogenesis, Narkilhati et al concluded DEVDfmk was not ultimately antiepileptogenic. However, some of the early results in this study were promising (Narkilahti et al, 2003a). Fine-tuning treatment regimes, targeting additional caspases activated during epileptogenesis, or a more complete approach (e.g., gene knockout) might yield definitive results. The availability of mouse models in which apoptotic death may be prominent offer novel paradigms for study and the opportunity for exploiting transgenic and knockout mice for the genes of interest.
Novel Functions for Apoptosis Regulators Relevant to Epileptogenesis
Several new lines of research are set to increase the focus of apoptosis research in epilepsy. As highlighted above, executioner caspases appear to be involved in nonlethal cell remodeling during epileptogenesis (Narkilahti et al, 2003b; Narkilahti and Pitkanen, 2005). The extrinsic pathway may also perform nonlethal roles of significance to epileptogenesis. Fas and FADD have been reported to perform roles in cell proliferation (Desbarats and Newell, 2000; Zhang et al, 1998a), while more recent data show activation of the Fas receptor can drive neurite extension through induction of p35 (Desbarats et al, 2003). Likewise, a paradigm shift is emerging in how we view the constitutive function of Bcl-2 family proteins and caspases. Traditionally, these proteins were considered to reside inactive until the arrival of a signal-to-kill but a broader role for several has emerged. Proapoptotic Bad has been shown to be part of the glycolysis machinery (Danial et al, 2003), its dephosphorylation and proapoptotic function emerging in response to glucose deprivation. In a detailed investigation of the role of the Bcl-2 family member Bak during postnatal development, Fannjiang et al (2003) showed that Bak−/− mice had an increase in the frequency of miniature excitatory postsynaptic currents and a decrease in the frequency of miniature inhibitory postsynaptic currents. The investigators concluded that Bak inhibited endogenous neuronal excitability and that the data were consistent with a change in the kinetics of neurotransmitter release. This observation is not isolated, and a role for Bcl-xl in synaptic function has also been proposed (Jonas et al, 2003). Other functions of Bcl-2 proteins may also influence neuronal excitability. In particular, the ability of Bcl-2 to modulate intracellular calcium levels and stores such as those in the ER, properties likely to impact neuronal function (Murphy et al, 1996). In light of these observations and taking the example of Bcl-xl influencing synaptic transmission, we might reevaluate the functional significance of the elevated Bcl-xl levels observed in the temporal cortex of patients with intractable epilepsy (Henshall et al, 2000b). If Bcl-2 family proteins regulate neuronal excitability then their involvement in seizures themselves and not just their sequelae becomes a possibility. Thus, in the future we may look at altered expression of apoptosis-regulatory proteins in epilepsy not just from the perspective of their influence on cell death and survival, but as underlying factors that will influence seizure thresholds and seizure frequency in epilepsy.
Considerations with Targeting Apoptosis Pathways in Patients
Defining roles for Bcl-2 and caspase proteins in intracellular ion homeostasis, neuronal excitability, and network remodeling brings questions of the potential side effects of manipulating these systems to treat epilepsy. When these pathways were considered ‘inactive’ until the arrival of a death signal, the likely side effects of blocking their function were minimal. However, as the list of nondeath functions for these two families expands so does the likelihood that intervention in these pathways, particularly the sort of protracted treatments likely necessary in an antiepileptogenesis regime, will lead to complications with normal brain function. For example, caspase inhibitors might impair certain types of memory acquisition (Dash et al, 2000). However, these effects are balanced against the benefits of targeting these pathways for neuroprotection that could equally serve to preserve such functions (Kotloski et al, 2002).
Another consideration is that both provoked (Parent et al, 1997) and spontaneous (Cha et al, 2004) seizures trigger neurogenesis. While the functional consequences of neurogenesis after seizures remain incompletely understood, significant numbers of new neurons die in the subsequent weeks by a caspase-dependent apoptotic pathway (Ekdahl et al, 2001). Accordingly, preventing caspase activation will increase numbers of immature neurons within the seizure-damaged brain.
Last, reports suggest that blocking apoptotic pathways may either result in induction of a necrosis program (Hartmann et al, 2001), or alternatively, leave behind nonfunctional neurons that provide no benefit to the damaged brain (Roy et al, 2002). Further studies are now required to evaluate the long-term effects of modulating these pathways and of the relative merits of neuroprotection mediated by targeting Bcl-2 family proteins or caspases.
Concluding Remarks
This review highlights current understanding of the molecular pathways of apoptosis as they relate to neuronal death after seizures and epileptogenesis. The caspases and Bcl-2 family proteins likely play a role in both processes, and modulation of these pathways in human TLE is now recognized. Questions still remain as to the functional significance of individual genes and the question of whether seizure-damaged neurons are undergoing ‘apoptosis’ remains controversial in epilepsy because the molecular machinery of apoptosis can be activated in damaged brain cells that do not subsequently display an apoptotic morphology during degeneration. Research on apoptosis pathways has evolved considerably in recent years as it encompasses developments in the field and reevaluates the importance of these for epilepsy. Principal interest remains on the potential for neuroprotection in epilepsy by targeting apoptosis as supported by the efficacy of caspase inhibitors and genetic manipulation of proapoptotic factors after seizures. However, data showing apoptosis-regulatory molecules are associated with restructuring sites in the hippocampus long after an initial precipitating injury have major implications for our understanding of the pathogenesis of epilepsy and approaches to antiepileptogenesis. Lastly, the emergence of functions for Bcl-2 family proteins that impact more broadly on neurophysiology has implications for network excitability in animal models and epilepsy patients. The clinical relevance of work on apoptosis pathways in epilepsy is set to expand from interest in their neuroprotective potential to encompass their role in remodeling and brain function that may yield neuroprotective and antiepileptogenic treatments for patients.